
Rescue of Low-Yield DNA Samples for Next-Generation Sequencing Using Vacuum Centrifugal Concentration in a Clinical Workflow

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Vacuum Centrifugation
2.3. Data Handling and Statistical Analysis
2.4. DNA Extraction
2.5. DNA Library Preparation and Sequencing
3. Results
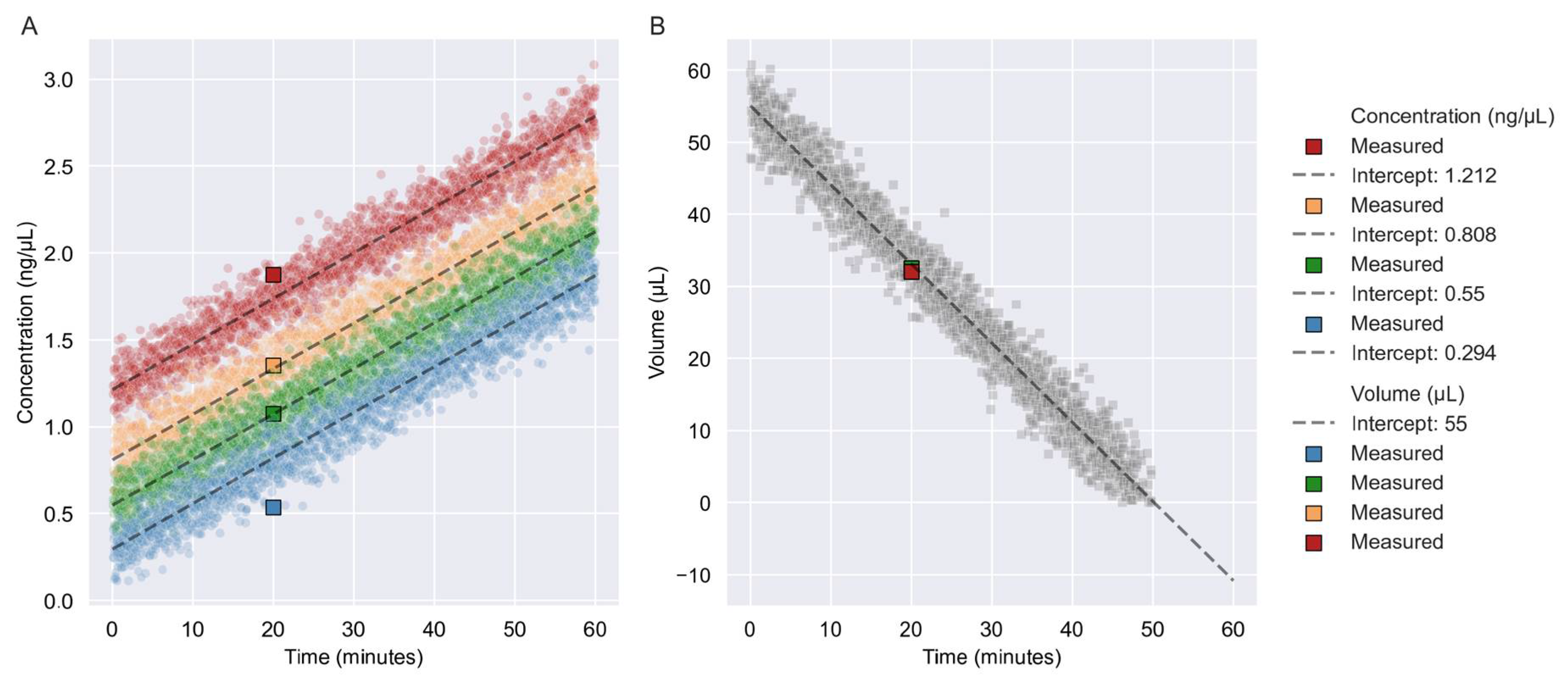
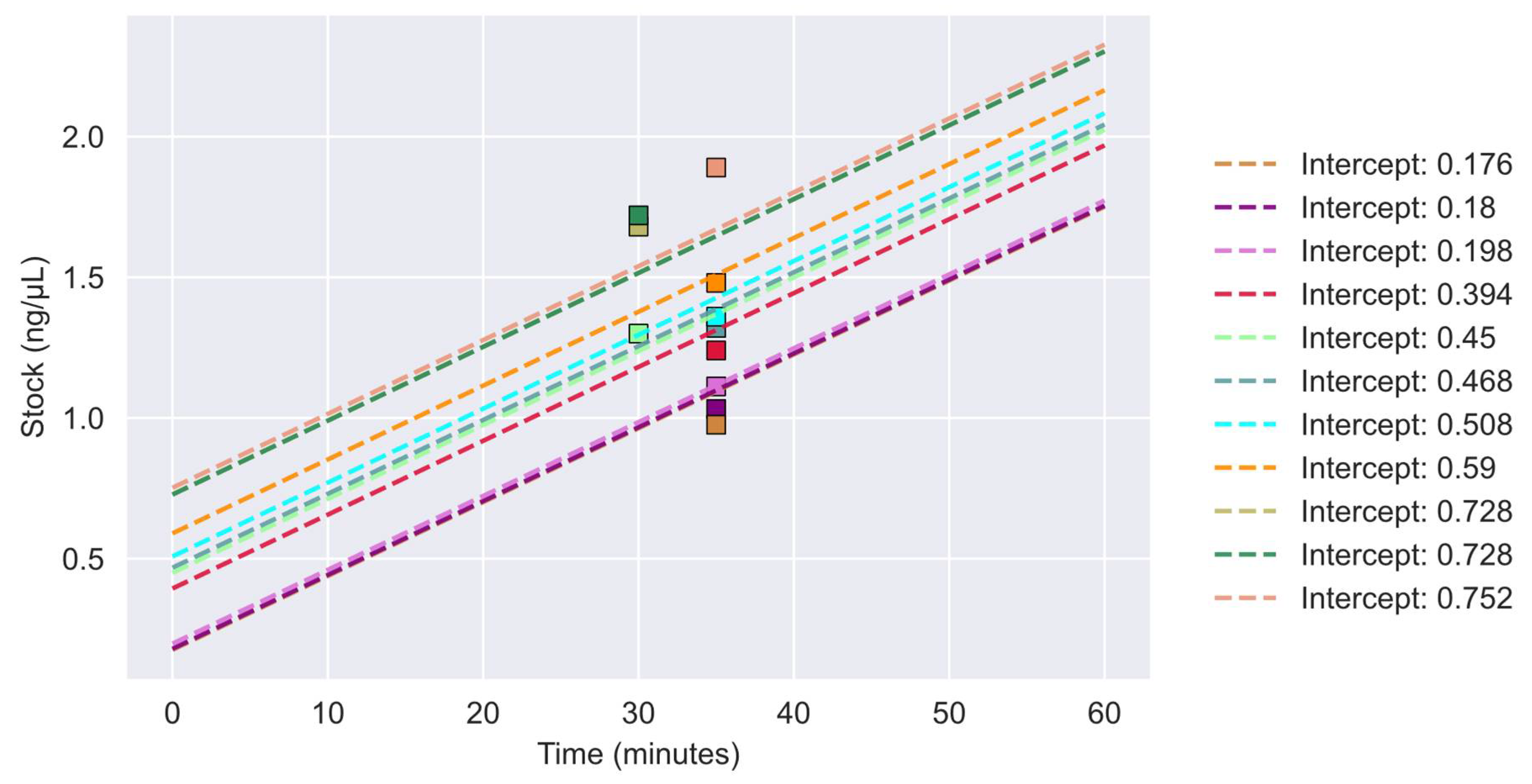
3.1. Prediction of Concentration and Volume
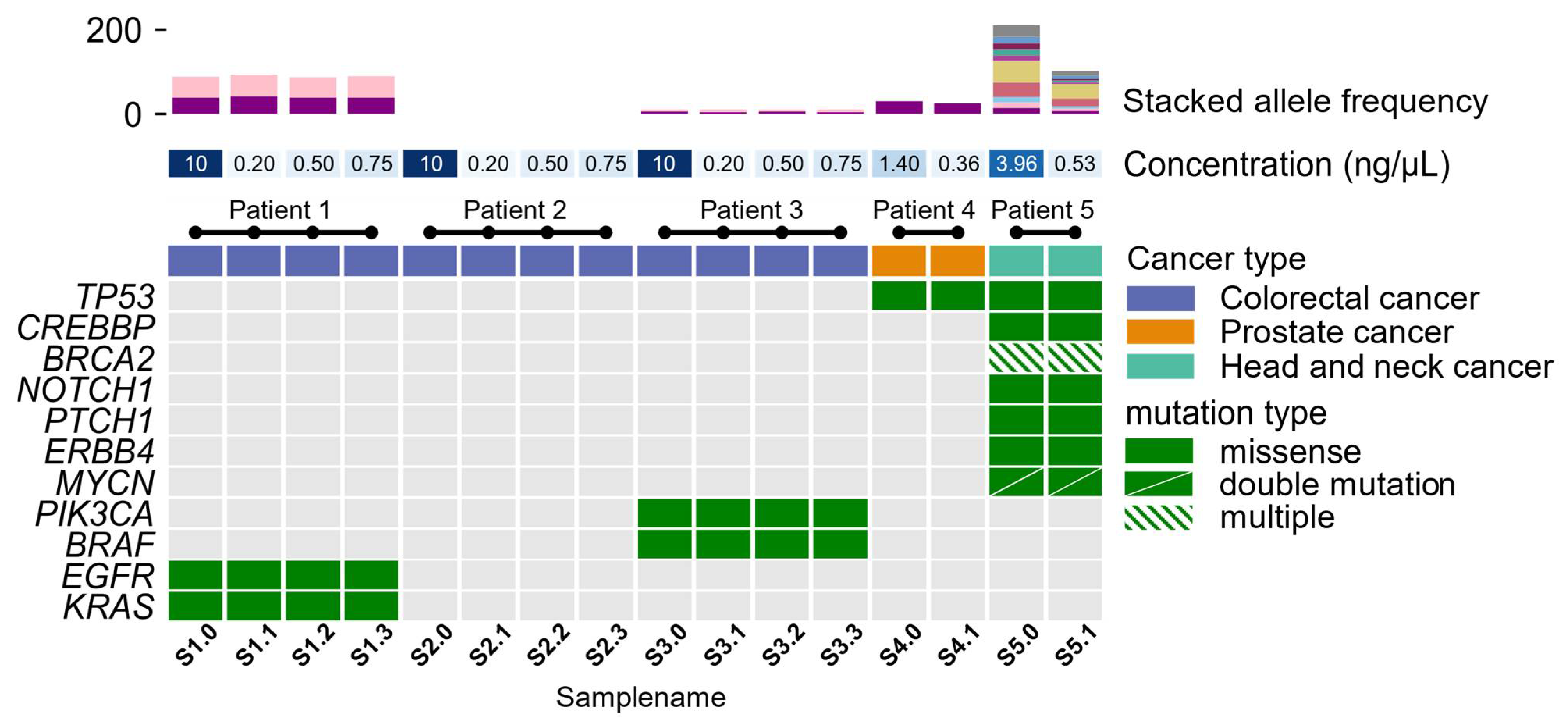
3.2. Molecular Profiling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morkel, M.; Riemer, P.; Bläker, H.; Sers, C. Similar but different: Distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget 2015, 6, 20785–20800. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, T.S.; de Oliveira, D.V.N.P.; Espersen, M.L.M.; Klarskov, L.L.; Skovrider-Ruminski, W.; Hogdall, E. Frequency and coexistence of KRAS, NRAS, BRAF and PIK3CA mutations and occurrence of MMR deficiency in Danish colorectal cancer patients. Apmis 2021, 129, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, Y.; Hirotsu, Y.; Amemiya, K.; Oyama, T.; Mochizuki, H.; Omata, M. Accurate detection of KRAS, NRAS and BRAF mutations in metastatic colorectal cancers by bridged nucleic acid-clamp real-time PCR. BMC Med. Genom. 2019, 12, 162. [Google Scholar] [CrossRef]
- Jensen, B.V.; Schou, J.V.; Yilmaz, M.; Johannesen, H.H.; Skougaard, K.; Linnemann, D.; Hogdall, E.V.; Larsen, F.O.; Johansen, J.S.; Pfeiffer, P.; et al. Cetuximab plus irinotecan administered biweekly with reduced infusion time to heavily pretreated patients with metastatic colorectal cancer and related RAS and BRAF-mutation status. Int. J. Cancer 2021, 148, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Garber, J.E. BRCA1/2 testing: Therapeutic implications for breast cancer management. Br. J. Cancer 2018, 119, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Rachmat, R.; Enyioma, S.; Ghose, A.; Revythis, A.; Boussios, S. Brca mutations in prostate cancer: Assessment, implications, and treatment considerations. Int. J. Mol. Sci. 2021, 22, 12628. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, I.; Betsou, F.; Mathieson, W. Does vacuum centrifugal concentration reduce yield or quality of nucleic acids extracted from FFPE biospecimens? Anal. Biochem. 2019, 566, 16–19. [Google Scholar] [CrossRef]
- Crosby, D.; Bhatia, S.; Brindle, K.M.; Coussens, L.M.; Dive, C.; Emberton, M.; Esener, S.; Fitzgerald, R.C.; Gambhir, S.S.; Kuhn, P.; et al. Early detection of cancer. Science 2022, 375, eaay9040. [Google Scholar] [CrossRef]
- Ferrer, I.; Armstrong, J.; Capellari, S.; Parchi, P.; Arzberger, T.; Bell, J.; Budka, H.; Ströbel, T.; Giaccone, G.; Rossi, G.; et al. Effects of formalin fixation, paraffin embedding, and time of storage on DNA preservation in brain tissue: A brainnet Europe study. Brain Pathol. 2007, 17, 297–303. [Google Scholar] [CrossRef]
- Hedegaard, J.; Thorsen, K.; Lund, M.K.; Hein, A.-M.K.; Hamilton-Dutoit, S.; Vang, S.; Nordentoft, I.K.; Birkenkamp-Demtröder, K.; Kruhøffer, M.; Hager, H.; et al. Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PLoS ONE 2014, 9, e98187. [Google Scholar] [CrossRef]
- Guyard, A.; Boyez, A.; Pujals, A.; Robe, C.; Van Nhieu, J.T.; Allory, Y.; Moroch, J.; Georges, O.; Fournet, J.-C.; Zafrani, E.-S.; et al. DNA degrades during storage in formalin-fixed and paraffin-embedded tissue blocks. Virchows Arch. 2017, 471, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Berra, C.M.; Torrezan, G.T.; de Paula, C.A.; Hsieh, R.; Lourenço, S.V.; Carraro, D.M. Use of uracil-DNA glycosylase enzyme to reduce DNA-related artifacts from formalin-fixed and paraffin-embedded tissues in diagnostic routine. Appl. Cancer Res. 2019, 39, 7. [Google Scholar] [CrossRef]
- Serizawa, M.; Yokota, T.; Hosokawa, A.; Kusafuka, K.; Sugiyama, T.; Tsubosa, Y.; Yasui, H.; Nakajima, T.; Koh, Y. The efficacy of uracil DNA glycosylase pretreatment in amplicon-based massively parallel sequencing with DNA extracted from archived formalin-fixed paraffin-embedded esophageal cancer tissues. Cancer Genet. 2015, 208, 415–427. [Google Scholar] [CrossRef]
- Do, H.; Dobrovic, A. Sequence artifacts in DNA from formalin-fixed tissues: Causes and strategies for minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Crowdis, J.; He, M.X.; Reardon, B.; van Allen, E.M. CoMut: Visualizing integrated molecular information with comutation plots. Bioinformatics 2020, 36, 4348–4349. [Google Scholar] [CrossRef]
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci. Rep. 2020, 10, 3501. [Google Scholar] [CrossRef]
- Quail, M.A.; Smith, M.; Coupland, P.; Otto, T.D.; Harris, S.R.; Connor, T.R.; Bertoni, A.; Swerdlow, H.P.; Gu, Y. A tale of three NGS sequencing platforms. BMC Genom. 2012, 13, 341. [Google Scholar]
- Vestergaard, L.K.; Oliveira, D.N.P.; Hoegdall, C.K.; Hoegdall, E.V. Next Generation Sequencing Technology in the Clinic and Its Challenges. Cancers 2021, 13, 1751. [Google Scholar] [CrossRef]
- Vestergaard, L.K.; de Oliveira, D.N.V.P.; Poulsen, T.S.; Høgdall, C.K.; Høgdall, E.V. OncomineTM Comprehensive Assay v3 vs. OncomineTM Comprehensive Assay Plus. Cancers 2021, 13, 5230. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Eefsen, R.L.; Simonsen, K.S.; Grundtvig, P.; Klarskov, L.; Chen, I.M.; Høgdall, D.; Jensen, B.V.; Lorentzen, T.; Poulsen, T.S.; Theile, S.; et al. Genomic landscape of treatment-refractory metastatic colorectal cancer. Acta Oncol. 2021, 60, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vestergaard, L.K.; Mikkelsen, N.S.; Oliveira, D.V.N.P.; Poulsen, T.S.; Hoegdall, E.V. Rescue of Low-Yield DNA Samples for Next-Generation Sequencing Using Vacuum Centrifugal Concentration in a Clinical Workflow. Reports 2023, 6, 23. https://doi.org/10.3390/reports6020023
Vestergaard LK, Mikkelsen NS, Oliveira DVNP, Poulsen TS, Hoegdall EV. Rescue of Low-Yield DNA Samples for Next-Generation Sequencing Using Vacuum Centrifugal Concentration in a Clinical Workflow. Reports. 2023; 6(2):23. https://doi.org/10.3390/reports6020023
Chicago/Turabian StyleVestergaard, Lau K., Nicolai S. Mikkelsen, Douglas V. N. P. Oliveira, Tim S. Poulsen, and Estrid V. Hoegdall. 2023. "Rescue of Low-Yield DNA Samples for Next-Generation Sequencing Using Vacuum Centrifugal Concentration in a Clinical Workflow" Reports 6, no. 2: 23. https://doi.org/10.3390/reports6020023