

Convergence of Pro-Stress and Pro-Inflammatory Signaling in the Central Noradrenergic System: Implications for Mood and Anxiety Disorders
{kind=link}
Abstract
:1. Introduction
2. The Stress Response and HPA Axis Dysregulation
3. The LC–NE System and Its Dysregulation under Chronic Stress
4. The Effects of Chronic Stress on Peripheral Inflammation
5. Microglia and the Induction of Neuroinflammation under Chronic Stress
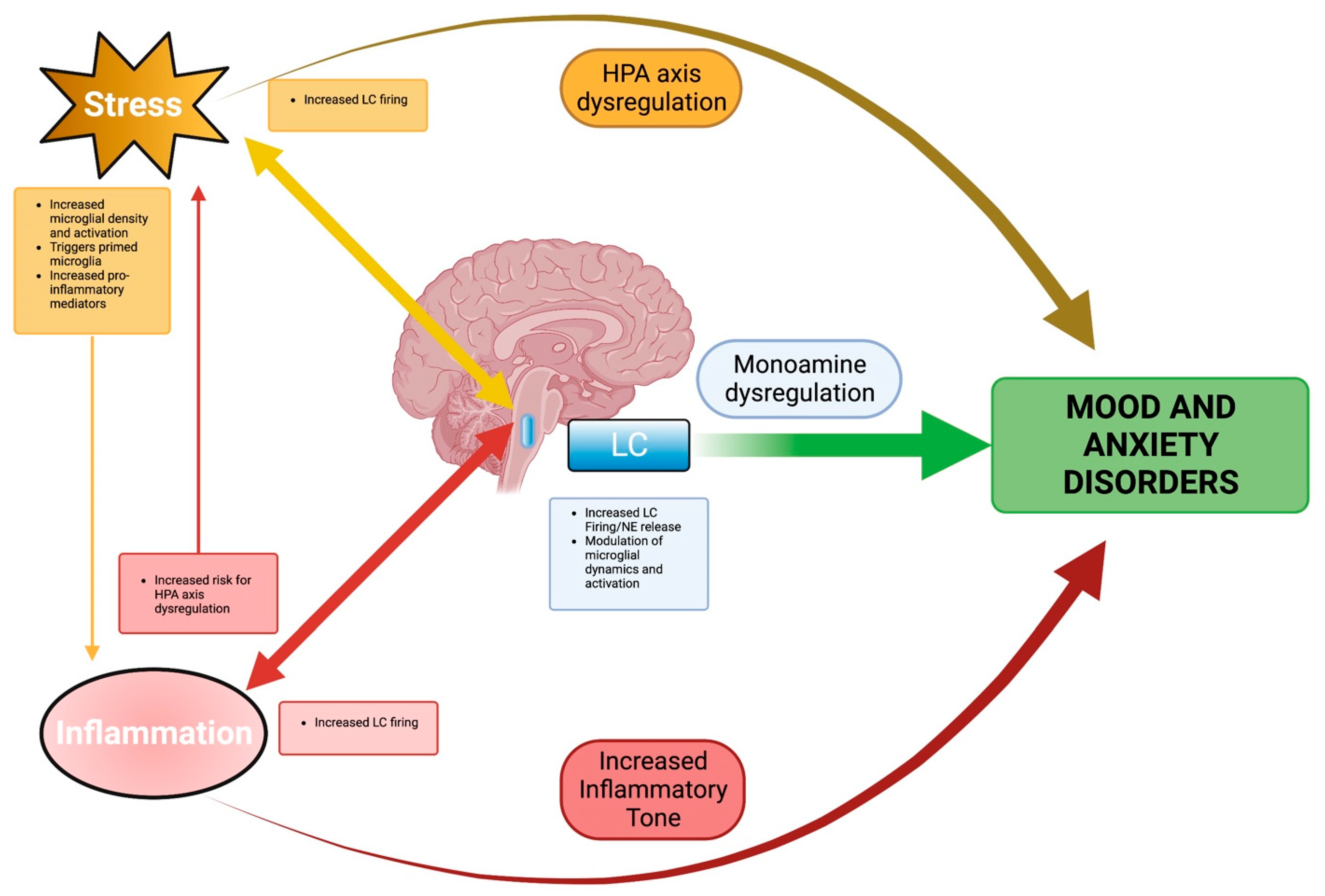
6. Integration of Stress and Neuroinflammation in LC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sekhon, S.; Gupta, V. Mood Disorder. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Kessler, R.C.; Berglund, P.; Chiu, W.T.; Demler, O.; Heeringa, S.; Hiripi, E.; Jin, R.; Pennell, B.E.; Walters, E.E.; Zaslavsky, A.; et al. The US National Comorbidity Survey Replication (NCS-R): Design and field procedures. Int. J. Methods Psychiatr. Res. 2004, 13, 69–92. [Google Scholar] [CrossRef]
- Craske, M.G.; Stein, M.B.; Eley, T.C.; Milad, M.R.; Holmes, A.; Rapee, R.M.; Wittchen, H.U. Anxiety disorders. Nat. Rev. Dis Prim. 2017, 3, 17024. [Google Scholar] [CrossRef]
- Szuhany, K.L.; Simon, N.M. Anxiety Disorders: A Review. JAMA 2022, 328, 2431–2445. [Google Scholar] [CrossRef] [PubMed]
- Hall, R.C.; Wise, M.G. The clinical and financial burden of mood disorders. Cost and outcome. Psychosomatics 1995, 36, S11–S18. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.E.; Sisitsky, T.; Kessler, R.C.; Finkelstein, S.N.; Berndt, E.R.; Davidson, J.R.; Ballenger, J.C.; Fyer, A.J. The economic burden of anxiety disorders in the 1990s. J. Clin. Psychiatry 1999, 60, 427–435. [Google Scholar] [CrossRef]
- Huang, K.L.; Su, T.P.; Chen, T.J.; Chou, Y.H.; Bai, Y.M. Comorbidity of cardiovascular diseases with mood and anxiety disorder: A population based 4-year study. Psychiatry Clin. Neurosci. 2009, 63, 401–409. [Google Scholar] [CrossRef]
- Simon, G.E.; Von Korff, M.; Saunders, K.; Miglioretti, D.L.; Crane, P.K.; Van Belle, G.; Kessler, R.C. Association Between Obesity and Psychiatric Disorders in the US Adult Population. Arch. Gen. Psychiatry 2006, 63, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Anker, J.J.; Kushner, M.G. Co-Occurring Alcohol Use Disorder and Anxiety: Bridging Psychiatric, Psychological, and Neurobiological Perspectives. Alcohol Res. 2019, 40, arcr.v40.1.03. [Google Scholar] [CrossRef] [PubMed]
- Turner, E.H.; Matthews, A.M.; Linardatos, E.; Tell, R.A.; Rosenthal, R. Selective Publication of Antidepressant Trials and Its Influence on Apparent Efficacy. N. Engl. J. Med. 2008, 358, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Fournier, J.C.; Derubeis, R.J.; Hollon, S.D.; Dimidjian, S.; Amsterdam, J.D.; Shelton, R.C.; Fawcett, J. Antidepressant Drug Effects and Depression Severity. JAMA 2010, 303, 47. [Google Scholar] [CrossRef]
- Khan, A.; Faucett, J.; Lichtenberg, P.; Kirsch, I.; Brown, W.A. A systematic review of comparative efficacy of treatments and controls for depression. PLoS ONE 2012, 7, e41778. [Google Scholar] [CrossRef]
- Johnson, D.A.W. Depression: Treatment compliance in general practice. Acta Psychiatr. Scand. 1981, 63, 447–453. [Google Scholar] [CrossRef]
- Altemus, M.; Sarvaiya, N.; Neill Epperson, C. Sex differences in anxiety and depression clinical perspectives. Front. Neuroendocrinol. 2014, 35, 320–330. [Google Scholar] [CrossRef]
- Kelly, M.M.; Tyrka, A.R.; Price, L.H.; Carpenter, L.L. Sex differences in the use of coping strategies: Predictors of anxiety and depressive symptoms. Depress. Anxiety 2008, 25, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Susukida, R.; Mojtabai, R.; Mendelson, T. Sex Differences in Help Seeking for Mood and Anxiety Disorders in the National Comorbidity Survey-Replication. Depress. Anxiety 2015, 32, 853–860. [Google Scholar] [CrossRef]
- Van Dijk, M.T.; Murphy, E.; Posner, J.E.; Talati, A.; Weissman, M.M. Association of Multigenerational Family History of Depression With Lifetime Depressive and Other Psychiatric Disorders in Children. JAMA Psychiatry 2021, 78, 778. [Google Scholar] [CrossRef]
- Angst, J.; Gamma, A.; Rössler, W.; Ajdacic, V.; Klein, D.N. Childhood adversity and chronicity of mood disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2011, 261, 21–27. [Google Scholar] [CrossRef] [PubMed]
- LeMoult, J.; Humphreys, K.L.; Tracy, A.; Hoffmeister, J.A.; Ip, E.; Gotlib, I.H. Meta-analysis: Exposure to Early Life Stress and Risk for Depression in Childhood and Adolescence. J. Am. Acad Child. Adolesc Psychiatry 2020, 59, 842–855. [Google Scholar] [CrossRef]
- Watson, S.; Owen, B.M.; Gallagher, P.; Hearn, A.J.; Young, A.H.; Ferrier, I.N. Family history, early adversity and the hypothalamic-pituitary-adrenal (HPA) axis: Mediation of the vulnerability to mood disorders. Neuropsychiatr. Dis. Treat. 2007, 3, 647–653. [Google Scholar]
- Young, E.A.; Abelson, J.L.; Curtis, G.C.; Nesse, R.M. Childhood adversity and vulnerability to mood and anxiety disorders. Depress. Anxiety 1997, 5, 66–72. [Google Scholar] [CrossRef]
- Leonard, B. Stress, norepinephrine and depression. J. Psychiatry Neurosci. 2001, 26, S11–S16. [Google Scholar] [CrossRef]
- Haapakoski, R.; Mathieu, J.; Ebmeier, K.P.; Alenius, H.; Kivimaki, M. Cumulative meta-analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav. Immun. 2015, 49, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Peruzzolo, T.L.; Pinto, J.V.; Roza, T.H.; Shintani, A.O.; Anzolin, A.P.; Gnielka, V.; Kohmann, A.M.; Marin, A.S.; Lorenzon, V.R.; Brunoni, A.R.; et al. Inflammatory and oxidative stress markers in post-traumatic stress disorder: A systematic review and meta-analysis. Mol. Psychiatry 2022, 27, 3150–3163. [Google Scholar] [CrossRef] [PubMed]
- Packard, A.E.B.; Egan, A.E.; Ulrich-Lai, Y.M. HPA Axis Interactions with Behavioral Systems. Compr. Physiol. 2016, 6, 1897–1934. [Google Scholar] [CrossRef]
- Russell, G.; Lightman, S. The human stress response. Nat. Rev. Endocrinol. 2019, 15, 525–534. [Google Scholar] [CrossRef]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, A.; Priftis, K.N. Regulation of the hypothalamic-pituitary-adrenal axis. Neuroimmunomodulation 2009, 16, 265–271. [Google Scholar] [CrossRef]
- Keller-Wood, M. Hypothalamic-Pituitary--Adrenal Axis-Feedback Control. Compr. Physiol. 2015, 5, 1161–1182. [Google Scholar] [CrossRef]
- Tsigos, C.; Kyrou, I.; Kassi, E.; Chrousos, G.P. Stress: Endocrine Physiology and Pathophysiology. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Karin, O.; Raz, M.; Tendler, A.; Bar, A.; Korem Kohanim, Y.; Milo, T.; Alon, U. A new model for the HPA axis explains dysregulation of stress hormones on the timescale of weeks. Mol. Syst. Biol. 2020, 16, e9510. [Google Scholar] [CrossRef]
- Barden, N. Implication of the hypothalamic–pituitary–adrenal axis in the physiopathology of depression. J. Psychiatry Neurosci. 2004, 29, 185. [Google Scholar]
- Juruena, M.F.; Eror, F.; Cleare, A.J.; Young, A.H. The Role of Early Life Stress in HPA Axis and Anxiety; Springer: Singapore, 2020; pp. 141–153. [Google Scholar]
- Nandam, L.S.; Brazel, M.; Zhou, M.; Jhaveri, D.J. Cortisol and Major Depressive Disorder-Translating Findings From Humans to Animal Models and Back. Front. Psychiatry 2019, 10, 974. [Google Scholar] [CrossRef] [PubMed]
- Poe, G.R.; Foote, S.; Eschenko, O.; Johansen, J.P.; Bouret, S.; Aston-Jones, G.; Harley, C.W.; Manahan-Vaughan, D.; Weinshenker, D.; Valentino, R.; et al. Locus coeruleus: A new look at the blue spot. Nat. Rev. Neurosci. 2020, 21, 644–659. [Google Scholar] [CrossRef]
- Benarroch, E.E. Locus coeruleus. Cell Tissue Res. 2018, 373, 221–232. [Google Scholar] [CrossRef]
- Van Egroo, M.; Koshmanova, E.; Vandewalle, G.; Jacobs, H.I.L. Importance of the locus coeruleus-norepinephrine system in sleep-wake regulation: Implications for aging and Alzheimer’s disease. Sleep Med. Rev. 2022, 62, 101592. [Google Scholar] [CrossRef]
- Szabadi, E. Functional neuroanatomy of the central noradrenergic system. J. Psychopharmacol. 2013, 27, 659–693. [Google Scholar] [CrossRef] [PubMed]
- Breton-Provencher, V.; Drummond, G.T.; Sur, M. Locus Coeruleus Norepinephrine in Learned Behavior: Anatomical Modularity and Spatiotemporal Integration in Targets. Front. Neural Circuits 2021, 15, 638007. [Google Scholar] [CrossRef]
- Jedema, H.P.; Grace, A.A. Corticotropin-Releasing Hormone Directly Activates Noradrenergic Neurons of the Locus Ceruleus Recorded In Vitro. J. Neurosci. 2004, 24, 9703–9713. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Foote, S.L.; Aston-Jones, G. Corticotropin-releasing factor activates noradrenergic neurons of the locus coeruleus. Brain Res. 1983, 270, 363–367. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Bloom, F. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1981, 1, 876–886. [Google Scholar] [CrossRef] [PubMed]
- McCall, J.G.; Al-Hasani, R.; Siuda, E.R.; Hong, D.Y.; Norris, A.J.; Ford, C.P.; Bruchas, M.R. CRH Engagement of the Locus Coeruleus Noradrenergic System Mediates Stress-Induced Anxiety. Neuron 2015, 87, 605–620. [Google Scholar] [CrossRef]
- Seki, K.; Yoshida, S.; Jaiswal, M.K. Molecular mechanism of noradrenaline during the stress-induced major depressive disorder. Neural Regen Res. 2018, 13, 1159–1169. [Google Scholar] [CrossRef]
- Borodovitsyna, O.; Flamini, M.D.; Chandler, D.J. Acute Stress Persistently Alters Locus Coeruleus Function and Anxiety-like Behavior in Adolescent Rats. Neuroscience 2018, 373, 7–19. [Google Scholar] [CrossRef]
- Naegeli, C.; Zeffiro, T.; Piccirelli, M.; Jaillard, A.; Weilenmann, A.; Hassanpour, K.; Schick, M.; Rufer, M.; Orr, S.P.; Mueller-Pfeiffer, C. Locus Coeruleus Activity Mediates Hyperresponsiveness in Posttraumatic Stress Disorder. Biol. Psychiatry 2018, 83, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Geracioti, T.D.; Baker, D.G.; Ekhator, N.N.; West, S.A.; Hill, K.K.; Bruce, A.B.; Schmidt, D.; Rounds-Kugler, B.; Yehuda, R.; Keck, P.E.; et al. CSF Norepinephrine Concentrations in Posttraumatic Stress Disorder. Am. J. Psychiatry 2001, 158, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.; Charney, S. Norepinephrine Dysfunction in Depression. J. Clin. Psychiatry 2000, 61, 16–24. [Google Scholar]
- Arango, V.; Underwood, M.D.; Mann, J.J. Fewer Pigmented Locus Coeruleus Neurons in Suicide Victims: Preliminary Results. Biol. Psychiatry 1969, 39, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Ordway, G.A.; Widdowson, P.S.; Smith, K.S.; Halaris, A. Agonist binding to alpha 2-adrenoceptors is elevated in the locus coeruleus from victims of suicide. J. Neurochem. 1994, 63, 617–624. [Google Scholar] [CrossRef]
- Ordway, G.A.; Schenk, J.; Stockmeier, C.A.; May, W.; Klimek, V. Elevated agonist binding to alpha2-adrenoceptors in the locus coeruleus in major depression. Biol. Psychiatry 2003, 53, 315–323. [Google Scholar] [CrossRef]
- Reichenberg, A.; Yirmiya, R.; Schuld, A.; Kraus, T.; Haack, M.; Morag, A.; Pollmächer, T. Cytokine-Associated Emotional and Cognitive Disturbances in Humans. Arch. Gen. Psychiatry 2001, 58, 445. [Google Scholar] [CrossRef]
- Kappelmann, N.; Lewis, G.; Dantzer, R.; Jones, P.B.; Khandaker, G.M. Antidepressant activity of anti-cytokine treatment: A systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol. Psychiatry 2018, 23, 335–343. [Google Scholar] [CrossRef]
- Vogelzangs, N.; Beekman, A.T.F.; De Jonge, P.; Penninx, B.W.J.H. Anxiety disorders and inflammation in a large adult cohort. Transl. Psychiatry 2013, 3, e249. [Google Scholar] [CrossRef] [PubMed]
- Bam, M.; Yang, X.; Zumbrun, E.E.; Zhong, Y.; Zhou, J.; Ginsberg, J.P.; Leyden, Q.; Zhang, J.; Nagarkatti, P.S.; Nagarkatti, M. Dysregulated immune system networks in war veterans with PTSD is an outcome of altered miRNA expression and DNA methylation. Sci. Rep. 2016, 6, 31209. [Google Scholar] [CrossRef]
- Jergović, M.; Bendelja, K.; Vidović, A.; Savić, A.; Vojvoda, V.; Aberle, N.; Rabatić, S.; Jovanovic, T.; Sabioncello, A. Patients with posttraumatic stress disorder exhibit an altered phenotype of regulatory T cells. Allergy Asthma Clin. Immunol. 2014, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Eraly, S.A.; Nievergelt, C.M.; Maihofer, A.X.; Barkauskas, D.A.; Biswas, N.; Agorastos, A.; O’Connor, D.T.; Baker, D.G. Assessment of Plasma C-Reactive Protein as a Biomarker of Posttraumatic Stress Disorder Risk. JAMA Psychiatry 2014, 71, 423. [Google Scholar] [CrossRef]
- Duque Ede, A.; Munhoz, C.D. The Pro-inflammatory Effects of Glucocorticoids in the Brain. Front. Endocrinol. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Topete, D.; Cidlowski, J.A. One Hormone, Two Actions: Anti- and Pro-Inflammatory Effects of Glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Yang, L.; He, C.; Zhou, J.W.; Li, W.Z.; Wu, W.N.; Chen, H.Q.; Yin, Y.Y. Chronic unpredictable mild stress accelerates lipopolysaccharide- induced microglia activation and damage of dopaminergic neurons in rats. Pharmacol. Biochem. Behav. 2019, 179, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.S.; Apple, C.G.; Kannan, K.B.; Funk, Z.M.; Plazas, J.M.; Efron, P.A.; Mohr, A.M. Chronic stress induces persistent low-grade inflammation. Am. J. Surg. 2019, 218, 677–683. [Google Scholar] [CrossRef]
- Frank, M.G.; Fonken, L.K.; Watkins, L.R.; Maier, S.F. Microglia: Neuroimmune-sensors of stress. Semin. Cell Dev. Biol. 2019, 94, 176–185. [Google Scholar] [CrossRef]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef]
- Andoh, M.; Koyama, R. Microglia regulate synaptic development and plasticity. Dev. Neurobiol. 2021, 81, 568–590. [Google Scholar] [CrossRef] [PubMed]
- Spiteri, A.G.; Wishart, C.L.; Pamphlett, R.; Locatelli, G.; King, N.J.C. Microglia and monocytes in inflammatory CNS disease: Integrating phenotype and function. Acta Neuropathol. 2022, 143, 179–224. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef]
- Cao, P.; Chen, C.; Liu, A.; Shan, Q.; Zhu, X.; Jia, C.; Peng, X.; Zhang, M.; Farzinpour, Z.; Zhou, W.; et al. Early-life inflammation promotes depressive symptoms in adolescence via microglial engulfment of dendritic spines. Neuron 2021, 109, 2573–2589.e2579. [Google Scholar] [CrossRef]
- Espinosa-Oliva, A.M.; de Pablos, R.M.; Villaran, R.F.; Arguelles, S.; Venero, J.L.; Machado, A.; Cano, J. Stress is critical for LPS-induced activation of microglia and damage in the rat hippocampus. Neurobiol. Aging 2011, 32, 85–102. [Google Scholar] [CrossRef]
- Kitayama, I.T.; Otani, M.; Murase, S. Contribution of the stress-induced degeneration of the locus coeruleus noradrenergic neurons to the pathophysiology of depression: A study on an animal model. Acta Neuropsychiatr. 2004, 16, 190–199. [Google Scholar] [CrossRef]
- Farooq, R.K.; Isingrini, E.; Tanti, A.; Le Guisquet, A.M.; Arlicot, N.; Minier, F.; Leman, S.; Chalon, S.; Belzung, C.; Camus, V. Is unpredictable chronic mild stress (UCMS) a reliable model to study depression-induced neuroinflammation? Behav. Brain Res. 2012, 231, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Du Preez, A.; Onorato, D.; Eiben, I.; Musaelyan, K.; Egeland, M.; Zunszain, P.A.; Fernandes, C.; Thuret, S.; Pariante, C.M. Chronic stress followed by social isolation promotes depressive-like behaviour, alters microglial and astrocyte biology and reduces hippocampal neurogenesis in male mice. Brain Behav. Immun. 2021, 91, 24–47. [Google Scholar] [CrossRef]
- Li, S.; Liao, Y.; Dong, Y.; Li, X.; Li, J.; Cheng, Y.; Cheng, J.; Yuan, Z. Microglial deletion and inhibition alleviate behavior of post-traumatic stress disorder in mice. J. Neuroinflamm. 2021, 18, 7. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Whitehead, C.; Surrao, K.; Pillai, A.; Madeshiya, A.; Li, Y.; Khodadadi, H.; Ahmed, A.O.; Turecki, G.; Baban, B.; et al. Type 1 interferon mediates chronic stress-induced neuroinflammation and behavioral deficits via complement component 3-dependent pathway. Mol. Psychiatry 2021, 26, 3043–3059. [Google Scholar] [CrossRef]
- Zhu, Y.; Klomparens, E.A.; Guo, S.; Geng, X. Neuroinflammation caused by mental stress: The effect of chronic restraint stress and acute repeated social defeat stress in mice. Neurol. Res. 2019, 41, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Song, A.Q.; Gao, B.; Fan, J.J.; Zhu, Y.J.; Zhou, J.; Wang, Y.L.; Xu, L.Z.; Wu, W.N. NLRP1 inflammasome contributes to chronic stress-induced depressive-like behaviors in mice. J. Neuroinflamm. 2020, 17, 178. [Google Scholar] [CrossRef]
- Muhie, S.; Gautam, A.; Meyerhoff, J.; Chakraborty, N.; Hammamieh, R.; Jett, M. Brain transcriptome profiles in mouse model simulating features of post-traumatic stress disorder. Mol. Brain 2015, 8, 14. [Google Scholar] [CrossRef]
- Wang, B.; Huang, X.; Pan, X.; Zhang, T.; Hou, C.; Su, W.J.; Liu, L.L.; Li, J.M.; Wang, Y.X. Minocycline prevents the depressive-like behavior through inhibiting the release of HMGB1 from microglia and neurons. Brain Behav. Immun. 2020, 88, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Liu, Y.Z.; Shen, X.L.; Wu, T.Y.; Zhang, T.; Wang, W.; Wang, Y.X.; Jiang, C.L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Li, S.; Lu, Y.; Li, X.; Liao, Y.; Peng, Z.; Li, Y.; Hou, L.; Yuan, Z.; Cheng, J. Stress-induced NLRP3 inflammasome activation negatively regulates fear memory in mice. J. Neuroinflamm. 2020, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Farooq, R.K.; Tanti, A.; Ainouche, S.; Roger, S.; Belzung, C.; Camus, V. A P2X7 receptor antagonist reverses behavioural alterations, microglial activation and neuroendocrine dysregulation in an unpredictable chronic mild stress (UCMS) model of depression in mice. Psychoneuroendocrinology 2018, 97, 120–130. [Google Scholar] [CrossRef]
- Wood, S.K.; Wood, C.S.; Lombard, C.M.; Lee, C.S.; Zhang, X.Y.; Finnell, J.E.; Valentino, R.J. Inflammatory Factors Mediate Vulnerability to a Social Stress-Induced Depressive-like Phenotype in Passive Coping Rats. Biol. Psychiatry 2015, 78, 38–48. [Google Scholar] [CrossRef]
- Koo, J.W.; Duman, R.S. Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci. Lett. 2009, 456, 39–43. [Google Scholar] [CrossRef]
- Kim, J.; Suh, Y.H.; Chang, K.A. Interleukin-17 induced by cumulative mild stress promoted depression-like behaviors in young adult mice. Mol. Brain 2021, 14, 11. [Google Scholar] [CrossRef]
- Borsody, M.K.; Weiss, J.M. Alteration of locus coeruleus neuronal activity by interleukin-1 and the involvement of endogenous corticotropin-releasing hormone. Neuroimmunomodulation 2002, 10, 101–121. [Google Scholar] [CrossRef]
- Kurosawa, N.; Shimizu, K.; Seki, K. The development of depression-like behavior is consolidated by IL-6-induced activation of locus coeruleus neurons and IL-1beta-induced elevated leptin levels in mice. Psychopharmacology 2016, 233, 1725–1737. [Google Scholar] [CrossRef]
- Pate, B.S.; Bouknight, S.J.; Harrington, E.N.; Mott, S.E.; Augenblick, L.M.; Smiley, C.E.; Morgan, C.G.; Calatayud, B.M.; Martínez-Muñiz, G.A.; Thayer, J.F.; et al. Site-Specific knockdown of microglia in the locus coeruleus regulates hypervigilant responses to social stress in female rats. Brain Behav. Immun. 2023, 109, 190–203. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, H.J.; Starkweather, A.; An, K.; Shim, I. Decreased Interleukin-4 Release from the Neurons of the Locus Coeruleus in Response to Immobilization Stress. Mediat. Inflamm. 2016, 2016, 3501905. [Google Scholar] [CrossRef] [PubMed]
- Gyoneva, S.; Traynelis, S.F. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J. Biol. Chem. 2013, 288, 15291–15302. [Google Scholar] [CrossRef]
- Liu, Y.U.; Ying, Y.; Li, Y.; Eyo, U.B.; Chen, T.; Zheng, J.; Umpierre, A.D.; Zhu, J.; Bosco, D.B.; Dong, H.; et al. Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci. 2019, 22, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Finnell, J.E.; Moffitt, C.M.; Hesser, L.A.; Harrington, E.; Melson, M.N.; Wood, C.S.; Wood, S.K. The contribution of the locus coeruleus-norepinephrine system in the emergence of defeat-induced inflammatory priming. Brain Behav. Immun. 2019, 79, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.D.; Campisi, J.; Sharkey, C.M.; Kennedy, S.L.; Nickerson, M.; Greenwood, B.N.; Fleshner, M. Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 2005, 135, 1295–1307. [Google Scholar] [CrossRef]
- Johnson, J.D.; Zimomra, Z.R.; Stewart, L.T. Beta-adrenergic receptor activation primes microglia cytokine production. J. Neuroimmunol. 2013, 254, 161–164. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Wu, L.J. How microglia sense and regulate neuronal activity. Glia 2021, 69, 1637–1653. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef]
- Weselek, G.; Keiner, S.; Fauser, M.; Wagenfuhr, L.; Muller, J.; Kaltschmidt, B.; Brandt, M.D.; Gerlach, M.; Redecker, C.; Hermann, A.; et al. Norepinephrine is a negative regulator of the adult periventricular neural stem cell niche. Stem Cells 2020, 38, 1188–1201. [Google Scholar] [CrossRef]
- Rakofsky, J.; Rapaport, M. Mood Disorders. Continuum 2018, 24, 804–827. [Google Scholar] [CrossRef]
- Bangasser, D.A.; Wiersielis, K.R.; Khantsis, S. Sex differences in the locus coeruleus-norepinephrine system and its regulation by stress. Brain Res. 2016, 1641, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Ohm, T.G.; Busch, C.; Bohl, J. Unbiased estimation of neuronal numbers in the human nucleus coeruleus during aging. Neurobiol. Aging 1997, 18, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Bohl, J.; Ohm, T.G. Spatial, temporal and numeric analysis of Alzheimer changes in the nucleus coeruleus. Neurobiol. Aging 1997, 18, 401–406. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, A.A.A.; Chandler, D.J. Convergence of Pro-Stress and Pro-Inflammatory Signaling in the Central Noradrenergic System: Implications for Mood and Anxiety Disorders. Neuroglia 2023, 4, 87-101. https://doi.org/10.3390/neuroglia4020007
Reyes AAA, Chandler DJ. Convergence of Pro-Stress and Pro-Inflammatory Signaling in the Central Noradrenergic System: Implications for Mood and Anxiety Disorders. Neuroglia. 2023; 4(2):87-101. https://doi.org/10.3390/neuroglia4020007
Chicago/Turabian StyleReyes, Arthur Anthony A., and Daniel J. Chandler. 2023. "Convergence of Pro-Stress and Pro-Inflammatory Signaling in the Central Noradrenergic System: Implications for Mood and Anxiety Disorders" Neuroglia 4, no. 2: 87-101. https://doi.org/10.3390/neuroglia4020007