1. Introduction
Stroke is defined as a neurological deficit resulting from an acute focal injury to the central nervous system (CNS) by a vascular cause [
1]. Approximately 85% of all strokes are ischemic in nature and occur when cerebral tissue fails to receive an adequate supply of oxygen [
2,
3]. According to the 2021 report from the American Heart Association, stroke was the leading cause of long-term disability and the second leading cause of death worldwide [
4]. The 2016 Global Burden of Disease data indicate that one-in-four people will have a stroke in their lifetime, with the expectation that stroke prevalence will continue to increase in the absence of preventative measures [
5,
6]. Despite the prevalence of ischemic stroke in causing death and disability, therapeutic interventions are relatively limited [
6,
7]. Currently, the only clinically approved method for treatment is the restoration of cerebral blood flow via intravenous administration of recombinant tissue-type plasminogen activator (rtPA) [
8,
9]. Unfortunately, rtPA has a narrow therapeutic time-window for administration and is associated with secondary cell death during the reperfusion period [
10]. Thus, the development of a novel treatment is still necessary.
Ischemia/reperfusion (I/R) injury is relatively common in stroke and occurs when the blood supply is reestablished after an ischemic event. Although reperfusion is necessary and often unavoidable, the subsequent restoration of blood flow triggers a pathologic cascade of molecular events involving oxidative stress, neuroinflammation, blood-brain barrier (BBB) disruption, excitotoxicity, and apoptosis [
11]. During early phases of I/R, the extent of cerebral damage is largely dependent on the extent of inflammation [
12]. The cycle of oxidative stress and neuroinflammation further increases the activation of deleterious signaling pathways implicated in the production of numerous pro-inflammatory cytokines that contribute to neuronal damage [
13,
14]. Following ischemic stroke, an irreversibly damaged core of tissue is surrounded by a salvageable penumbra, which is particularly vulnerable to neuronal death [
10]. In the hours and days following ischemia, the core territory expands in a process commonly referred to as the ischemic cascade [
15]. Focal ischemia induces a strong inflammatory response that begins within a few hours of inception and involves the activation of surrounding glial cells, such as microglia and astrocytes [
11]. At the time of peak inflammation, astrocytes typically undergo an “activation” process, wherein they begin to influence various physiological and behavioral responses [
16].
Astrocytes are the most prominent type of glial cell in the human brain and serve a critical role in several CNS functions, such as regulation of cerebral blood flow, formation of the BBB, neuronal metabolism, and defense against oxidative stress [
9]. Given that astrocytes are essential for numerous functions of the CNS and considering that their dysfunction is associated with many neurological diseases, astrocytes may be a promising target for stroke therapy. In the ischemic brain, among other pathologies, activated astrocytes undergo a process called astrogliosis, a defensive mechanism by which various molecular, functional, and morphologic changes occur [
17]. Astrogliosis is a unique cytological state that presents as protective glial scarring and results from a confluence of factors including, but not limited to, gene expression, cytokine effects, and neuronal communication [
18]. This ensuing glial scar causes astrocytes to proliferate, become hypertrophied, and upregulate expression of the intermediate filament glial fibrillary acidic protein (GFAP) [
3], which is expressed proportionally to the degree of reactivity [
17]. Hence, to understand the complex interplay between neuroprotection and deleterious inflammation, astrocytes must be understood as a central mediator within the post-stroke state.
The role of astrocytes in ischemic stroke is extremely complex and controversial. Accordingly, as peak astrocyte reactivity coincides with the peak inflammatory response [
19], understanding the time course of reactivity following ischemic stroke may provide an opportunity to target glial cells at times of peak inflammation. During ischemic conditions, the consequences of astrocytes’ activity can be either harmful or beneficial to surrounding neurons, depending at least in part on the time point post-stroke [
20]. This biphasic nature of post-ischemic astrocyte activity has been recently described as either protective or destructive based on two distinct phenotypes, which are denoted as A1 (harmful) or A2 (protective) [
21]. In the acute phase of stroke, reactive astrocytes display neuroprotective effects; however, in the post-acute and chronic phases, they may be detrimental to cerebral tissue [
3]. For example, microglia have been demonstrated to encourage the proliferation of the A1 astrocyte phenotype following stroke, which, through immunological mechanisms (such as the upregulation of complement cascade genes), ultimately lead to neurotoxic effects and neuronal cell death [
21].
Since reactive astrocytes appear to be harmful at later stages following ischemic stroke, their migration toward the affected brain region in the acute stages could prove to be a time-sensitive opportunity for various therapeutic interventions that target astrocytes. The lack of stroke treatment and prevention options illustrates the necessity to investigate novel interventions. Although previous studies have investigated the time course of reactive astrocytes following stroke, to our knowledge, this is the first study to examine the relationship between GFAP immunoreactivity at multiple timepoints after I/R using a 90-min middle cerebral artery occlusion (MCAo) stroke model. Therefore, the aim of the present study was to determine the temporal profile of astrocyte activation following a 90-min ischemic stroke in rats. These findings may offer a useful background for designing future studies to elucidate the optimal timepoint for therapeutic intervention of reactive astrocytes, allowing for further discourse regarding the potential therapies and cellular modalities surrounding this ubiquitous pathology.
2. Materials and Methods
2.1. Animals
Thirty-four male and female Sprague Dawley rats (250–300 g; Charles River, MA, USA) were used in the current study. Animals were housed in a 12-h light cycle with food and water ad libitum. Rats were randomly assigned to a stroke group (n = 22) or control group (n = 12). Animals in the stroke group underwent a 90-min MCAo surgery, whereas animals from the control group received a sham surgery in which the external carotid artery was cauterized. Following surgery, both stroke and control animals were grouped based on when euthanasia was performed following ischemic stroke, which was used to determine the temporal profile of astrocyte activation.
Sub-groups included day 2 (
n = 10), day 4 (
n = 11), and day 7 (
n = 13) post-surgery. Thus, there were 6 groups in total, as summarized in
Table 1. Inclusion criteria for MCAo animals were determined after confirmation of a successful ischemic stroke following hematoxylin and eosin (H&E) analysis. One animal failed to meet the criteria for an infarct volume of >20%, and three other animals died during the surgery and were therefore excluded. All procedures in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Central Michigan University under the IACUC protocol #2021-735.
2.2. Surgical Procedures
A 90-min ischemic stroke was induced using the MCAo intraluminal suture model following previously described methods [
22]. In brief, animals were anesthetized using an intraperitoneal injection of ketamine (100 mg/mL: 80 mg/kg; Henry Schein Animal Health, Dublin, OH, USA) and xylazine (20 mg/mL; 10 mg/kg; Henry Schein Animal Health, Dublin, OH, USA). Laser Doppler (Moor Instruments, Wilmington, DE, USA) was used to continuously measure blood flow to the right MCA territory. Rats were initially placed into a stereotaxic frame, and the laser Doppler wire was anchored to the cranium approximately 1.0 mm posterior and 5.0 mm lateral to bregma. The rat was removed from the stereotaxic frame once a baseline reading was obtained and placed supine onto a heating pad for surgery.
For the MCAo surgical procedure, a small incision was made in the neck to expose the right common carotid artery. Next, the external carotid artery was cauterized and cut, leaving a stump of the vessel. For the control (sham) group, the incision was subsequently sutured, and the animals were placed into recovery. For the stroke animals, microvascular clips were positioned on the common carotid and internal carotid arteries, and a small incision was made in the remaining stump of the external carotid artery to insert a 4–0 nylon suture (filament tip diameter: 0.39 mm; filament length: 2–3 mm). The clip from the internal carotid artery was removed and the suture was slowly advanced through the vessel intracranially until a reduction of blood flow (>40%) to the right MCA region was confirmed by the laser Doppler, indicating that the filament was blocking the origin of the MCA. After removing the clip from the common carotid artery, laser Doppler blood flow was checked again to verify the occlusion of the MCA. Following 90 min of ischemia, the filament was removed, and the external carotid artery was cauterized. Next, the incision in the neck was sutured close, and the animals were placed into recovery. Following surgery, lidocaine was applied to the head and neck of the animal to reduce discomfort, and the rats were subsequently monitored during the next five days.
2.3. Tissue Processing
Animals were euthanized at either 2 days (n = 10), 4 days (n = 11), or 7 days (n = 13) following surgery by an overdose (120 mg/kg) of sodium pentobarbital (Fatal Plus, Vortech Pharmaceuticals, Dearborn, MI, USA). Rats were transcardially perfused with 0.1 M phosphate-buffered saline (PBS, pH 7.4) and then 4% paraformaldehyde (PFA; diluted in 0.1 M PBS, pH 7.4). Extracted brains were immersed in 4% PFA for 48 h and then dehydrated successively in 10%, 20%, and 30% sucrose solutions for 72 h at 4 °C. Brains were then flash-frozen using 2-methylbutane on dry ice (Sigma Aldrich; St. Louis, MO) and stored at –80 °C until sectioning. Coronal brain sections at 30 µm were obtained using a cryostat (Cryocut 1800, Leica, Vibratome, St. Louis, MO, USA).
2.4. Infarct Volume Calculation
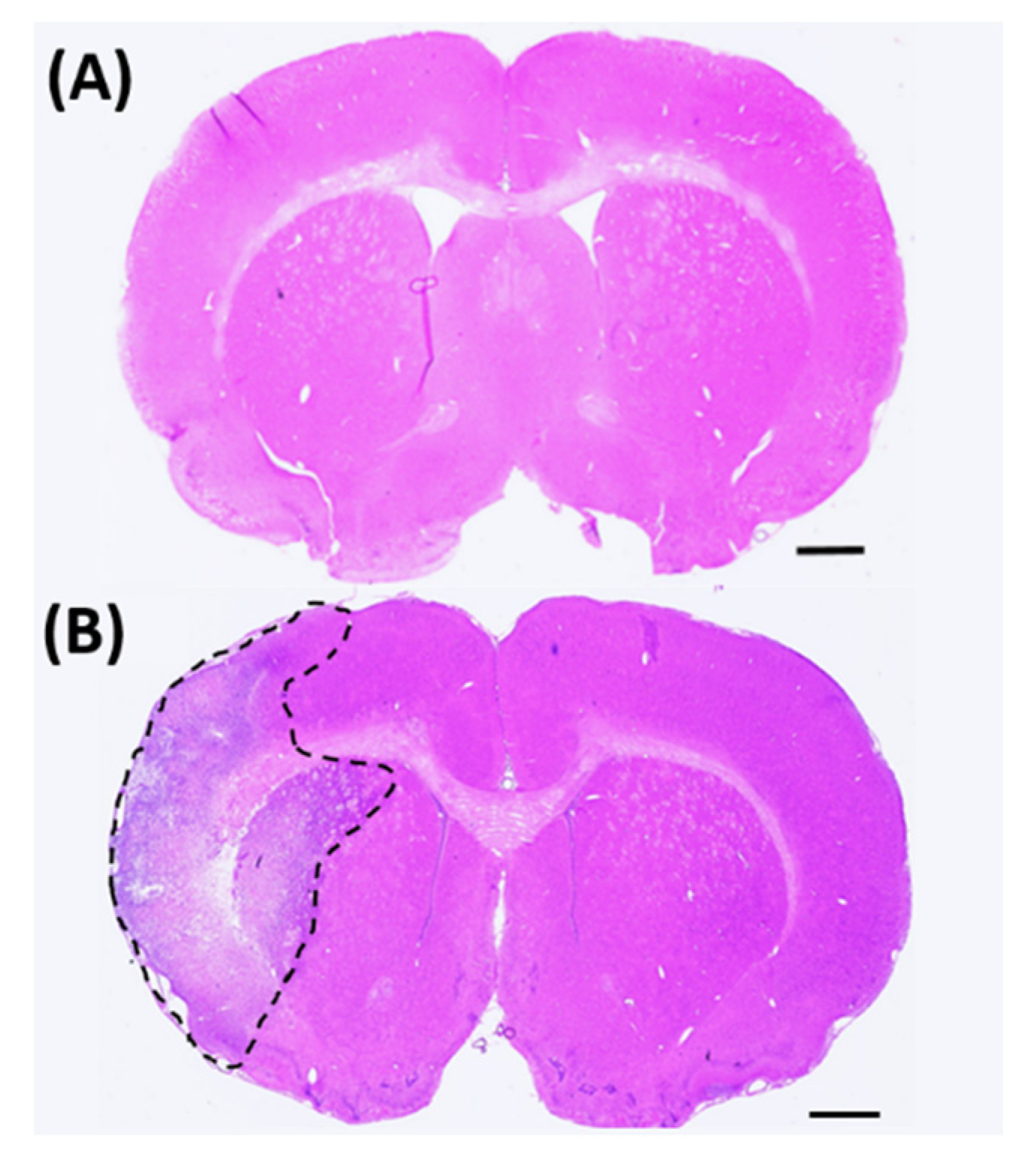
To assess tissue damage, 16 coronal brain sections for each animal were stained with H&E (
Figure 1) and scanned using a Nikon Coolscan IV scanner (Nikon, Melville, NY, USA). The infarct volume of each section was determined using ImageJ software (NIH, Rockville, MD, USA) and calculated using Swanson’s indirect method [
23]. Brain infarct volume was calculated as:
where
AC = contralateral hemisphere;
ANI = non-infarcted ipsilateral hemisphere.
2.5. Immunohistochemistry
To identify reactive astrocytes, free-floating brain sections were blocked with 10% normal goat serum (diluted in 0.3% PBS-T) for 1 h, followed by a 20-h incubation period with a GFAP primary antibody (rabbit anti-GFAP, 1:3000, Abcam, Cambridge, UK; diluted in 0.3% PBS-T). Sections were then incubated for 1 h with a secondary antibody (goat anti-rabbit, Alexa Fluor 594, 1:300 diluted in 0.3% PBS-T). Sections were washed three times between incubations with 0.1 M PBS for 15 min. All reactions took place at room temperature. Slices were mounted to SuperfrostTM Plus microscope slides and cover-slipped with premium cover glass (Fisher Scientific, Hampton, NH, USA) using fluoromount aqueous mounting medium (Sigma-Aldrich, St. Louis, MO, USA).
2.6. Tissue Imaging and GFAP Quantification
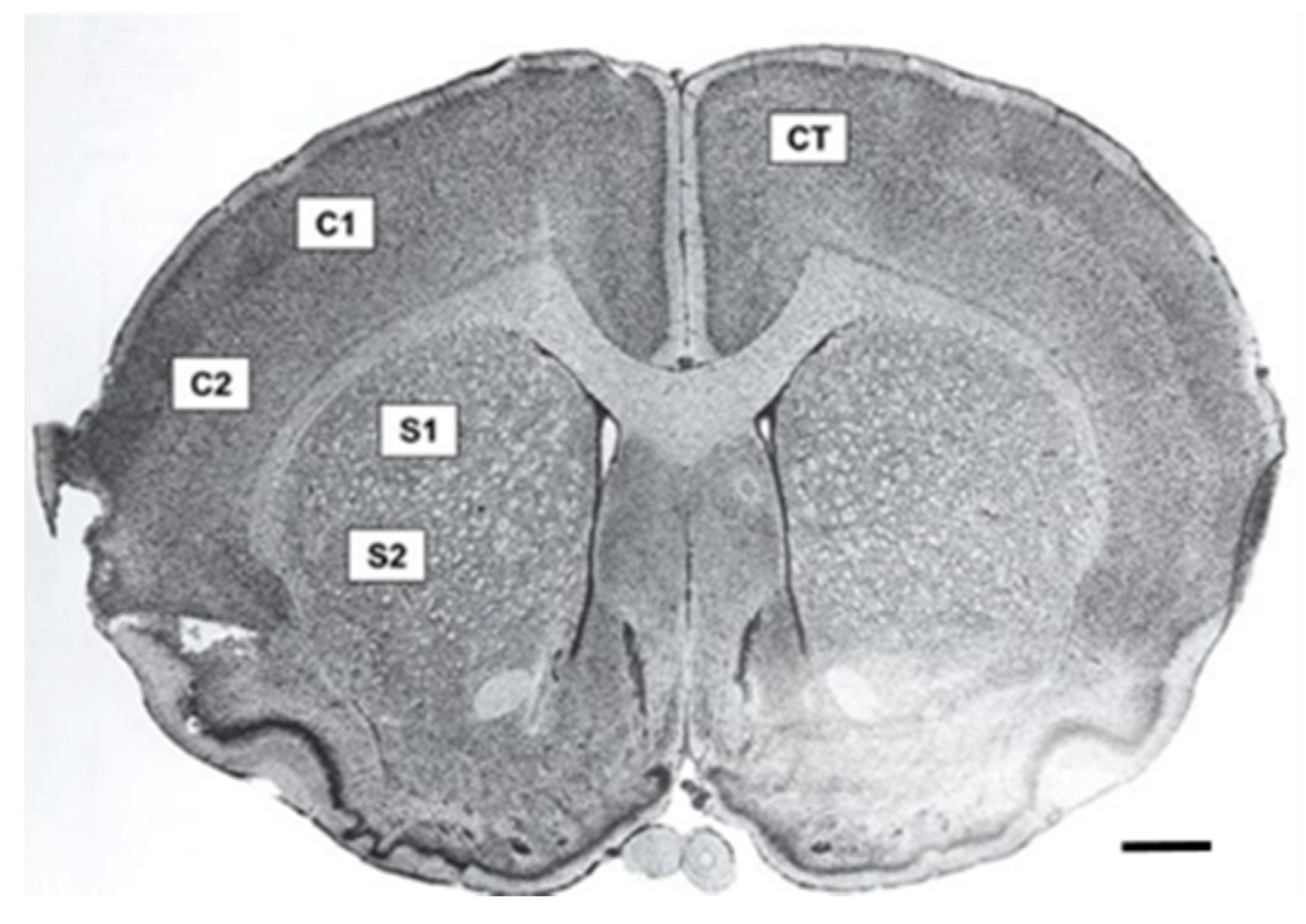
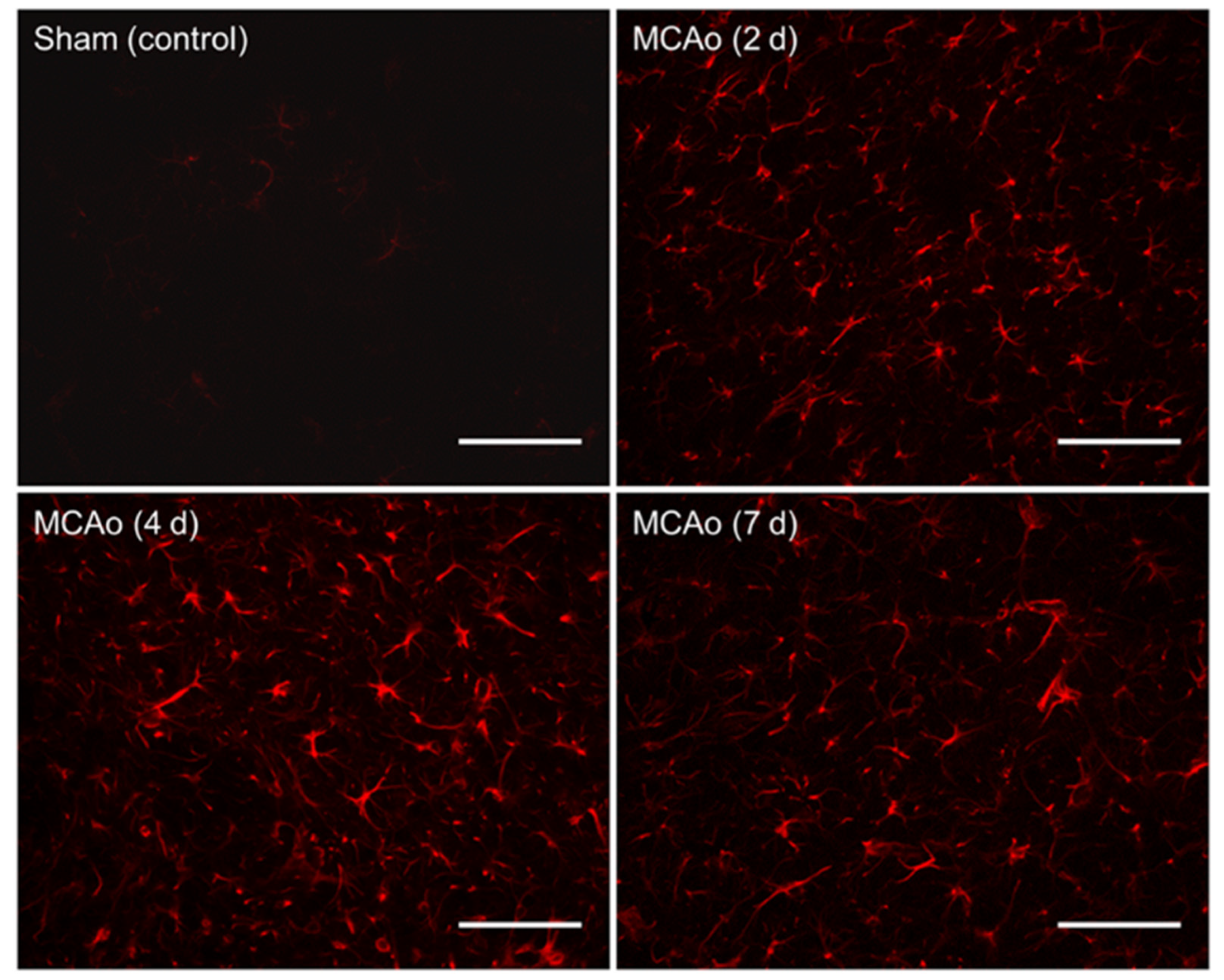
Fluorescence microscopy was performed using a Zeiss Axio Imager M2 (Gottingen, Germany). Z-stack photomicrographs (25 µm at 2.5 µm intervals, 40× objective) were obtained at an exposure time of 100 m/s and 100% light intensity. Photomicrographs were later compressed into a 2-D image. Five equally spaced tissue sections (intervals of 0.24 mm) were analyzed from each brain. To ensure consistency between animals, the section located at +0.2 mm bregma was considered the middle slice for each subject. Two successive slices located directly anterior, and two directly posterior, at intervals of 0.24 mm, were then obtained. Five images were taken for each tissue section in specific regions of interest. These included two images in the penumbral region of the cortex in the ipsilateral (affected) hemisphere, two images in the penumbra of the striatum in the ipsilateral (affected) hemisphere, and one image in the cortex of the contralateral (unaffected) hemisphere, which served as a control (
Figure 2). Thus, a total of 25 images were obtained and analyzed for each animal. Similar regions were analyzed in the sham surgery brain.
Quantification of reactive astrocytes was performed using densitometry. Z-stack photomicrographs were compressed into a 2-D image, which then underwent background correction and despeckle function. GFAP reactivity was analyzed with ImageJ software and quantified using a previously described automated thresholding algorithm [
24]. For densitometry analysis, ImageJ was used to compute the optical density of GFAP in each area. Relative density was calculated using the following equation as described by Fifield and Vanderluit [
7]:
2.7. Statistical Analysis
Statistical analyses were conducted via IBM SPSS Statistics for Windows, version 28 (IBM Corp., Armonk, NY, USA), which consisted of one-way and two-way ANOVAs with interactions of interest being further analyzed by Pearson’s correlation testing. If Levene’s test for equality of variances showed heterogeneity, Welch’s ANOVA was used. Post-hoc analyses were conducted using either Tukey honestly significant difference (HSD) in cases of homogeneity of variance or Games–Howell test when heterogeneity was shown. p < 0.05 was considered significant for all univariate analyses, with a Bonferroni-adjusted p-value of 0.025 being used as warranted in multivariate analyses.
4. Discussion
Astrocytes are the most prominent glial cell in the CNS and represent a major mediator of acute neurological diseases [
25]. Neuroinflammation and other conditions, such as ischemic stroke, present with astrocyte activation, which typically correlates with the peak inflammatory response [
19]. Astrocytes usually become reactive in the initial 2–7 days post-stroke and subsequently lead to the formation of a glial scar roughly 2 weeks following the original insult [
26]. During this period, astrocytes proliferate and show increased expression of the structural protein GFAP [
18,
20]. In the acute phase of neuroinflammation, astrocytes remain neuroprotective, but in the post-acute and chronic phases, astrocytes can both positively or negatively affect neuronal regrowth and plasticity [
3]. Despite their secretion of various neurotrophic factors and their maintenance of proper ionic regulation and homeostasis, activated astrocytes eventually initiate inflammatory responses that lead to adverse effects, such as neuronal cell death [
27,
28]. As astrocyte activation continues, positive feedback of increased inflammation leads to a further rise in neuroinflammation, either through astrocyte action on other immune cells or the inherent inflammatory potential of astrocytes [
17]. Collectively, these result in the variety of conditions seen in the neuroinflammatory state, where factors promoting survival compete with the detrimental products of astrocyte activation, cell death, and the introduction of immunological cells.
To aid in their supportive role, astrocytes may be assisted pharmacologically to promote neuroprotection and mitigate potentially damaging cascades. The literature has established that induced MCAo ischemic cores typically affect the striatal region of the brain first, followed by the destruction of adjacent structures such as the cortex [
29,
30]. The impact of the 90-min MCAo utilized in the present study supports this notion, as significant differences in GFAP immunoreactivity in the striatal region were seen between euthanization groups, whereas in the cortical region, there were not. Therefore, the timing of such events must be known, wherein by determining the temporal profile of peak astrocyte activation, one can better understand this cascade and focus on interventions to achieve optimal recovery. Although previous research has investigated the temporal profile of reactive astrocytes, to our knowledge, this is the first study to explore the relationship of GFAP immunoreactivity at various timepoints following ischemic stroke utilizing a 90-min MCAo model. Using the photothrombotic stroke method, Nowicka and colleagues (2008) found that extensive astrogliosis transpired as early as 4 h after insult and that GFAP expression was most pronounced at days 4 and 7 [
30]. Another study using the MCAo model found that GFAP-positive astrocytes accumulated in the penumbra 3–6 h after insult and reached a peak level at 1 h–3 days after reperfusion [
31]. These results contrast with our findings, which demonstrated that peak levels of GFAP occurred at 4 days after the MCAo surgery. However, it should be noted that our study used a 90-min ischemic stroke model, whereas the previous study used a 60-min MCAo. Nevertheless, we found that GFAP immunoreactivity was significantly higher at 4 days post-stroke compared to the other timepoints used, which included 2 days and 7 days after ischemia and subsequent reperfusion.
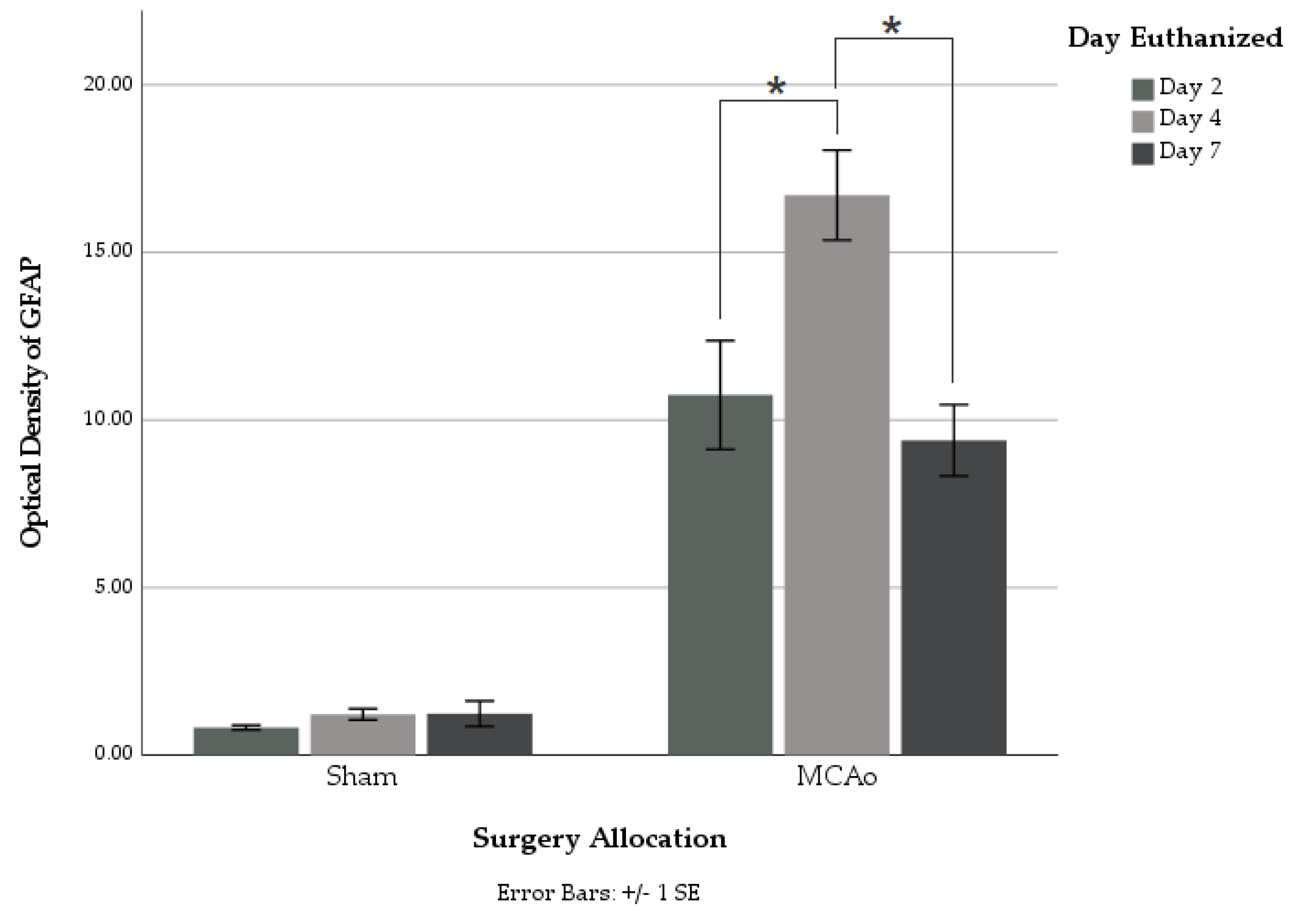
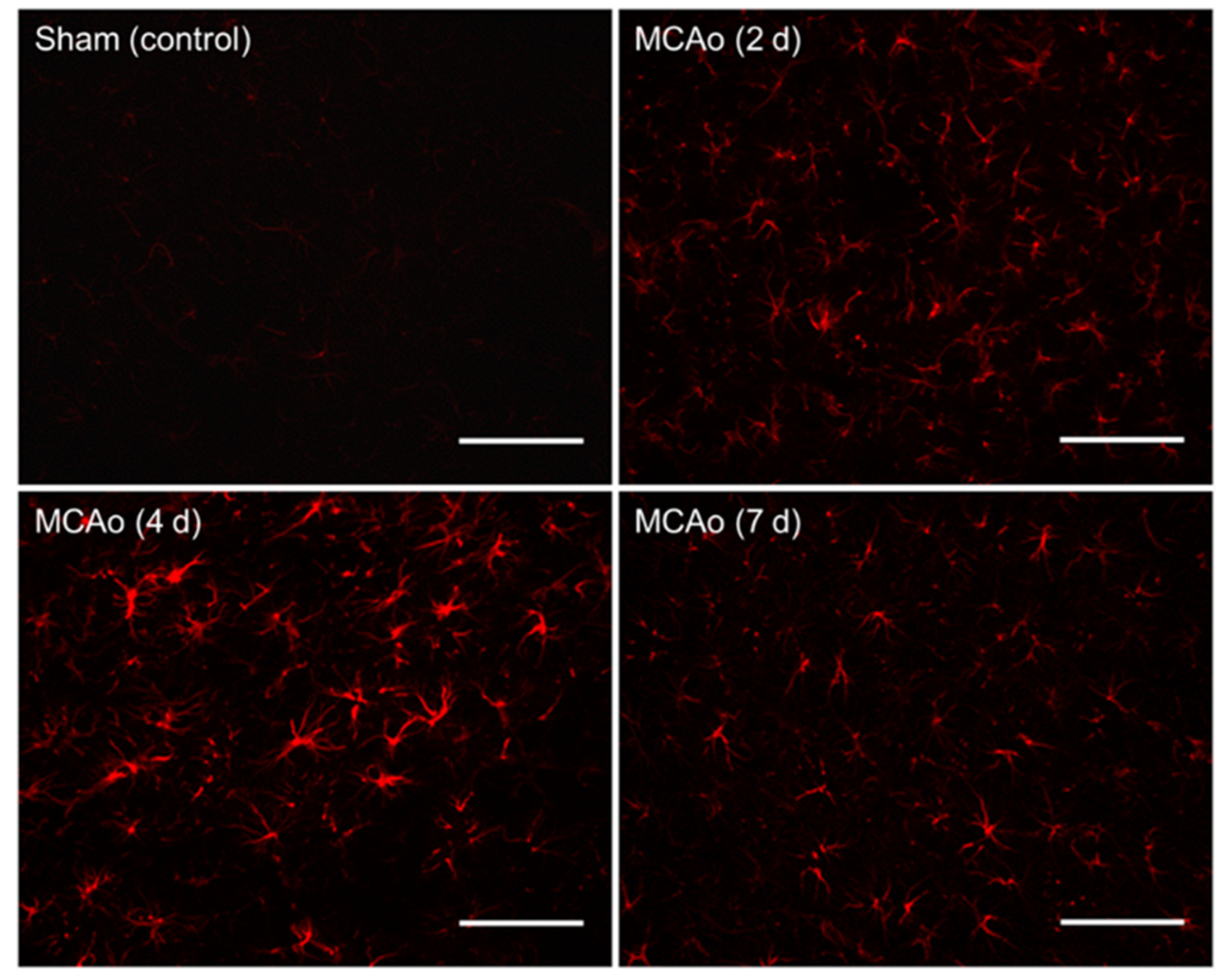
Utilizing GFAP immunohistochemical staining, we showed a non-linear relationship between GFAP reactivity and the euthanasia timepoint, which was found to peak at 4 days following surgery. Sham animals displayed no significant difference in GFAP staining between different euthanasia day groups. Within the MCAo groups, significantly greater amounts of overall GFAP immunoreactivity were found in those euthanized on day 4 as compared to those on both days 2 and 7 post-stroke (
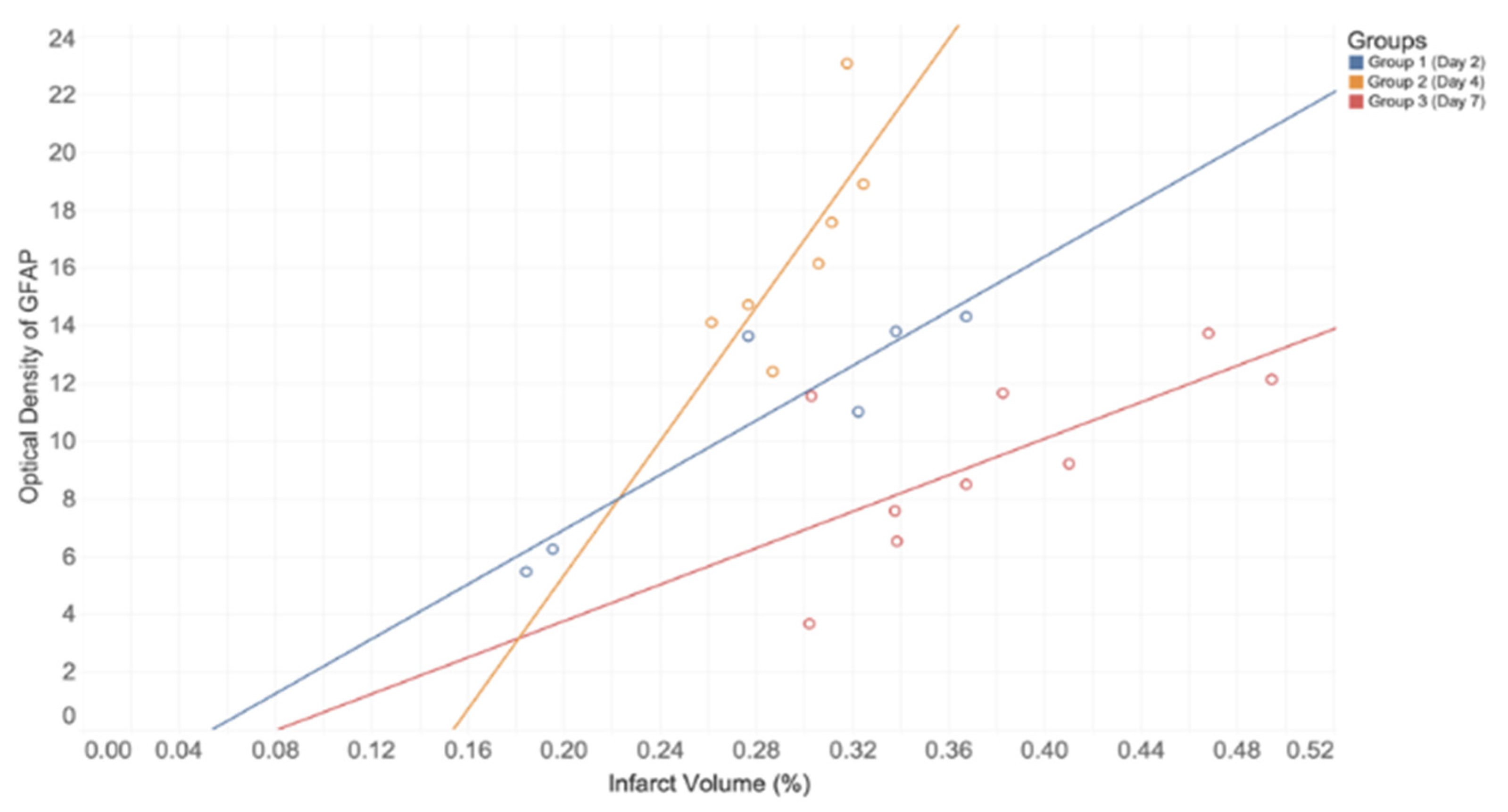
Figure 3). This suggests a timeline of events within astrocyte activation following ischemic injury. In addition, a strong positive correlation was found between infarct volume and GFAP immunoreactivity on each postoperative day of euthanization. This directly supports conclusions made from a recent review, which describes this correlation to be seen in both animal and human studies [
32]. Moreover, infarct volume was greater for those groups euthanized at later time points, with the greatest difference being shown between Group 2 and Group 3 (
Table 3). We postulate that this increased tissue loss can be attributed to normal chronological effects of post-stroke complications in the absence of any therapeutic intervention. These findings have numerous implications, as they not only demonstrate the potential peak in astrocyte immunoreactivity but they also suggest that this peak has relevance for the pathology and treatment of acute neuroinflammatory conditions.
This insight is directly related to the penetrance of pharmacological substances within the post-stroke brain. Through the relationship between astrocyte activation and systemic factors, such as the potential increase in permeability of the BBB and its connection to rising inflammation, one could mitigate the damage caused within inflamed tissue while potentially maximizing supportive effects [
33]. Within the BBB, the production of astrocytic prostaglandin E2 and pro-inflammatory cytokines leads to increased permeability, a condition known as “leakiness” [
34]. The increased potential for a variety of therapeutics to enter the brain under “leaky” conditions has shown utility for treating acute neurological insults and psychiatric conditions [
35,
36]. This leakiness offers a window of enhanced drug delivery, where conventional molecules, such as adenosine, coupled with modern therapeutics, such as nano-platforms, are being investigated as interventions for treating such conditions [
37]. By increasing BBB permeability, pharmaceutical interventions that would not be delivered under normal homeostatic conditions can now be offered as relevant therapeutics. Further relationships, such as that between peak GFAP presence and peak permeability, can serve to highlight this potential. Therefore, by determining the temporality of neuroinflammation through monitoring GFAP, we believe that further research can aim to refine the window for effective novel therapeutics in the treatment of acute neurologic conditions.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}