
Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis


Abstract
:Highlights
- We reviewed the latest advances in aberrant molecular events and pathological alterations in different cell populations in idiopathic pulmonary fibrosis.
- We comprehensively summarized the major inducers and signaling pathways of idiopathic pulmonary fibrosis.
- It is of great significance to understand the pathological mechanism of idiopathic pulmonary fibrosis.
- To provide new inspiration for the prevention and treatment of idiopathic pulmonary fibrosis.
Simple Summary
Abstract
1. Introduction
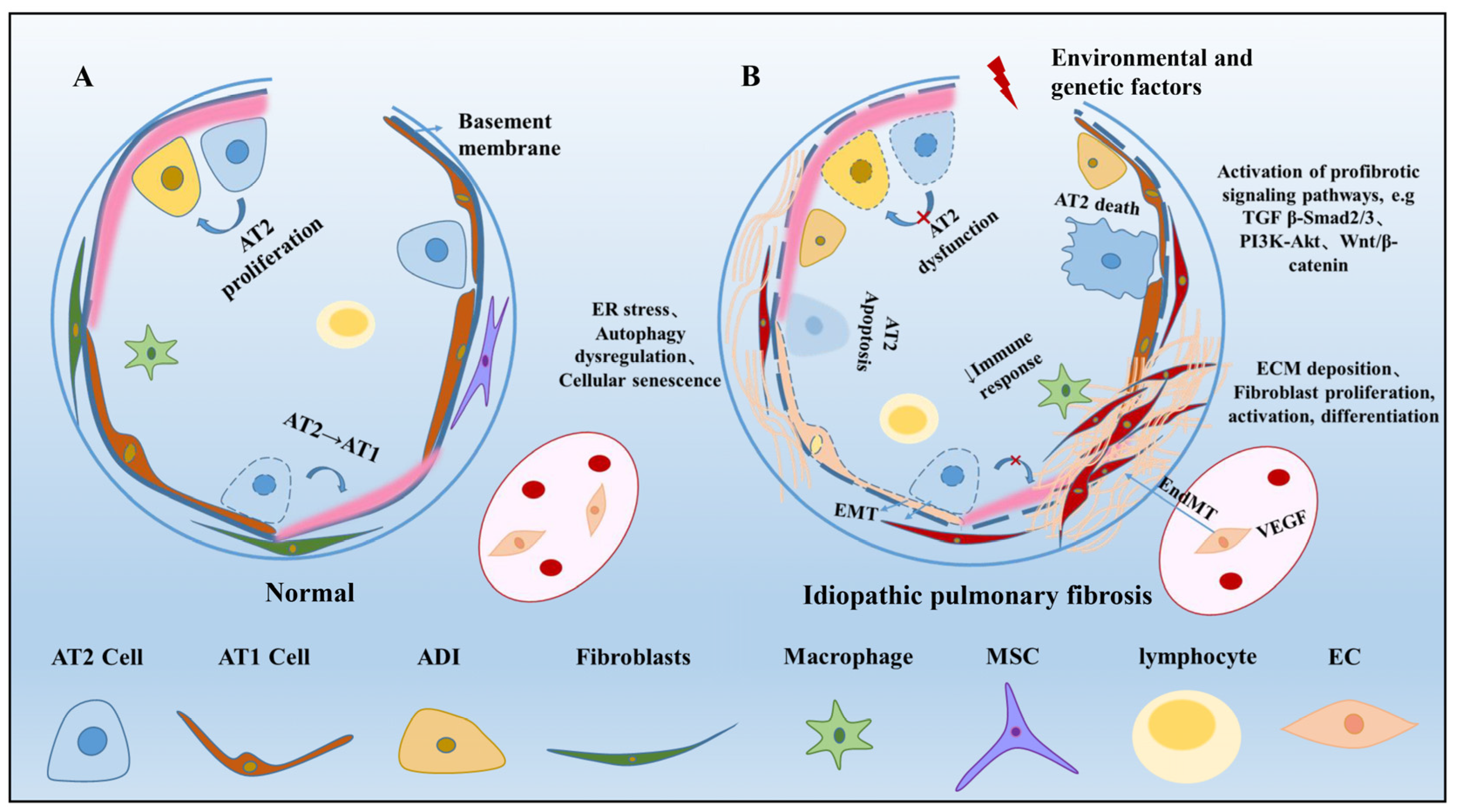
2. Alveolar Epithelial Cells in Pulmonary Fibrosis
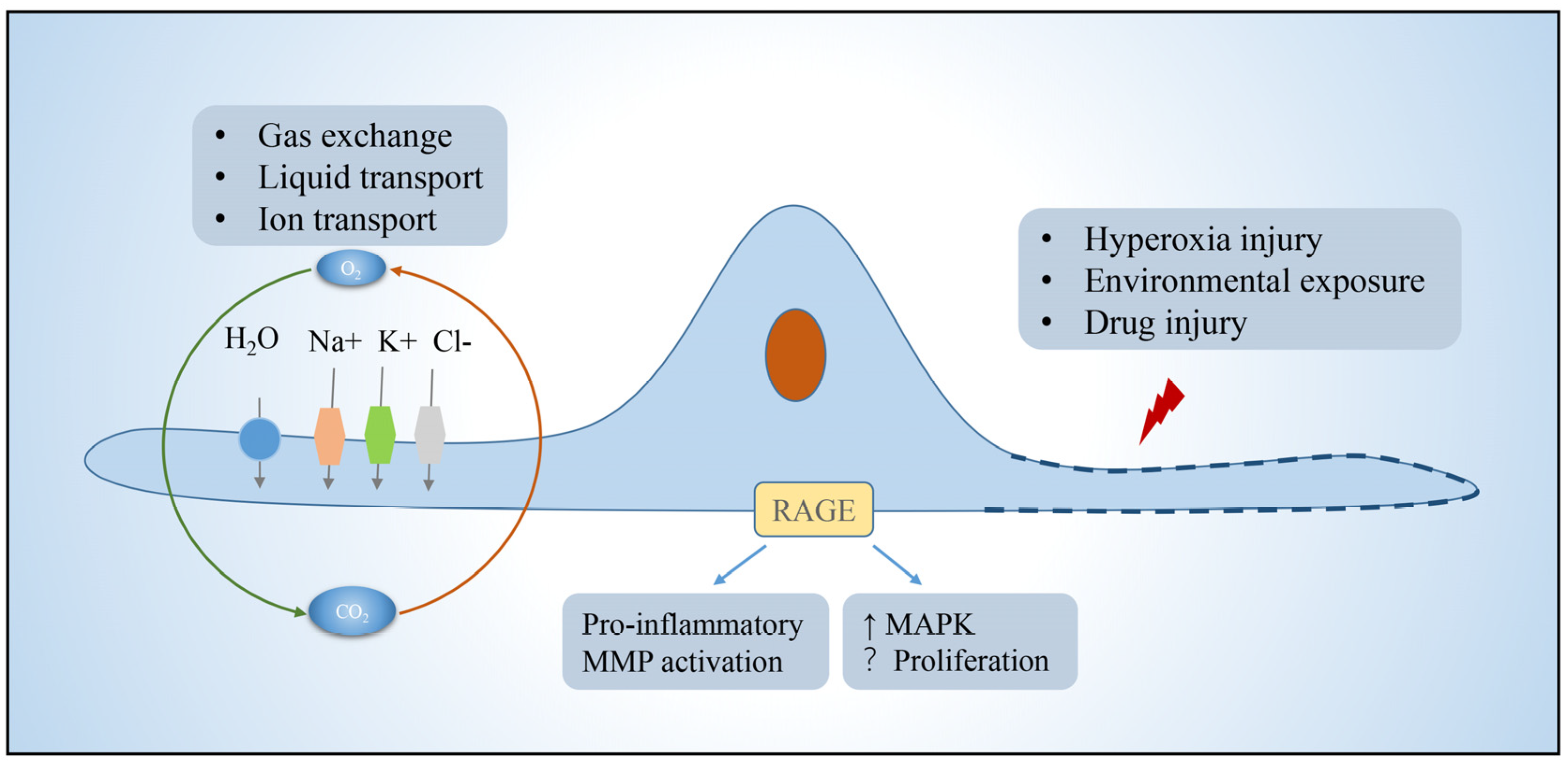
2.1. Alveolar Epithelial Type I Cells
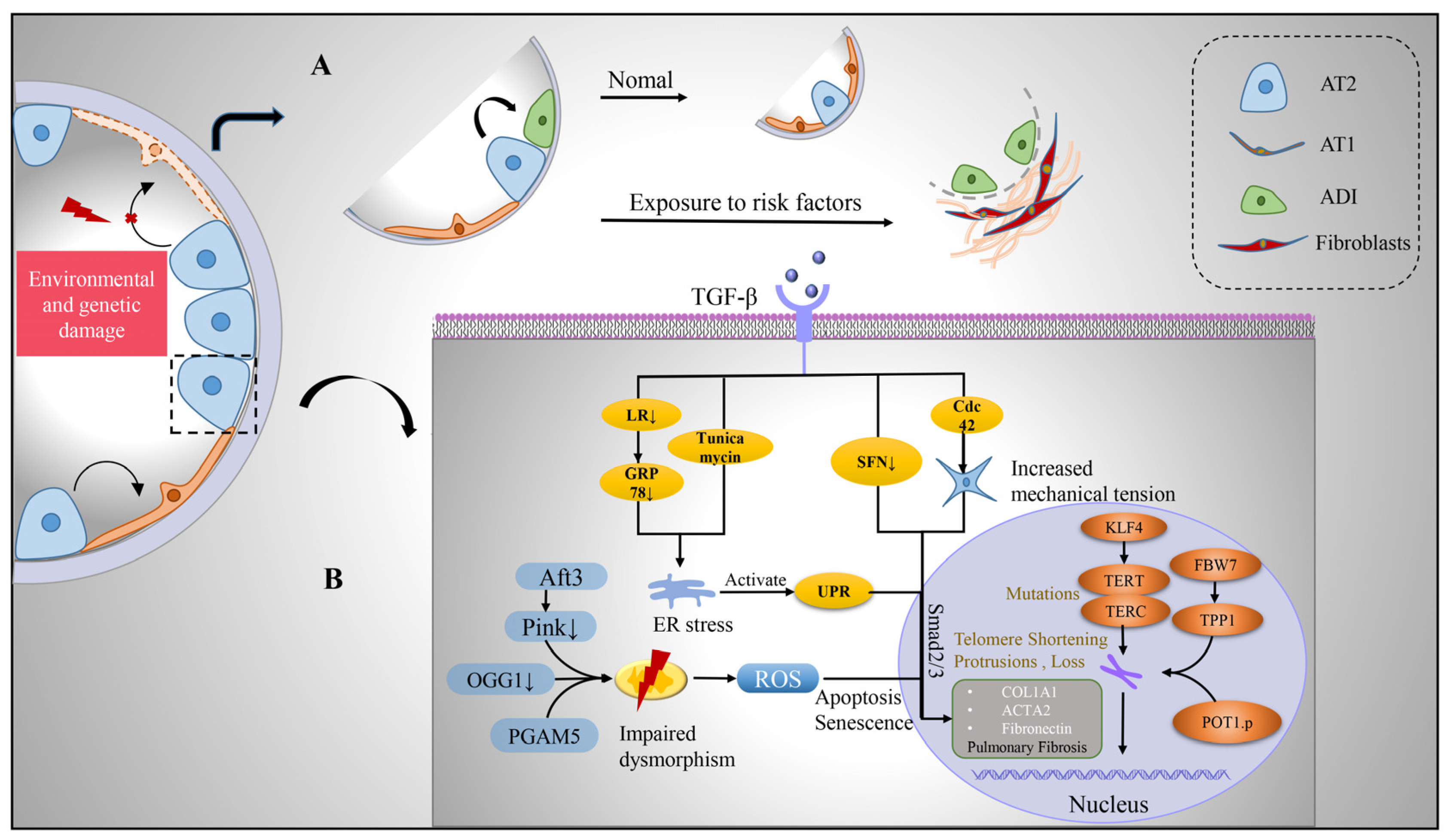
2.2. Alveolar Epithelial Type II Cells
2.2.1. Protein Homeostasis Destruction
2.2.2. Mitochondrial Damage
2.2.3. Telomere Shortening
2.3. Abnormal Basaloid Cells
3. Niche Cells in Pulmonary Fibrosis
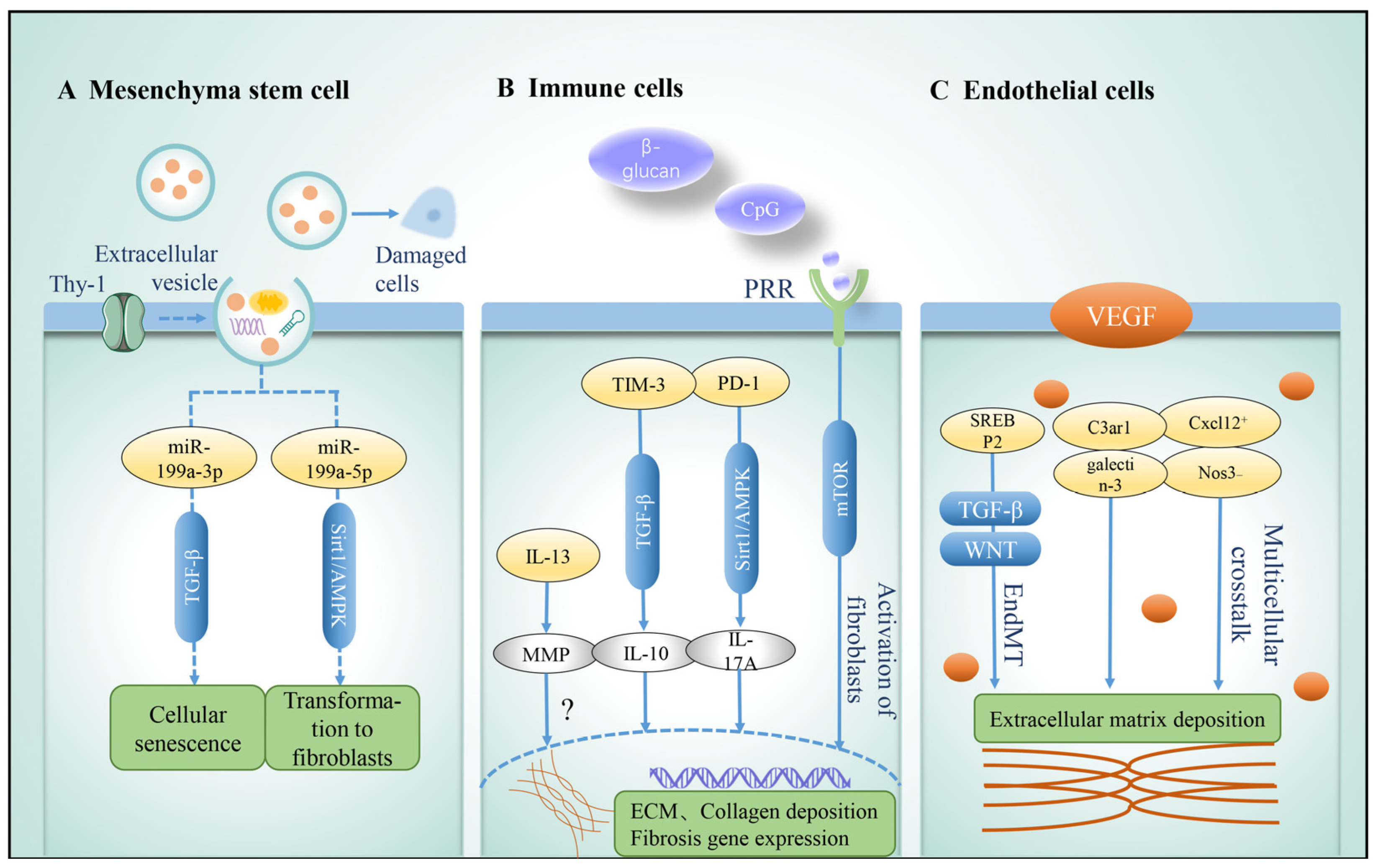
3.1. Mesenchyma Stem Cell
3.2. Fibroblasts
3.2.1. Telomere Shortening
3.2.2. Metabolic Abnormality
3.2.3. Mitochondrial Damage
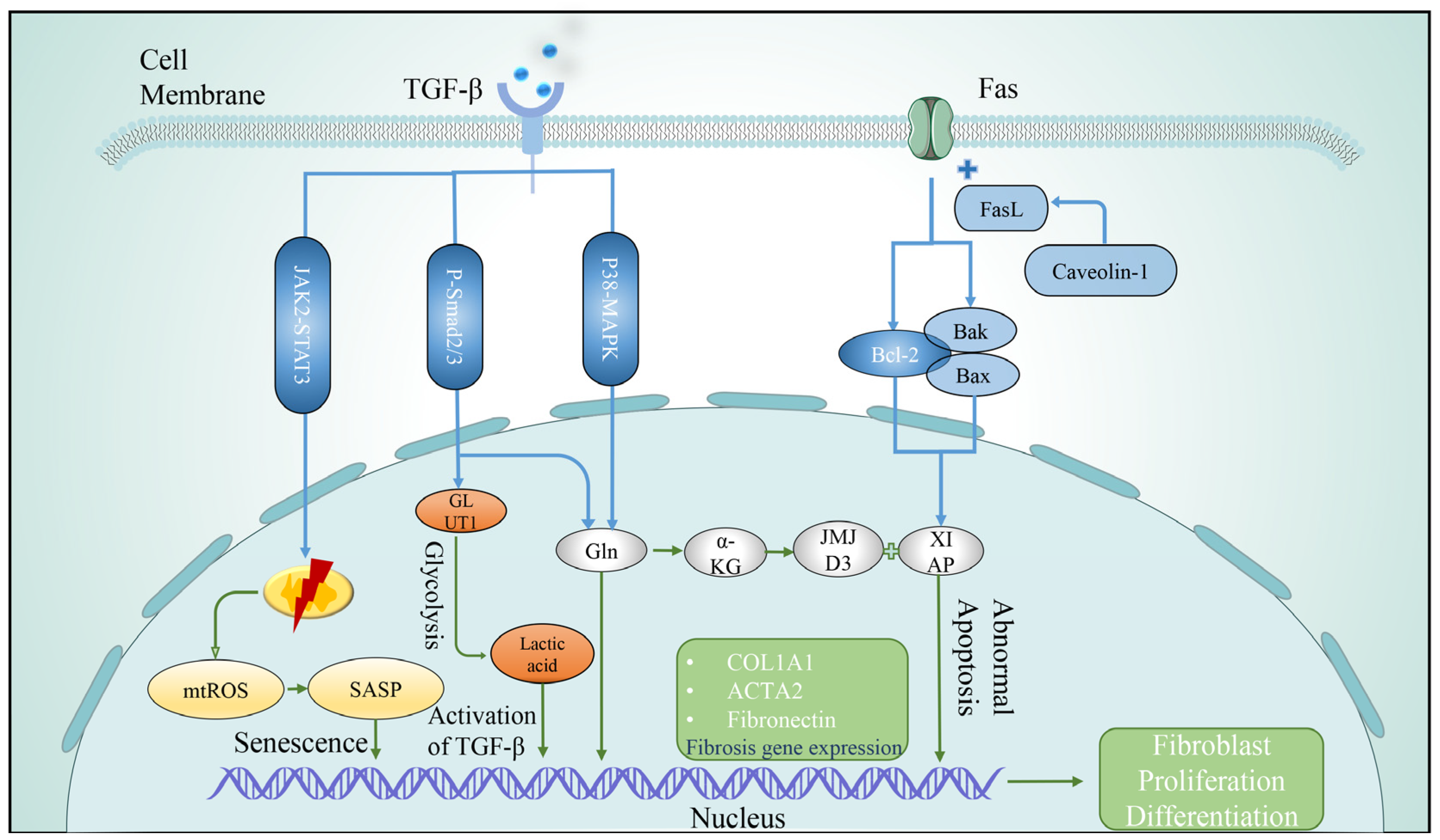
3.2.4. Apoptosis
3.2.5. Autophagy
3.2.6. Cellular Senescence
3.3. Immune Cells
3.3.1. Macrophages
3.3.2. Lymphocytes
3.3.3. T Cells
3.3.4. B Cells
3.4. Endothelial Cells
4. Cellular Crosstalk
5. Discussion and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef]
- Petnak, T.; Lertjitbanjong, P.; Thongprayoon, C.; Moua, T. Impact of Antifibrotic Therapy on Mortality and Acute Exacerbation in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. Chest 2021, 160, 1751–1763. [Google Scholar] [CrossRef]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574. [Google Scholar] [CrossRef]
- Brunnemer, E.; Walscher, J.; Tenenbaum, S.; Hausmanns, J.; Schulze, K.; Seiter, M.; Heussel, C.P.; Warth, A.; Herth, F.J.F.; Kreuter, M. Real-World Experience with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Respiration 2018, 95, 301–309. [Google Scholar] [CrossRef]
- Barratt, S.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558. [Google Scholar] [CrossRef] [Green Version]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Neumark, N.; Cosme, C., Jr.; Rose, K.A.; Kaminski, N. The Idiopathic Pulmonary Fibrosis Cell Atlas. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L887–L893. [Google Scholar] [CrossRef]
- Peyser, R.; MacDonnell, S.; Gao, Y.; Cheng, L.; Kim, Y.; Kaplan, T.; Ruan, Q.; Wei, Y.; Ni, M.; Adler, C.; et al. Defining the Activated Fibroblast Population in Lung Fibrosis Using Single-Cell Sequencing. Am. J. Respir. Cell Mol. Biol. 2019, 61, 74–85. [Google Scholar] [CrossRef]
- Serezani, A.P.M.; Pascoalino, B.D.; Bazzano, J.M.R.; Vowell, K.N.; Tanjore, H.; Taylor, C.J.; Calvi, C.L.; McCall, A.S.; Bacchetta, M.D.; Shaver, C.M.; et al. Multiplatform Single-Cell Analysis Identifies Immune Cell Types Enhanced in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 67, 50–60. [Google Scholar] [CrossRef]
- Liu, X.; Qin, X.; Qin, H.; Jia, C.; Yuan, Y.; Sun, T.; Chen, B.; Chen, C.; Zhang, H. Characterization of the heterogeneity of endothelial cells in bleomycin-induced lung fibrosis using single-cell RNA sequencing. Angiogenesis 2021, 24, 809–821. [Google Scholar] [CrossRef]
- Wang, Z.; Chai, C.; Wang, R.; Feng, Y.; Huang, L.; Zhang, Y.; Xiao, X.; Yang, S.; Zhang, Y.; Zhang, X. Single-cell transcriptome atlas of human mesenchymal stem cells exploring cellular heterogeneity. Clin. Transl. Med. 2021, 11, e650. [Google Scholar] [CrossRef]
- Zepp, J.A.; Morrisey, E.E. Cellular crosstalk in the development and regeneration of the respiratory system. Nat. Rev. Mol. Cell. Biol. 2019, 20, 551–566. [Google Scholar] [CrossRef]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef]
- Dobbs, L.G.; Johnson, D.M.; Vanderbilt, J.; Allen, L.; Gonzalez, R. The great big alveolar TI cell: Evolving concepts and paradigms. Cell. Physiol. Biochem. 2010, 25, 55–62. [Google Scholar] [CrossRef]
- Wong, M.H.; Chapin, O.C.; Johnson, M.D. LPS-stimulated cytokine production in type I cells is modulated by the renin-angiotensin system. Am. J. Respir. Cell Mol. Biol. 2012, 46, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Stogsdill, M.P.; Stogsdill, J.A.; Bodine, B.G.; Fredrickson, A.C.; Sefcik, T.L.; Wood, T.T.; Kasteler, S.D.; Reynolds, P.R. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 49, 128–134. [Google Scholar] [CrossRef]
- Weibel, E.R. On the tricks alveolar epithelial cells play to make a good lung. Am. J. Respir. Crit. Care Med. 2015, 191, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp. Mol. Pathol. 1975, 22, 142–150. [Google Scholar] [CrossRef]
- Song, M.J.; Davidovich, N.; Lawrence, G.G.; Margulies, S.S. Superoxide mediates tight junction complex dissociation in cyclically stretched lung slices. J. Biomech. 2016, 49, 1330–1335. [Google Scholar] [CrossRef]
- Queisser, M.A.; Kouri, F.M.; Konigshoff, M.; Wygrecka, U.; Schubert, M.; Eickelberg, O.; Preissnel, K.T. Loss of RAGE in pulmonary fibrosis: Molecular relations to functional changes in pulmonary cell types. Am. J. Respir. Cell Mol. Biol. 2008, 39, 337–345. [Google Scholar] [CrossRef]
- Shirasawa, M.; Fujiwara, N.; Hirabayashi, S.; Ohno, H.; Iida, J.; Makita, K.; Hata, Y. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9, 165–174. [Google Scholar] [CrossRef]
- Ding, H.; Ji, X.; Chen, R.; Ma, T.; Tang, Z.; Fen, Y.; Cai, H. Antifibrotic properties of receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2015, 35, 34–41. [Google Scholar] [CrossRef]
- He, M.; Kubo, H.; Ishizawa, K.; Hegab, A.E.; Yamamoto, Y.; Yamamoto, H.; Yamaya, M. The role of the receptor for advanced glycation end-products in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1427–L1436. [Google Scholar] [CrossRef] [Green Version]
- Shivshankar, P.; Brampton, C.; Miyasato, S.; Kasper, M.; Thannickal, V.J.; Le Saux, C.J. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am. J. Respir. Cell Mol. Biol. 2012, 47, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412. [Google Scholar] [CrossRef] [Green Version]
- Zepp, J.A.; Morley, M.P.; Loebel, C.; Kremp, M.M.; Chaudhry, F.N.; Basil, M.C.; Leach, J.P.; Liberti, D.C.; Niethamer, T.K.; Ying, Y.; et al. Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 2021, 371, eabc3172. [Google Scholar] [CrossRef]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.J.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef] [Green Version]
- Penkala, I.J.; Liberti, D.C.; Pankin, J.; Sivakumar, A.; Kremp, M.M.; Jayachandran, S.; Katzen, J.; Leach, J.P.; Windmueller, R.; Stolz, K.; et al. Age-dependent alveolar epithelial plasticity orchestrates lung homeostasis and regeneration. Cell Stem Cell 2021, 28, 1775–1789.e5. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhu, X.; Hua, Y.; Zhao, Q.; Wang, K.; Zhen, L.; Wang, G.; Lu, J.; Luo, A.; Cho, C.W.; et al. YY1 mediates TGF-beta1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir. Res. 2019, 20, 249. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef]
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13. [Google Scholar] [CrossRef] [Green Version]
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Romer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79. [Google Scholar] [CrossRef]
- Shochet, G.E.; Israeli-Shani, L.; Kains, I.; Wand, O.; Shitrit, D. MiR-608 overexpression in idiopathic pulmonary fibrosis (IPF). BMC Pulm. Med. 2021, 21, 1. [Google Scholar]
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Proteostatic and Metabolic Control of Stemness. Cell Stem Cell 2017, 20, 593–608. [Google Scholar] [CrossRef] [Green Version]
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1alpha triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 2018, 8, 17939. [Google Scholar] [CrossRef] [Green Version]
- Delbrel, E.; Uzunhan, Y.; Soumare, A.; Gille, T.; Marchant, D.; Planes, C.; Boncoeur, E. ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. Int. J. Mol. Sci. 2019, 20, 1299. [Google Scholar] [CrossRef] [PubMed]
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538. [Google Scholar] [CrossRef] [Green Version]
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-linked Role for Endoplasmic Reticulum Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 198–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, L.; Xiang, Z.; Ren, Y.; Zheng, X.; Zhao, Q.; Zhou, Q.; Zhou, Y.; Xu, L.; Wang, Y. Microcystin-LR ameliorates pulmonary fibrosis via modulating CD206(+) M2-like macrophage polarization. Cell Death Dis. 2020, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, V.K.; Lebrecht, D.; Nicholson, A.G.; Wells, A.; Bhayani, H.; Gazdhar, A.; Tamm, M.; Venhoff, N.; Geiser, T.; Walker, U.A. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis. Sci. Rep. 2019, 9, 5500. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.S.; Song, J.W.; Chi, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef]
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Rachek, L.; Yeldandi, A.; Piseaux-Aillon, R.; Ciesielski, M.J.; Ridge, K.M.; Gottardi, C.J.; Lam, A.P.; et al. Mitochondrial 8-oxoguanine DNA glycosylase mitigates alveolar epithelial cell PINK1 deficiency, mitochondrial DNA damage, apoptosis, and lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1084–L1096. [Google Scholar] [CrossRef]
- Ganzleben, I.; He, G.-W.; Günther, C.; Prigge, E.-S.; Richter, K.; Rieker, R.J.; Mougiakakos, D.; Neurath, M.F.; Becker, C. PGAM5 is a key driver of mitochondrial dysfunction in experimental lung fibrosis. Cell. Mol. Life Sci. 2019, 76, 4783–4794. [Google Scholar] [CrossRef]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; Werneck-De-Castro, J.P.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Piñeiro-Hermida, S.; Autilio, C.; Martínez, P.; Bosch, F.; Pérez-Gil, J.; Blasco, M.A. Telomerase treatment prevents lung profibrotic pathologies associated with physiological aging. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Lee, J.S.; La, J.; Aziz, S.; Dobrinskikh, E.; Brownell, R.; Jones, K.D.; Achtar-Zadeh, N.; Green, G.; Elicker, B.M.; Golden, J.A.; et al. Molecular markers of telomere dysfunction and senescence are common findings in the usual interstitial pneumonia pattern of lung fibrosis. Histopathology 2021, 79, 67–76. [Google Scholar] [CrossRef]
- Snetselaar, R.; Van Batenburg, A.A.; Van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; Van Der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; Van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE 2017, 12, e0189467. [Google Scholar] [CrossRef] [Green Version]
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680. [Google Scholar] [CrossRef] [Green Version]
- Kropski, J.A.; Blackwell, T.S. Progress in Understanding and Treating Idiopathic Pulmonary Fibrosis. Annu. Rev. Med. 2019, 70, 211–224. [Google Scholar] [CrossRef]
- Arish, N.; Petukhov, D.; Wallach-Dayan, S.B. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int. J. Mol. Sci. 2019, 20, 2996. [Google Scholar] [CrossRef] [Green Version]
- Kelich, J.; Aramburu, T.; van der Vis, J.J.; Showe, L.; Kossenkov, A.; van der Smagt, J.; Massink, M.; Schoemaker, A.; Hennekam, E.; Veltkamp, M.; et al. Telomere dysfunction implicates POT1 in patients with idiopathic pulmonary fibrosis. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef]
- Wang, L.; Chen, R.; Li, G.; Wang, Z.; Liu, J.; Liang, Y. FBW7 Mediates Senescence and Pulmonary Fibrosis through Telomere Uncapping. Cell Metab. 2020, 32, 860–877.e9. [Google Scholar] [CrossRef]
- Wang, H.; Xu, H.; Lyu, W.; Xu, Q.; Fan, S.; Chen, H.; Wang, D.; Chen, J.; Dai, J. KLF4 regulates TERT expression in alveolar epithelial cells in pulmonary fibrosis. Cell Death Dis. 2022, 13, 435. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef]
- Cao, H.; Chen, X.; Hou, J.; Wang, C.; Xiang, Z.; Shen, Y.; Han, X. The Shh/Gli signaling cascade regulates myofibroblastic activation of lung-resident mesenchymal stem cells via the modulation of Wnt10a expression during pulmonary fibrogenesis. Lab. Investig. 2020, 100, 363–377. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Wang, H.; Huang, T.; Yao, W.; Li, J.; Zhang, X. Single-cell RNA-seq highlights heterogeneity in human primary Wharton’s jelly mesenchymal stem/stromal cells cultured in vitro. Stem Cell Res. Ther. 2020, 11, 149. [Google Scholar] [CrossRef] [Green Version]
- Cahill, E.F.; Kennelly, H.; Carty, F.; Mahon, B.P.; English, K. Hepatocyte Growth Factor Is Required for Mesenchymal Stromal Cell Protection Against Bleomycin-Induced Pulmonary Fibrosis. Stem Cells Transl. Med. 2016, 5, 1307–1318. [Google Scholar] [CrossRef]
- Wang, X.; Gao, J.-L.; Zhao, M.-M.; Zhu, H.-X.; Tian, Y.-X.; Jun-Ling, G.; Jiang, X.-H.; Yu, L.; Tian, J.-R.; Cui, J.-Z. Therapeutic effects of conditioned medium from bone marrow-derived mesenchymal stem cells on epithelial-mesenchymal transition in A549 cells. Int. J. Mol. Med. 2018, 41, 659–668. [Google Scholar] [CrossRef]
- Yang, S.; Liu, P.; Jiang, Y.; Wang, Z.; Dai, H.; Wang, C. Therapeutic Applications of Mesenchymal Stem Cells in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2021, 9, 639657. [Google Scholar] [CrossRef]
- Lan, Y.-W.; Theng, S.-M.; Huang, T.-T.; Choo, K.-B.; Chen, C.-M.; Kuo, H.-P.; Chong, K.-Y. Oncostatin M-Preconditioned Mesenchymal Stem Cells Alleviate Bleomycin-Induced Pulmonary Fibrosis Through Paracrine Effects of the Hepatocyte Growth Factor. Stem Cells Transl. Med. 2017, 6, 1006–1017. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.L.; Lau, S.N.; Leaw, B.; Nguyen, H.P.T.; Salamonsen, L.A.; Saad, M.I.; Chan, S.T.; Zhu, D.; Krause, M.; Kim, C.; et al. Amnion Epithelial Cell-Derived Exosomes Restrict Lung Injury and Enhance Endogenous Lung Repair. Stem Cells Transl. Med. 2018, 7, 180–196. [Google Scholar] [CrossRef] [Green Version]
- Shentu, T.P.; Huang, T.S.; Cernelc-Kohan, M.; Chan, J.; Wong, S.S.; Espinoza, C.R.; Tan, C.; Gramaglia, I.; van der Heyde, H.; Chien, S.; et al. Thy-1 dependent uptake of mesenchymal stem cell-derived extracellular vesicles blocks myofibroblastic differentiation. Sci. Rep. 2017, 7, 18052. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Han, Q.; Hong, Y.; Li, W.; Gong, G.; Cui, J.; Mao, M.; Liang, X.; Hu, B.; Li, X.; et al. Inhibition of miR-199a-5p rejuvenates aged mesenchymal stem cells derived from patients with idiopathic pulmonary fibrosis and improves their therapeutic efficacy in experimental pulmonary fibrosis. Stem Cell Res. Ther. 2021, 12, 147. [Google Scholar] [CrossRef]
- Willis, G.R.; Fernandez-Gonzalez, A.; Anastas, J.; Vitali, S.H.; Liu, X.; Ericsson, M.; Kwong, A.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal Stromal Cell Exosomes Ameliorate Experimental Bronchopulmonary Dysplasia and Restore Lung Function through Macrophage Immunomodulation. Am. J. Respir. Crit. Care Med. 2018, 197, 104–116. [Google Scholar] [CrossRef]
- Luo, X.-Y.; Meng, X.-J.; Cao, D.-C.; Wang, W.; Zhou, K.; Li, L.; Guo, M.; Wang, P. Transplantation of bone marrow mesenchymal stromal cells attenuates liver fibrosis in mice by regulating macrophage subtypes. Stem Cell Res. Ther. 2019, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Nassiri, S.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019, 4, e128060. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Ding, L.; Wang, L.; Cao, Y.; Zhu, H.; Lu, J.; Li, X.; Song, T.; Hu, Y.; Dai, J. Umbilical cord-derived mesenchymal stem cells on scaffolds facilitate collagen degradation via upregulation of MMP-9 in rat uterine scars. Stem Cell Res. Ther. 2017, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Chu, K.-A.; Wang, S.-Y.; Yeh, C.-C.; Fu, T.-W.; Fu, Y.-S.; Ko, T.-L.; Chiu, M.-M.; Chen, T.-H.; Tsai, P.-J. Reversal of bleomycin-induced rat pulmonary fibrosis by a xenograft of human umbilical mesenchymal stem cells from Wharton’s jelly. Theranostics 2019, 9, 6646–6664. [Google Scholar] [CrossRef]
- Cardenes, N.; Alvarez, D.; Sellares, J.; Peng, Y.; Corey, C.; Wecht, S.; Nouraie, S.M.; Shanker, S.; Sembrat, J.; Bueno, M.; et al. Senescence of bone marrow-derived mesenchymal stem cells from patients with idiopathic pulmonary fibrosis. Stem Cell Res. Ther. 2018, 9, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustos, M.L.; Huleihel, L.; Kapetanaki, M.G.; Lino-Cardenas, C.L.; Mroz, L.; Ellis, B.M.; McVerry, B.J.; Richards, T.J.; Kaminski, N.; Cerdenes, N.; et al. Aging mesenchymal stem cells fail to protect because of impaired migration and antiinflammatory response. Am. J. Respir. Crit. Care Med. 2014, 189, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Periera-Simon, S.; Xia, X.; Catanuto, P.; Coronado, R.; Kurtzberg, J.; Bellio, M.; Lee, Y.S.; Khan, A.; Smith, R.; Elliot, S.J.; et al. Anti-fibrotic effects of different sources of MSC in bleomycin-induced lung fibrosis in C57BL6 male mice. Respirology 2021, 26, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Averyanov, A.; Koroleva, I.; Konoplyannikov, M.; Revkova, V.; Lesnyak, V.; Kalsin, V.; Danilevskaya, O.; Nikitin, A.; Sotnikova, A.; Kotova, S.; et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl. Med. 2020, 9, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Blokland, K.E.C.; Waters, D.W.; Schuliga, M.; Read, J.; Pouwels, S.D.; Grainge, C.L.; Jaffar, J.; Westall, G.; Mutsaers, S.E.; Prêle, C.M.; et al. Senescence of IPF Lung Fibroblasts Disrupt Alveolar Epithelial Cell Proliferation and Promote Migration in Wound Healing. Pharmaceutics 2020, 12, 389. [Google Scholar] [CrossRef]
- Schiller, H.B.; Fernandez, I.E.; Burgstaller, G.; Schaab, C.; Scheltema, R.A.; Schwarzmayr, T.; Strom, T.M.; Eickelberg, O.; Mann, M. Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair. Mol. Syst. Biol. 2015, 11, 819. [Google Scholar] [CrossRef]
- Booth, A.J.; Hadley, R.; Cornett, A.M.; Dreffs, A.A.; Matthes, S.A.; Tsui, J.L.; Weiss, K.; Horowitz, J.C.; Fiore, V.F.; Barker, T.H.; et al. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 2012, 186, 866–876. [Google Scholar] [CrossRef] [Green Version]
- Decaris, M.L.; Gatmaitan, M.; FlorCruz, S.; Luo, F.; Li, K.; Holmes, W.E.; Hellerstein, M.K.; Turner, S.M.; Emson, C.L. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol. Cell. Proteomics 2014, 13, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Boyd, D.F.; Allen, E.K.; Randolph, A.G.; Guo, X.J.; Weng, Y.; Sanders, C.J.; Bajracharya, R.; Lee, N.K.; Guy, C.S.; Vogel, P.; et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 2020, 587, 466–471. [Google Scholar] [CrossRef]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed]
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-beta and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 22, 6107. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhang, J.; Shi, L.; Ning, Y.; Zhu, Y.; Chen, S.; Yang, M.; Chen, J.; Zhou, G.-W.; Li, Q. NOGO-B promotes EMT in lung fibrosis via MMP14 mediates free TGF-beta1 formation. Oncotarget 2017, 8, 71024–71037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Wen, L.; Shi, Q.F.; Gao, F.; Huang, B.; Meng, J.; Hu, C.P.; Wang, C.M. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-kappaB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020, 11, 978. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.W.; Zhang, X.; Lin, S.C.; Lin, Y.C.; Li, C.H.; Akhrymuk, I.; Lin, S.H.; Lin, C.C. Atractylodin Suppresses TGF-beta-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. Int. J. Mol. Sci. 2021, 22, 11152. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ward, C.; Eapen, M.S.; Myers, S.; Hallgren, O.; Levine, H.; Sohal, S.S. Epithelial-mesenchymal transition, a spectrum of states: Role in lung development, homeostasis, and disease. Dev. Dyn. 2018, 247, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Ye, L.; Du, F.; Ye, M.; Li, C.; Zhu, X.; Wang, Q.; Jiang, H.; Liu, Z.; Ma, J.; et al. Iron metabolism regulation of epithelial-mesenchymal transition in idiopathic pulmonary fibrosis. Ann. Transl. Med. 2021, 9, 1755. [Google Scholar] [CrossRef]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S.P. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef] [Green Version]
- Bueno, M.; Calyeca, J.; Rojas, M.; Mora, A.L. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020, 33, 101509. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef] [PubMed]
- M’Kacher, R.; Jaillet, M.; Colicchio, B.; Vasarmidi, E.; Mailleux, A.; Dieterlen, A.; Kannengiesser, C.; Borie, C.; Oudrhiri, N.; Junker, S.; et al. Lung Fibroblasts from Idiopathic Pulmonary Fibrosis Patients Harbor Short and Unstable Telomeres Leading to Chromosomal Instability. Biomedicines 2022, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ullenbruch, M.; Choi, Y.Y.; Yu, H.; Ding, L.; Xaubet, A.; Pereda, J.; Feghali-Bostwick, C.A.; Bitterman, P.B.; Henke, C.A.; et al. Telomerase and telomere length in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 260–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roque, W.; Romero, F. Cellular metabolomics of pulmonary fibrosis, from amino acids to lipids. Am. J. Physiol. Cell Physiol. 2021, 320, C689–C695. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, B.; Azuelos, I.; Anastasiou, D.; Chambers, R.C. Fibrometabolism—An emerging therapeutic frontier in pulmonary fibrosis. Sci. Signal. 2021, 14, eaay1027. [Google Scholar] [CrossRef]
- Zhao, Y.D.; Yin, L.; Archer, S.; Lu, C.; Zhao, G.; Yao, Y.; Wu, L.; Hsin, M.; Waddell, T.K.; Keshavjee, S.; et al. Metabolic heterogeneity of idiopathic pulmonary fibrosis: A metabolomic study. BMJ Open Respir. Res. 2017, 4, e000183. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, R.B.; O’Leary, E.M.; Witt, L.J.; Tian, Y.; Gökalp, G.A.; Meliton, A.Y.; Dulin, N.O.; Mutlu, G.M. Glutamine Metabolism Is Required for Collagen Protein Synthesis in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 597–606. [Google Scholar] [CrossRef]
- Bai, L.; Bernard, K.; Tang, X.; Hu, M.; Horowitz, J.C.; Thannickal, V.J.; Sanders, Y.Y. Glutaminolysis Epigenetically Regulates Antiapoptotic Gene Expression in Idiopathic Pulmonary Fibrosis Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 60, 49–57. [Google Scholar] [CrossRef]
- Bärnthaler, T.; Theiler, A.; Zabini, D.; Trautmann, S.; Stacher-Priehse, E.; Lanz, I.; Klepetko, W.; Sinn, K.; Flick, H.; Scheidl, S.; et al. Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J. Allergy Clin. Immunol. 2020, 145, 818–833.e11. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Moon, S.-J.; Lee, S.E.; Hwang, G.W.; Yoo, H.J.; Song, J.W. The arachidonic acid metabolite 11,12-epoxyeicosatrienoic acid alleviates pulmonary fibrosis. Exp. Mol. Med. 2021, 53, 864–874. [Google Scholar] [CrossRef]
- Li, J.M.; Yang, D.C.; Oldham, J.; Linderholm, A.; Zhang, J.; Liu, J.; Kenyon, N.J.; Chen, C.H. Therapeutic targeting of argininosuccinate synthase 1 (ASS1)-deficient pulmonary fibrosis. Mol. Ther. 2021, 29, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.T.; Lakshmi, S.P.; Zhang, Y.; Reddy, R.C. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J. 2014, 28, 5299–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, L.; Huang, Y.; Luo, M.; Wang, H.; Jiang, Z.; Zheng, J.; Yang, Z.; Chen, Z.; Zhang, C.; et al. Pharmaceutical targeting of succinate dehydrogenase in fibroblasts controls bleomycin-induced lung fibrosis. Redox Biol. 2021, 46, 102082. [Google Scholar] [CrossRef] [PubMed]
- Malsin, E.S.; Kamp, D.W. The mitochondria in lung fibrosis: Friend or foe? Transl. Res. 2018, 202, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Burgess, J.K.; Mutsaers, S.E.; Prele, C.M.; Schuliga, M.; Grainge, C.L.; Knight, D.A. Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L162–L172. [Google Scholar] [CrossRef] [Green Version]
- Luis-Garcia, E.R.; Becerril, C.; Salgado-Aguayo, A.; Aparicio-Trejo, O.E.; Romero, Y.; Flores-Soto, E.; Mendoza-Milla, C.; Montano, M.; Chagoya, V.; Pedraza-Chaverri, J.; et al. Mitochondrial Dysfunction and Alterations in Mitochondrial Permeability Transition Pore (mPTP) Contribute to Apoptosis Resistance in Idiopathic Pulmonary Fibrosis Fibroblasts. Int. J. Mol. Sci. 2021, 22, 7870. [Google Scholar] [CrossRef]
- Ryu, C.; Sun, H.; Gulati, M.; Herazo-Maya, J.D.; Chen, Y.; Osafo-Addo, A.; Brandsdorfer, C.; Winkler, J.; Blaul, C.; Faunce, J.; et al. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 1571–1581. [Google Scholar] [CrossRef]
- Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1 alpha repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef] [Green Version]
- Leung, J.Y.; Wilson, H.L.; Voltzke, K.J.; Williams, L.A.; Lee, H.J.; Wobker, S.E.; Kim, W.Y. Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B.V. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [Green Version]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Wei, L.; Schuliga, M.; Jaffar, J.; Westall, G.P.; Hansbro, P.M.; Prele, C.M.; Mutsaers, S.E.; et al. STAT3 Regulates the Onset of Oxidant-induced Senescence in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Predescu, S.A.; Zhang, J.; Bardita, C.; Patel, M.; Godbole, V.; Predescu, D.N. Mouse Lung Fibroblast Resistance to Fas-Mediated Apoptosis Is Dependent on the Baculoviral Inhibitor of Apoptosis Protein 4 and the Cellular FLICE-Inhibitory Protein. Front. Physiol. 2017, 8, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.-I.; Groshong, S.D.; Frankel, S.K.; Edelman, B.L.; Cosgrove, G.P.; Terry-Powers, J.L.; Remigio, L.K.; Curran-Everett, D.; Brown, K.K.; Cool, C.D.; et al. Compartmentalized expression of c-FLIP in lung tissues of patients with idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 42, 140–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013, 1, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, K.; Pan, T.; Mou, Y.; Zhang, L.; Xiong, W.; Xu, Y.; Yu, J.; Wang, Y. Scutellarein inhibits BLM-mediated pulmonary fibrosis by affecting fibroblast differentiation, proliferation, and apoptosis. Ther. Adv. Chronic Dis. 2020, 11, 2040622320940185. [Google Scholar] [CrossRef] [PubMed]
- Araya, J.; Kojima, J.; Takasaka, N.; Ito, S.; Fujii, S.; Hara, H.; Yanagisawa, H.; Kobayashi, K.; Tsurushige, C.; Kawaishi, M.; et al. Insufficient autophagy in idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L56–L69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surolia, R.; Li, F.J.; Wang, Z.; Li, H.; Dsouza, K.; Thomas, V.; Mirov, S.; Pérez-Sala, D.; Athar, M.; Thannickal, V.J.; et al. Vimentin intermediate filament assembly regulates fibroblast invasion in fibrogenic lung injury. JCI Insight 2019, 4, e123253. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Xu, Q.; Wan, H.; Xing, S.; Shang, C.; Gao, Y.; He, Z. Lipopolysaccharide promotes lung fibroblast proliferation through autophagy inhibition via activation of the PI3K-Akt-mTOR pathway. Lab. Investig. 2019, 99, 625–633. [Google Scholar] [CrossRef]
- Gui, X.; Chen, H.; Cai, H.; Sun, L.; Gu, L. Leptin promotes pulmonary fibrosis development by inhibiting autophagy via PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 498, 660–666. [Google Scholar] [CrossRef]
- Wang, Y.; Sui, Y.; Lian, A.; Han, X.; Liu, F.; Zuo, K.; Liu, M.; Sun, W.; Wang, Z.; Liu, Z.; et al. Elongation factor-2 kinase acts downstream of p38 MAPK to regulate proliferation, apoptosis and autophagy in human lung fibroblasts. Exp. Cell Res. 2018, 363, 291–298. [Google Scholar] [CrossRef]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escriva, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wang, X.; Yu, H.; Wang, C.; Liu, Y.; Zhao, R.; Zhang, G. Beclin 1, LC3, and p62 expression in paraquat-induced pulmonary fibrosis. Hum. Exp. Toxicol. 2019, 38, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Cherubini, E.; Scozzi, D.; Pietrangeli, V.; Tabbì, L.; Raffa, S.; Leone, L.; Visco, V.; Torrisi, M.R.; Bruno, P.; et al. Decreased expression of autophagic beclin 1 protein in idiopathic pulmonary fibrosis fibroblasts. J. Cell. Physiol. 2013, 228, 1516–1524. [Google Scholar] [CrossRef]
- Baek, A.R.; Hong, J.; Song, K.S.; Jang, A.S.; Kim, D.J.; Chin, S.S.; Park, S.W. Spermidine attenuates bleomycin-induced lung fibrosis by inducing autophagy and inhibiting endoplasmic reticulum stress (ERS)-induced cell death in mice. Exp. Mol. Med. 2020, 52, 2034–2045. [Google Scholar] [CrossRef]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Ghavami, S.; Yeganeh, B.; Zeki, A.A.; Shojaei, S.; Kenyon, N.J.; Ott, S.; Samali, A.; Patterson, J.; Alizadeh, J.; Moghadam, A.R.; et al. Autophagy and the unfolded protein response promote profibrotic effects of TGF-beta(1) in human lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L493–L504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.M.; Zhang, J.; Zhang, Y.; Fei, C.; Wang, L.; Yi, Z.W.; Zhang, Z.Q. Interleukin-18 promotes fibroblast senescence in pulmonary fibrosis through down-regulating Klotho expression. Biomed. Pharmacother. 2019, 113, 108756. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. CCN2 induces cellular senescence in fibroblasts. J. Cell Commun. Signal. 2017, 11, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, H.; Hou, J.; Wang, H.; Zheng, Y.; Li, H.; Cai, H.; Han, X.; Dai, J. Epithelial cell senescence induces pulmonary fibrosis through Nanog-mediated fibroblast activation. Aging 2019, 12, 242–259. [Google Scholar] [CrossRef]
- Morimoto, K.; Janssen, W.J.; Terada, M. Defective efferocytosis by alveolar macrophages in IPF patients. Respir. Med. 2012, 106, 1800–1803. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Shouval, D.S.; Biswas, A.; Goettel, J.A.; McCann, K.; Conaway, E.; Redhu, N.S.; Mascanfroni, I.D.; Al Adham, Z.; Lavoie, S.; Ibourk, M.; et al. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 2014, 40, 706–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zigmond, E.; Bernshtein, B.; Friedlander, G.; Walker, C.R.; Yona, S.; Kim, K.-W.; Brenner, O.; Krauthgamer, R.; Varol, C.; Müller, W.; et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 2014, 40, 720–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Jiao, Y.; Fan, E.K.; Scott, M.J.; Li, Y.; Li, S.; Billiar, T.R.; Wilson, M.A.; Shi, X.; Fan, J. Aging-Impaired Filamentous Actin Polymerization Signaling Reduces Alveolar Macrophage Phagocytosis of Bacteria. J. Immunol. 2017, 199, 3176–3186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.K.; Smith, C.A.; Sakamoto, K.; Kaminski, N.; Koff, J.L.; Goldstein, D.R. Aging Impairs Alveolar Macrophage Phagocytosis and Increases Influenza-Induced Mortality in Mice. J. Immunol. 2017, 199, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Liu, Q.; Adrianto, I.; Wu, X.; Glassbrook, J.; Khalasawi, N.; Yin, C.; Yi, Q.; Dong, Z.; Geissmann, F.; et al. Histone deacetylase 3 controls lung alveolar macrophage development and homeostasis. Nat. Commun. 2020, 11, 3822. [Google Scholar] [CrossRef]
- Aran, D.; Looney, A.P.; Liu, L.; Wu, E.; Fong, V.; Hsu, A.; Chak, S.; Naikawadi, R.P.; Wolters, P.J.; Abate, A.R.; et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019, 20, 163–172. [Google Scholar] [CrossRef]
- Madala, S.K.; Pesce, J.T.; Ramalingam, T.R.; Wilson, M.S.; Minnicozzi, S.; Cheever, A.W.; Thompson, R.W.; Mentink-Kane, M.M.; Wynn, T.A. Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J. Immunol. 2010, 184, 3955–3963. [Google Scholar] [CrossRef] [Green Version]
- Pellicoro, A.; Aucott, R.L.; Ramachandran, P.; Robson, A.J.; Fallowfield, J.A.; Snowdon, V.K.; Hartland, S.N.; Vernon, M.; Duffield, J.S.; Benyon, R.C.; et al. Elastin accumulation is regulated at the level of degradation by macrophage metalloelastase (MMP-12) during experimental liver fibrosis. Hepatology 2012, 55, 1965–1975. [Google Scholar] [CrossRef]
- Ucero, A.C.; Bakiri, L.; Roediger, B.; Suzuki, M.; Jimenez, M.; Mandal, P.; Braghetta, P.; Bonaldo, P.; Paz-Ares, L.; Fustero-Torre, C.; et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J. Clin. Investig. 2019, 129, 3293–3309. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.I.; Ibarra, L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.S.; Kearley, J.; Copenhaver, A.M.; Sanden, C.; Mori, M.; Yu, L.; Pritchard, G.H.; Berlin, A.A.; Hunter, C.A.; Bowler, R.; et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat. Immunol. 2016, 17, 626–635. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuka, Y.; Yaku, A.; Handa, T.; Vandenbon, A.; Hikichi, Y.; Motomura, Y.; Sato, A.; Yoshinaga, M.; Tanizawa, K.; Watanabe, K.; et al. Profibrotic function of pulmonary group 2 innate lymphoid cells is controlled by regnase-1. Eur. Respir. J. 2021, 57, 2000018. [Google Scholar] [CrossRef]
- Hrusch, C.L.; Manns, S.T.; Bryazka, D.; Casaos, J.; Bonham, M.C.; Jaffery, M.R.; Blaine, K.M.; Mills, K.A.M.; Verhoef, A.; Adegunsoye, A.O.; et al. ICOS protects against mortality from acute lung injury through activation of IL-5(+) ILC2s. Mucosal Immunol. 2018, 11, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Aoki, A.; Morimoto, Y.; Kiuchi, M.; Okano, M.; Nakayama, T. The immunopathology of lung fibrosis: Amphiregulin-producing pathogenic memory T helper-2 cells control the airway fibrotic responses by inducing eosinophils to secrete osteopontin. Semin. Immunopathol. 2019, 41, 339–348. [Google Scholar] [CrossRef]
- Yombo, D.J.K.; Odayar, V.; Gupta, N.; Jegga, A.G.; Madala, S.K. The Protective Effects of IL-31RA Deficiency During Bleomycin-Induced Pulmonary Fibrosis. Front. Immunol. 2021, 12, 645717. [Google Scholar] [CrossRef]
- Celada, L.J.; Kropski, J.A.; Herazo-Maya, J.D.; Luo, W.; Creecy, A.; Abad, A.T.; Chioma, O.S.; Lee, G.; Hassell, N.E.; Shaginurova, G.I.; et al. PD-1 up-regulation on CD4(+) T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-beta1 production. Sci. Transl. Med. 2018, 10, eaar8356. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Gong, L.; Li, Z.; Chen, H.; Xu, M.; Rong, R.; Zhang, Y.; Zhu, Q. Antifibrotic effect of Gancao Ganjiang decoction is mediated by PD-1/TGF-beta1/IL-17A pathway in bleomycin-induced idiopathic pulmonary fibrosis. J. Ethnopharmacol. 2021, 281, 114522. [Google Scholar] [CrossRef]
- Wang, Y.; Kuai, Q.; Gao, F.; Wang, Y.; He, M.; Zhou, H.; Han, G.; Jiang, X.; Ren, S.; Yu, Q. Overexpression of TIM-3 in Macrophages Aggravates Pathogenesis of Pulmonary Fibrosis in Mice. Am. J. Respir. Cell Mol. Biol. 2019, 61, 727–736. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.E.; Ahangari, F.; Li, Q.; Jain, S.; Verleden, S.E.; Herazo-Maya, J.; Vukmirovic, M.; Deiuliis, G.; Tzouvelekis, A.; Tanabe, N.; et al. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight 2019, 4, e131597. [Google Scholar] [CrossRef] [PubMed]
- Heukels, P.; Van Hulst, J.A.C.; Van Nimwegen, M.; Boorsma, C.E.; Melgert, B.N.; Von Der Thusen, J.H.; Blink, B.V.D.; Hoek, R.A.S.; Miedema, J.R.; Neys, S.F.H.; et al. Enhanced Bruton’s tyrosine kinase in B-cells and autoreactive IgA in patients with idiopathic pulmonary fibrosis. Respir. Res. 2019, 20, 232. [Google Scholar] [CrossRef] [PubMed]
- McDonough, J.E.; Kaminski, N.; Thienpont, B.; Hogg, J.C.; Vanaudenaerde, B.M.; Wuyts, W.A. Gene correlation network analysis to identify regulatory factors in idiopathic pulmonary fibrosis. Thorax 2019, 74, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.F.; Egan, A.M.; Shaughnessy, G.F.; Anderson, D.K.; Kottom, T.J.; Dasari, H.; Van Keulen, V.P.; Aubry, M.C.; Yi, E.S.; Limper, A.H.; et al. Antifibrotics Modify B-Cell-induced Fibroblast Migration and Activation in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2021, 64, 722–733. [Google Scholar] [CrossRef]
- Cho, S.J.; Stout-Delgado, H.W. Aging and Lung Disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef] [Green Version]
- Galambos, C.; Ng, Y.-S.; Ali, A.; Noguchi, A.; Lovejoy, S.; D’Amore, P.A.; Demello, D.E. Defective pulmonary development in the absence of heparin-binding vascular endothelial growth factor isoforms. Am. J. Respir. Cell Mol. Biol. 2002, 27, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Murray, L.A.; Habiel, D.M.; Hohmann, M.; Camelo, A.; Shang, H.; Zhou, Y.; Coelho, A.L.; Peng, X.; Gulati, M.; Crestani, B.; et al. Antifibrotic role of vascular endothelial growth factor in pulmonary fibrosis. JCI Insight 2017, 2, e92192. [Google Scholar] [CrossRef] [Green Version]
- Barratt, S.L.; Blythe, T.; Jarrett, C.; Ourradi, K.; Shelley-Fraser, G.M.; Day, J.; Qiu, Y.; Harper, S.; Maher, T.M.; Oltean, S.; et al. Differential Expression of VEGF-A(xxx) Isoforms Is Critical for Development of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, S.; Morrell, C.N.; Tarango, C.; Thomas, G.D.; Yuhanna, I.S.; Girardi, G.; Herz, J.; Urbanus, R.T.; de Groot, P.G.; Thorpe, P.E.; et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via beta2GPI and apoER2. J. Clin. Investig. 2011, 121, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory modeling of endothelial-to-mesenchymal transition reveals galectin-3 as a mediator in pulmonary fibrosis. Cell Death Dis. 2021, 12, 327. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Zhang, J.; Miao, Y.; He, M.; Kang, J.; Huang, H.-Y.; Chou, C.-H.; Huang, T.-S.; Hong, H.-C.; Su, S.-H.; et al. Role of endothelial cells in pulmonary fibrosis via SREBP2 activation. JCI Insight 2021, 6, e125635. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.A.; Fisher, A.J.; Borthwick, L.A. The Role of Epithelial Damage in the Pulmonary Immune Response. Cells 2021, 10, 2763. [Google Scholar] [CrossRef]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.; Heins, H.; Na, C.-L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwara, M.I.; Green, N.J.; Borthwick, L.A.; Mann, J.; Mayer-Barber, K.D.; Barron, L.; Corris, P.A.; Farrow, S.N.; Wynn, T.A.; Fisher, A.J.; et al. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014, 7, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Chun, W.; Lee, H.J.; Min, J.H.; Kim, S.M.; Seo, J.Y.; Ahn, K.S.; Oh, S.R. The Role of Macrophages in the Development of Acute and Chronic Inflammatory Lung Diseases. Cells 2021, 10, 897. [Google Scholar] [CrossRef]
- Smadja, D.M.; Mauge, L.; Nunes, H.; d’Audigier, C.; Juvin, K.; Borie, R.; Carton, Z.; Bertil, S.; Blanchard, A.; Crestani, B.; et al. Imbalance of circulating endothelial cells and progenitors in idiopathic pulmonary fibrosis. Angiogenesis 2013, 16, 147–157. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The MSC: An injury drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | Function and Mechanism | Effects in Lung Disease | Ref. |
|---|---|---|---|
| AT1 | Gas exchange, Ion and liquid transport, Congenital immunity | Compositional gas barrier, Involved in inflammation | [15,16,17] |
| AT2 | Self-renewal and differentiation, Damage repair | Impaired stem cell function, Pro-fibrotic signaling, ER stress, Telomere attrition, Mitochondrial dysfunction, Differentiate into fibroblasts | [31,33,39,40,41,46,47,54,55,56] |
| Abnormal basaloid cells | Transdifferentiation | Alveolar bronchogenic phenotype | [64,65] |
| MSC | Self-renewal and differentiation, Damage repair, Immunoregulation | Secreted bioactive molecules, Inhibited fibroblast activation, Inhibited cell apoptosis, Reduced epithelial-mesenchymal transition | [67,68,69,70,71] |
| Fibroblasts | Tissue repair | ECM deposition, Differentiate into myoblasts, Telomere shortening, Metabolic abnormality, Mitochondrial damage, Resistance to apoptosis, Autophagy, Cellular senescence | [86,87,88,89,92,93,94,95,96,99,100,104,114,115,122,123,126] |
| Immune Cells | Damage repair, Immunoregulation | Promote the release of inflammatory factors, ECM deposition/Degradation of ECM | [141,142,153,154,157,162,163,164] |
| ECs | Angiogenesis, Damage repair, Immunoregulation | Anti-inflammation, Inhibited cell apoptosis/Differentiate into fibroblasts | [92,94,166,167] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Wang, J. Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis. Adv. Respir. Med. 2023, 91, 26-48. https://doi.org/10.3390/arm91010005
Zhang Y, Wang J. Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis. Advances in Respiratory Medicine. 2023; 91(1):26-48. https://doi.org/10.3390/arm91010005
Chicago/Turabian StyleZhang, Yihang, and Jiazhen Wang. 2023. "Cellular and Molecular Mechanisms in Idiopathic Pulmonary Fibrosis" Advances in Respiratory Medicine 91, no. 1: 26-48. https://doi.org/10.3390/arm91010005