

Inter-Population Genetic Diversity and Clustering of Merozoite Surface Protein-1 (pkmsp-1) of Plasmodium knowlesi Isolates from Malaysia and Thailand
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Clearance
2.2. Plasmodium Species Confirmation
2.3. DNA Extraction
2.4. Amplification of pkmsp-1 Gene
2.5. Cloning and Sequencing of pkmsp-1
2.6. Genetic Diversity and Natural Selection Analysis
2.7. Phylogenetic Inference
3. Results
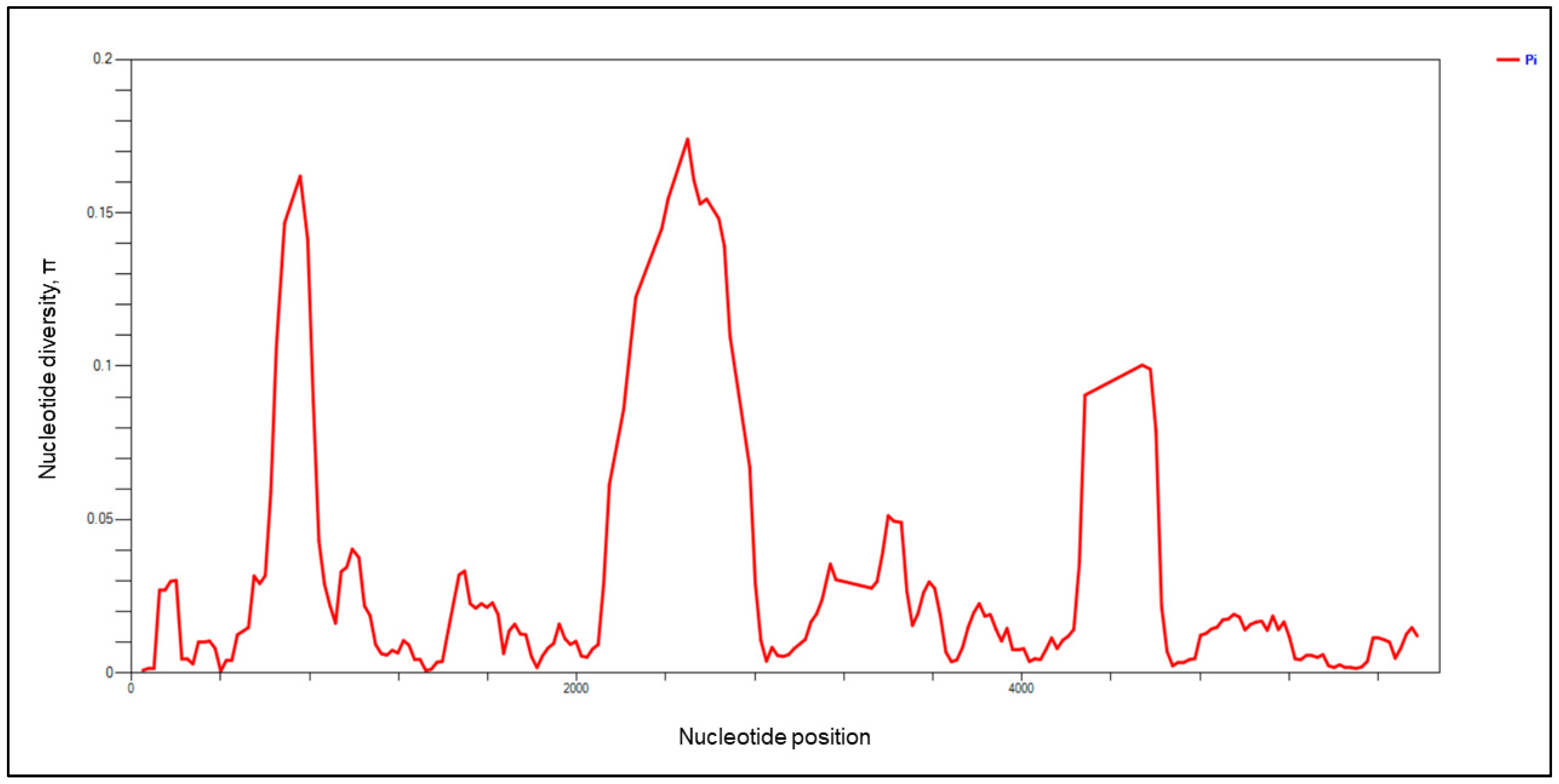
3.1. Genetic Diversity and Natural Selection of pkmsp-1
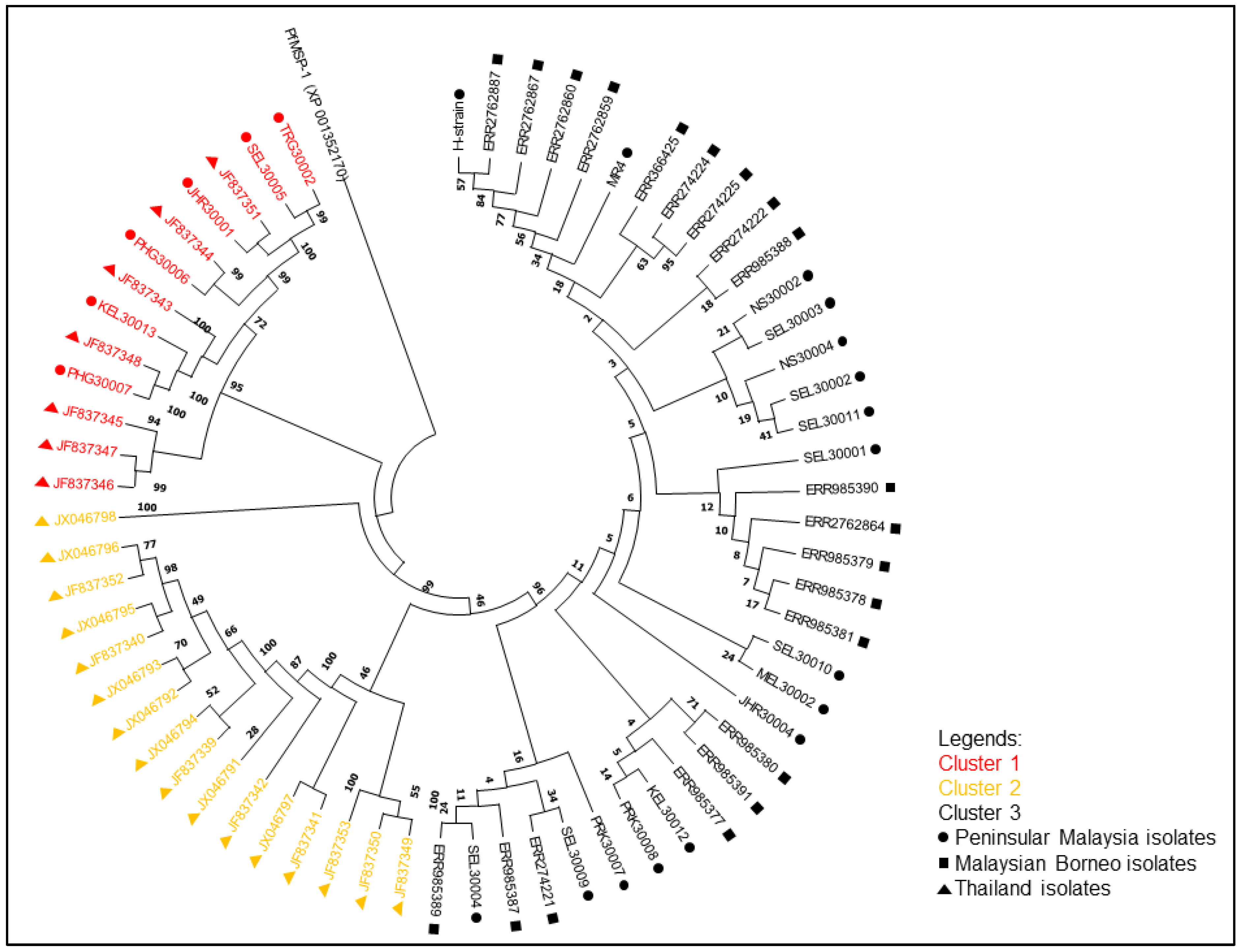
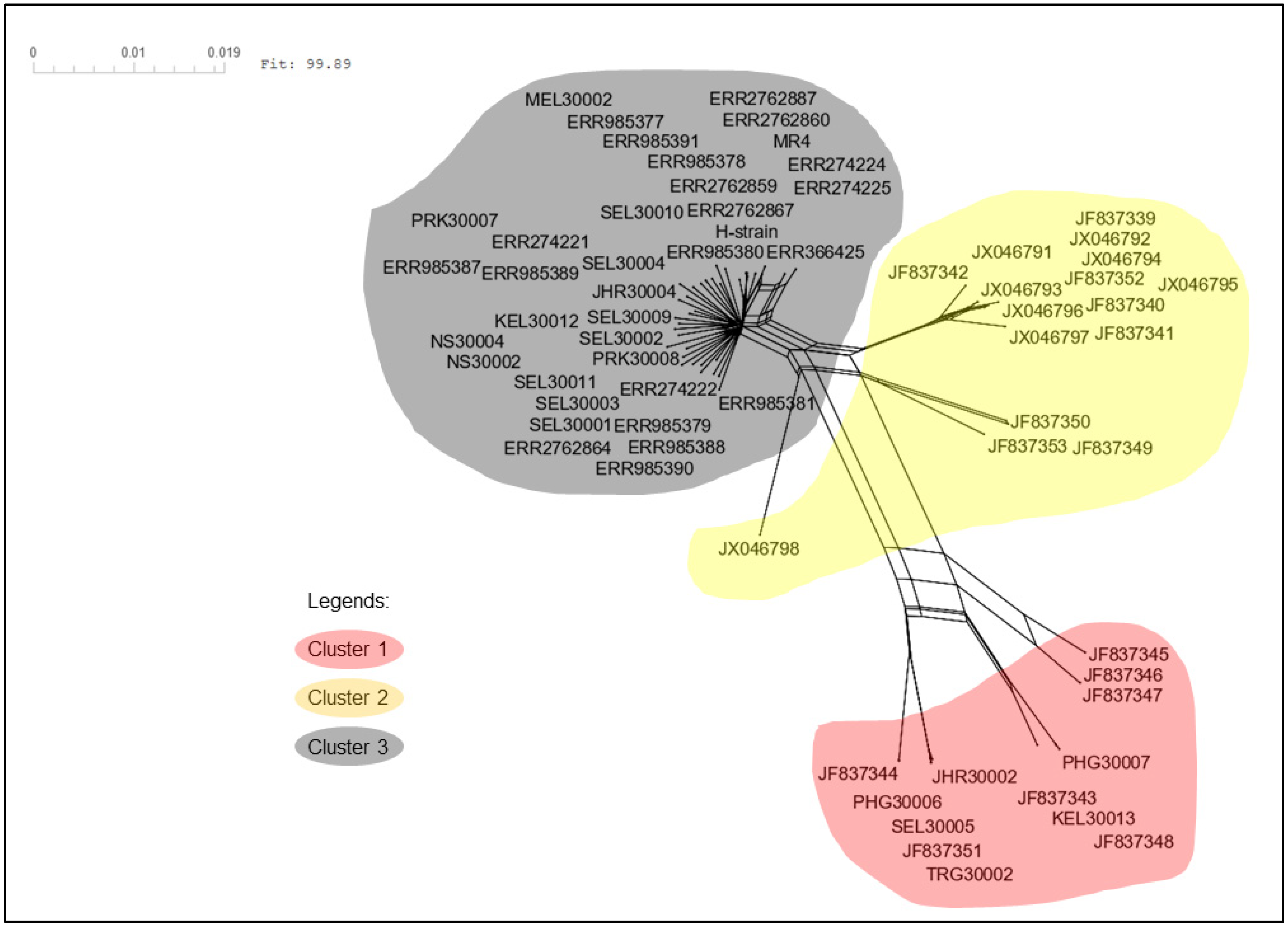
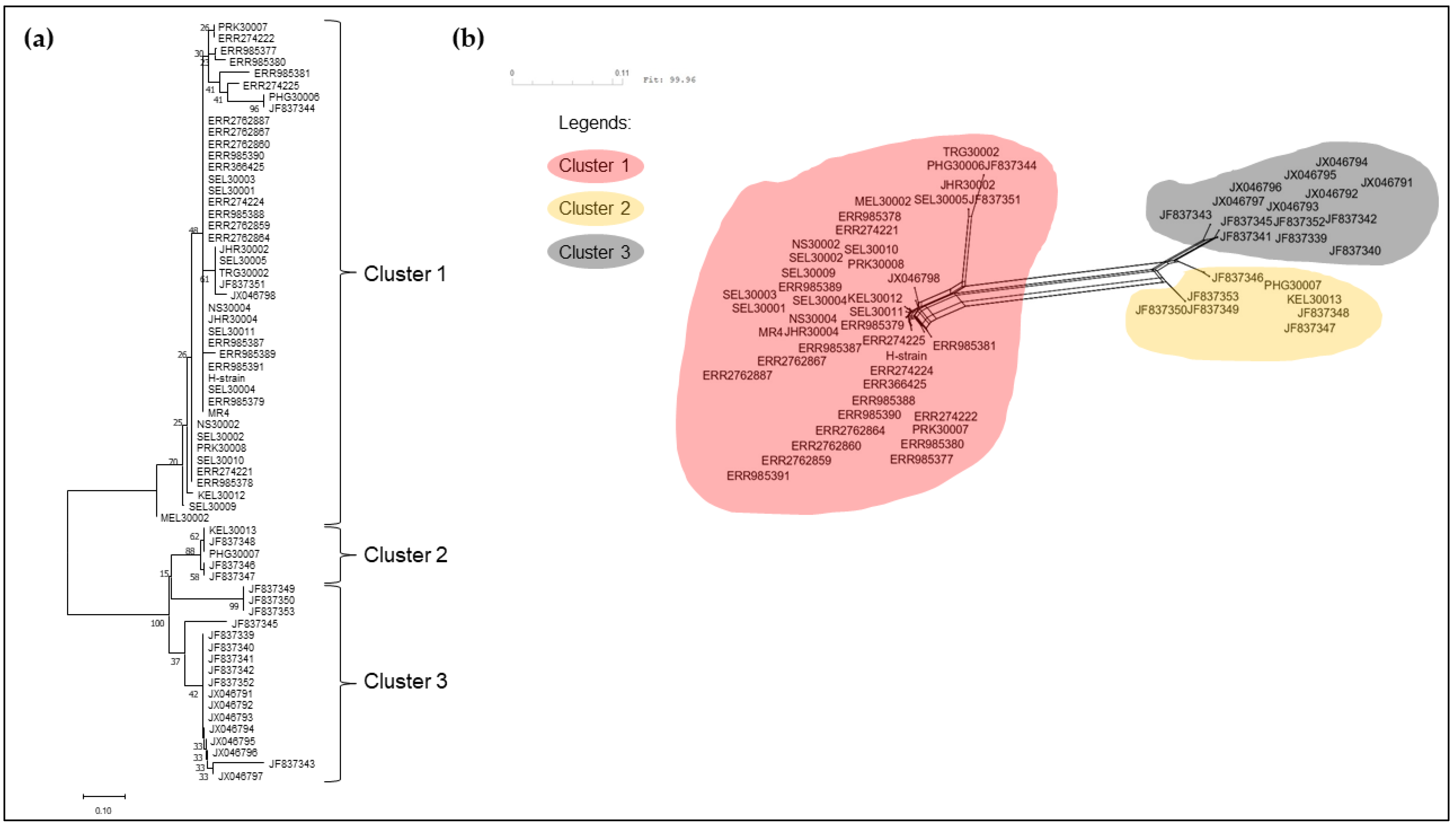
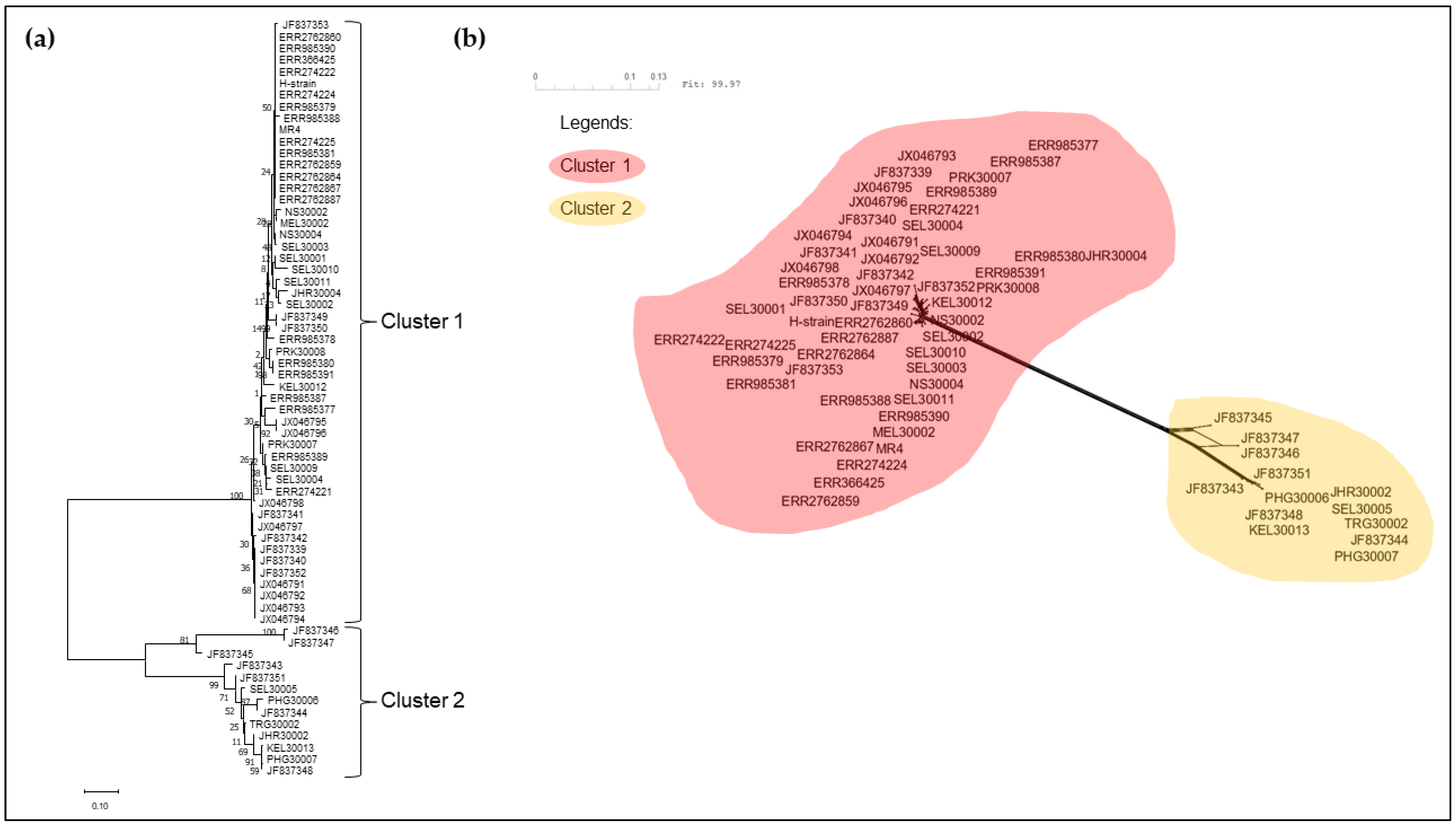
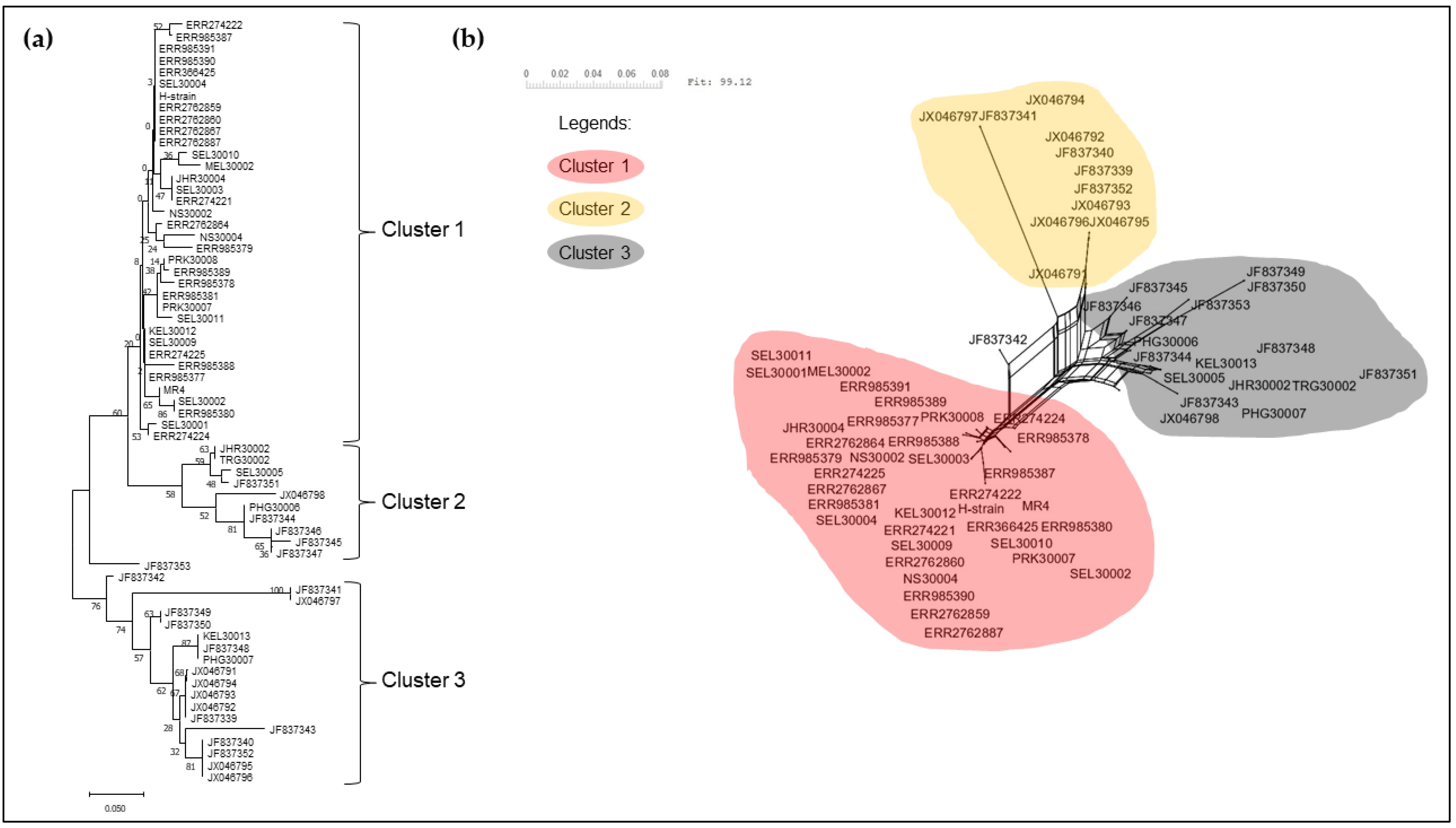
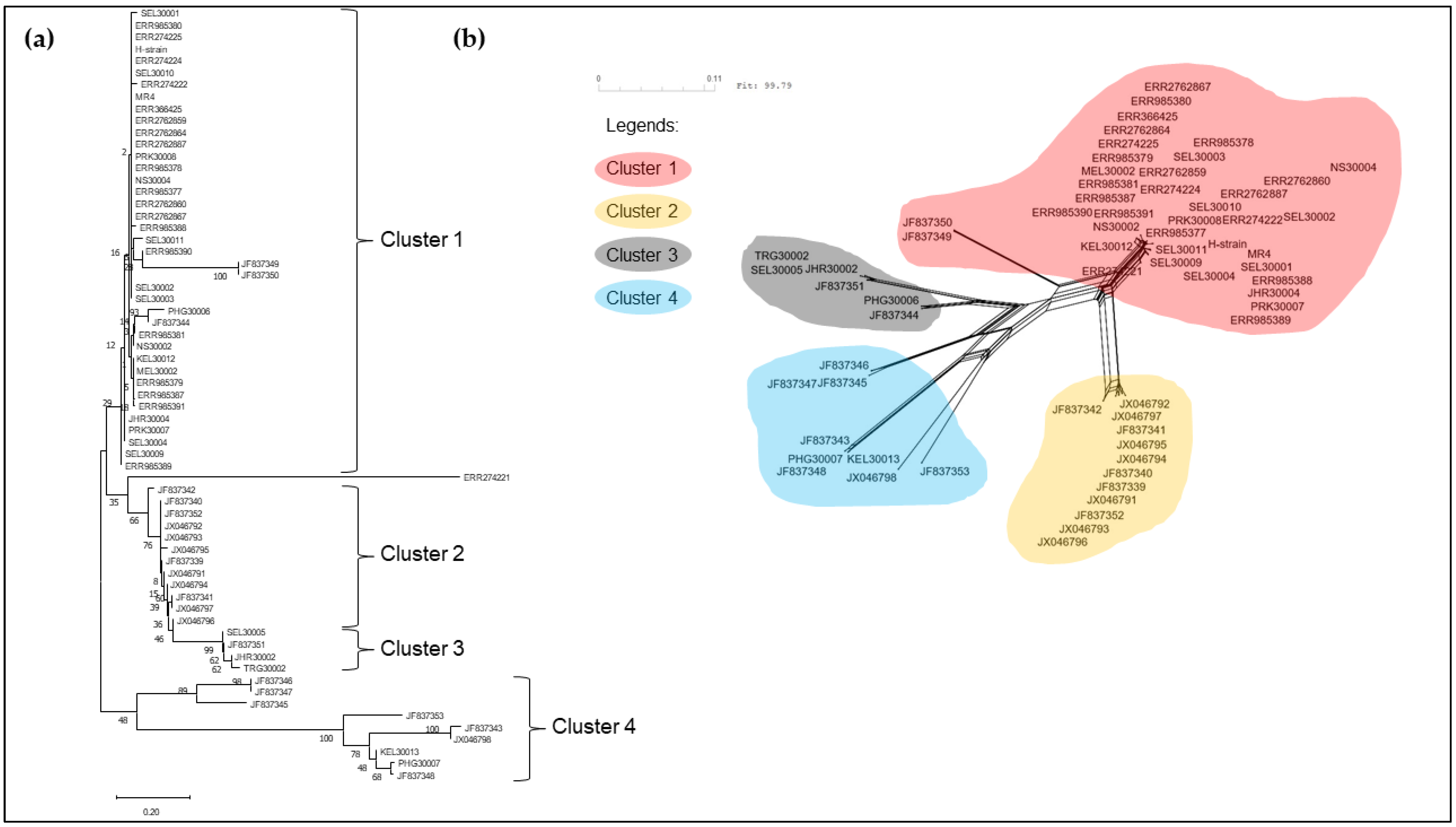
3.2. Phylogenetic Analysis of pkmsp-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2022; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2022; Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed on 9 March 2023).
- Koh, G.J.; Ismail, P.K.; Koh, D. Occupationally Acquired Plasmodium knowlesi Malaria in Brunei Darussalam. Saf. Health Work. 2019, 10, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Ngernna, S.; Rachaphaew, N.; Thammapalo, S.; Prikchoo, P.; Kaewnah, O.; Manopwisedjaroen, K.; Phumchuea, K.; Suansomjit, C.; Roobsoong, W.; Sattabongkot, J.; et al. Case report: Case series of human Plasmodium knowlesi infection on the Southern border of Thailand. Am. J. Trop. Med. Hyg. 2019, 101, 1397–1401. [Google Scholar] [CrossRef] [PubMed]
- Lubis, I.N.D.; Wijaya, H.; Lubis, M.; Lubis, C.P.; Divis, P.C.S.; Beshir, K.B.; Sutherland, C.J. Contribution of Plasmodium knowlesi to multispecies human Malaria infections in North Sumatera, Indonesia. J. Infect. Dis. 2017, 215, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Iwagami, M.; Nakatsu, M.; Khattignavong, P.; Soundala, P.; Lorphachan, L.; Keomalaphet, S.; Xangsayalath, P.; Kawai, S.; Hongvanthong, B.; Brey, P.T.; et al. First case of human infection with Plasmodium knowlesi in Laos. PLoS Negl. Trop. Dis. 2018, 12, e0006244. [Google Scholar] [CrossRef] [PubMed]
- Vythilingam, I.; Wong, M.L.; Wan-Yussof, W.S. Current status of Plasmodium knowlesi vectors: A public health concern? Parasitology 2018, 145, 32–40. [Google Scholar] [CrossRef]
- Sinton, J.A.; Mulligan, H.W. A critical review of the literature relating to the identification of the malaria parasites recorded from monkeys of the families Ceropithecidae and Colobidae. Rec. Malar. Surv. India 1932, 3, 357–380. [Google Scholar]
- Daneshvar, C.; Davis, T.M.; Cox-Singh, J.; Rafa’ee, M.Z.; Zakaria, S.K.; Divis, P.C.; Singh, B. Clinical and laboratory features of human Plasmodium knowlesi infection. Clin. Infect. Dis. 2009, 49, 852–860. [Google Scholar] [CrossRef]
- Fornace, K.M.; Herman, L.S.; Abidin, T.R.; Chua, T.H.; Daim, S.; Lorenzo, P.J.; Grignard, L.; Nuin, N.A.; Ying, L.T.; Grigg, M.J.; et al. Exposure and infection to Plasmodium knowlesi in case study communities in Northern Sabah, Malaysia and Palawan, The Philippines. PLoS Negl. Trop. Dis. 2018, 12, e0006432. [Google Scholar] [CrossRef]
- Kotepui, M.; Kotepui, K.U.; Milanez, G.D.; Masangkay, F.R. Prevalence of severe Plasmodium knowlesi infection and risk factors related to severe complications compared with non-severe P. knowlesi and severe P. falciparum malaria: A systematic review and meta-analysis. Infect. Dis. Poverty 2020, 9, 106. [Google Scholar] [CrossRef]
- Kaur, C.; Pramanik, A.; Kumari, K.; Mandage, R.; Dinda, A.K.; Sankar, J.; Bagga, A.; Agarwal, S.K.; Sinha, A.; Singh, G.; et al. Renal detection of Plasmodium falciparum, Plasmodium vivax and Plasmodium knowlesi in malaria associated acute kidney injury: A retrospective case-control study. BMC Res. Notes 2020, 13, 37. [Google Scholar] [CrossRef]
- Chin, A.Z.; Avoi, R.; Atil, A.; Awang Lukman, K.; Syed Abdul Rahim, S.S.; Ibrahim, M.Y.; Ahmed, K.; Jeffree, M.S. Risk factor of Plasmodium knowlesi infection in Sabah Borneo Malaysia, 2020: A population-based case-control study. PLoS ONE. 2021, 16, e0257104. [Google Scholar] [CrossRef]
- Fornace, K.M.; Abidin, T.R.; Alexander, N.; Brock, P.; Grigg, M.J.; Murphy, A.; William, T.; Menon, J.; Drakeley, C.J.; Cox, J. Association between landscape factors and spatial patterns of Plasmodium knowlesi infections in Sabah, Malaysia. Emerg. Infect. Dis. 2016, 22, 201–208. [Google Scholar] [CrossRef]
- World Health Organization. Malaria Policy Advisory Group (MPAG) Meeting. Available online: https://www.who.int/news-room/events/detail/2021/04/13/default-calendar/19th-meeting-of-the-malaria-policy-advisory-group (accessed on 16 March 2023).
- Noordin, N.R.; Lee, P.Y.; Mohd Bukhari, F.D.; Fong, M.Y.; Abdul Hamid, M.H.; Jelip, J.; Mudin, R.N.; Lau, Y.L. Prevalence of asymptomatic and/or low-density malaria infection among high-risk groups in Peninsular Malaysia. Am. J. Trop. Med. Hyg. 2020, 103, 1107–1110. [Google Scholar] [CrossRef]
- Imwong, M.; Madmanee, W.; Suwannasin, K.; Kunasol, C.; Peto, T.J.; Tripura, R.; von Seidlein, L.; Nguon, C.; Davoeung, C.; Day, N.P.J.; et al. Asymptomatic natural human infections with the simian malaria parasites Plasmodium cynomolgi and Plasmodium knowlesi. J. Infect. Dis. 2019, 219, 695–702. [Google Scholar] [CrossRef]
- Aikawa, M.; Miller, L.H.; Johnson, J.; Rabbege, J. Erythrocyte entry by malarial parasites. J. Cell Biol. 1978, 77, 72–82. [Google Scholar] [CrossRef]
- Holder, A.A.; Freeman, R.R. Biosynthesis and processing of a Plasmodium falciparum schizont antigen recognized by immune serum and a monoclonal antibody. J. Exp. Med. 1982, 156, 1528–1538. [Google Scholar] [CrossRef]
- Pachebat, J.A.; Kadekoppala, M.; Grainger, M.; Dluzewski, A.R.; Gunaratne, R.S.; Scott-Finnigan, T.J.; Ogun, S.A.; Ling, I.T.; Bannister, L.H.; Taylor, H.M.; et al. Extensive proteolytic processing of the malaria parasite merozoite surface protein 7 during biosynthesis and parasite release from erythrocytes. Mol. Biochem. Parasitol. 2007, 151, 59–69. [Google Scholar] [CrossRef]
- Trucco, C.; Fernandez-Reyes, D.; Howell, S.; Stafford, W.H.; Scott-Finnigan, T.J.; Grainger, M.; Ogun, S.A.; Taylor, W.R.; Holder, A.A. The merozoite surface protein 6 gene codes for a 36 kDa protein associated with the Plasmodium falciparum merozoite surface protein-1 complex. Mol. Biochem. Parasitol. 2001, 112, 91–101. [Google Scholar] [CrossRef]
- Holder, A.A.; Blackman, M.J.; Burghaus, P.A.; Chappel, J.A.; Ling, I.T.; McCallum-Deighton, N.; Shai, S. A malaria merozoite surface protein (MSP1)-structure, processing and function. Mem. Inst. Oswaldo. Cruz. 1992, 87, 37–42. [Google Scholar] [CrossRef]
- Blackman, M.J.; Dennis, E.D.; Hirst, E.M.A.; Kocken, C.H.; Scott-Finnigan, T.J.; Thomas, A.W. Plasmodium knowlesi: Secondary processing of the malaria merozoite surface protein-1. Exp. Parasitol. 1996, 83, 229–239. [Google Scholar] [CrossRef]
- Holder, A.A. The carboxy-terminus of merozoite surface protein 1: Structure, specific antibodies and immunity to malaria. Parasitology 2009, 136, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Bergmann-Leitner, E.S.; Duncan, E.H.; Angov, E. MSP-1p42-specific antibodies affect growth and development of intra-erythrocytic parasites of Plasmodium falciparum. Malar. J. 2009, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.W.; Fowkes, F.J.; Gilson, P.R.; Elliott, S.R.; Tavul, L.; Michon, P.; Dabod, E.; Siba, P.M.; Mueller, I.; Crabb, B.S.; et al. Quantifying the importance of MSP1-19 as a target of growth-inhibitory and protective antibodies against Plasmodium falciparum in humans. PLoS ONE 2011, 6, e27705. [Google Scholar] [CrossRef] [PubMed]
- Yap, N.J.; Vythilingam, I.; Hoh, B.P.; Goh, X.T.; Muslim, A.; Ngui, R.; Rajoo, Y.; Choy, S.H.; William, T.; Yeo, T.W.; et al. Genetic polymorphism and natural selection in the C-terminal 42 kDa region of merozoite surface protein-1 (MSP-1) among Plasmodium knowlesi samples from Malaysia. Parasites Vectors 2018, 11, 626. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Chu, K.B.; Vythilingam, I.; Quan, F.S. Within-population genetic diversity and population structure of Plasmodium knowlesi merozoite surface protein 1 gene from geographically distinct regions of Malaysia and Thailand. Malar. J. 2018, 17, 442. [Google Scholar] [CrossRef]
- Viriyakosol, S.; Siripoon, N.; Petcharapirat, C.; Petcharapirat, P.; Jarra, W.; Thaithong, S.; Brown, K.N.; Snounou, G. Genotyping of Plasmodium falciparum isolates by the polymerase chain reaction and potential uses in epidemiological studies. Bull. World Health Organ. 1995, 73, 85–95. [Google Scholar]
- Ntoumi, F.; Ngoundou-Landji, J.; Luty, A.J.; Dubreuil, G.; Millet, P. Allelic polymorphism of Plasmodium falciparum MSP-2 gene in blood samples from Gabonese children. Bull. Soc. Pathol. Exot. 2001, 94, 183–187. [Google Scholar]
- Opute, A.O.; Akinkunmi, J.A.; Funsho, A.O.; Obaniyi, A.K.; Anifowoshe, A.T. Genetic diversity of Plasmodium falciparum isolates in Nigeria. A review. Egypt J. Med. Hum. Genet. 2022, 23, 129. [Google Scholar] [CrossRef]
- Snounou, G.; Zhu, X.; Siripoon, N.; Jarra, W.; Thaithong, S.; Brown, K.N.; Viriyakosol, S. Biased distribution of msp1 and msp2 allelic variants in Plasmodium falciparum populations in Thailand. Trans. R Soc. Trop. Med. Hyg. 1999, 93, 369–374. [Google Scholar] [CrossRef]
- Soares, L.A.; Evangelista, J.; Orlandi, P.P.; Almeida, M.E.; de Sousa, L.P.; Chaves, Y.; Barbosa-Filho, R.; Lacerda, M.V.; Mariuba, L.A.; Nogueira, P.A. Genetic diversity of MSP1 Block 2 of Plasmodium vivax isolates from manaus (Central Brazilian Amazon). J. Immunol. Res. 2014, 2014, 14–16. [Google Scholar] [CrossRef]
- Fola, A.A.; Harrison, G.L.A.; Hazairin, M.H.; Barnadas, C.; Hetzel, M.W.; Iga, J.; Siba, P.M.; Mueller, I.; Barry, A.E. Higher complexity of infection and genetic diversity of Plasmodium vivax than Plasmodium falciparum across all malaria transmission zones of Papua New Guinea. Am. J. Trop. Med. Hyg. 2017, 96, 630–641. [Google Scholar] [CrossRef]
- Abukari, Z.; Okonu, R.; Nyarko, S.B.; Lo, A.C.; Dieng, C.C.; Salifu, S.P.; Gyan, B.A.; Lo, E.; Amoah, L.E. The diversity, multiplicity of infection and population structure of P. falciparum parasites circulating in asymptomatic carriers living in high and low malaria transmission settings of Ghana. Genes 2019, 10, 434. [Google Scholar] [CrossRef]
- Putaporntip, C.; Thongaree, S.; Jongwutiwes, S. Differential sequence diversity at merozoite surface protein-1 locus of Plasmodium knowlesi from humans and macaques in Thailand. Infect. Genet. Evol. 2013, 18, 213–219. [Google Scholar] [CrossRef]
- Snounou, G.; Viriyakosol, S.; Jarra, W.; Thaithong, S.; Brown, K. Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol. Biochem. Parasitol. 1993, 58, 283–292. [Google Scholar] [CrossRef]
- Imwong, M.; Tanomsing, N.; Pukrittayakamee, S.; Day, N.P.J.; White, N.J.; Snounou, G. Spurious amplification of a Plasmodium vivax small-subunit RNA gene by use of primers currently used to detect P. knowlesi. J. Clin. Microbiol. 2009, 47, 4173–4175. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Hudson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Sondo, P.; Derra, K.; Rouamba, T.; Nakanabo Diallo, S.; Taconet, P.; Kazienga, A.; Ilboudo, H.; Tahita, M.C.; Valéa, I.; Sorgho, H. Determinants of Plasmodium falciparum multiplicity of infection and genetic diversity in Burkina Faso. Parasites Vectors 2020, 13, 427. [Google Scholar] [CrossRef]
- Somé, A.F.; Bazié, T.; Zongo, I.; Yerbanga, R.S.; Nikiéma, F.; Neya, C.; Taho, L.K.; Ouédraogo, J.B. Plasmodium falciparum msp1 and msp2 genetic diversity and allele frequencies in parasites isolated from symptomatic malaria patients in Bobo-Dioulasso, Burkina Faso. Parasites Vectors 2018, 11, 323. [Google Scholar] [CrossRef]
- Usman-Yamman, H.; Omalu, C.J.I.; Abubakar, A.; Abolarinwa, S.O.; Eke, S.S.; Otuu, C.A. Genetic diversity of Plasmodium falciparum isolates in Minna, North Central Nigeria inferred by PCR genotyping of merozoite surface protein 1 and 2. Infect. Genet. Evol. 2021, 96, 105143. [Google Scholar] [CrossRef]
- Jamil, K.F.; Pratama, N.R.; Marantina, S.S.; Harapan, H.; Kurniawan, M.R.; Zanaria, T.M.; Hutagalung, J.; Rozi, I.E.; Asih, P.B.S.; Supargiyono; et al. Allelic diversity of merozoite surface protein genes (msp1 and msp2) and clinical manifestations of Plasmodium falciparum malaria cases in Aceh, Indonesia. Malar. J. 2021, 20, 182. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.A.; Lau, Y.L.; Quan, F.S. Diversity and natural selection on the thrombospondin-related adhesive protein (TRAP) gene of Plasmodium knowlesi in Malaysia. Malar. J. 2018, 17, 274. [Google Scholar] [CrossRef]
- Chua, C.Y.; Lee, P.C.; Lau, T.Y. Analysis of polymorphisms and selective pressures on ama1 gene in Plasmodium knowlesi isolates from Sabah, Malaysia. J. Genet. 2017, 96, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Lau, Y.L.; Jelip, J.; Ooi, C.H.; Cheong, F.W. Genetic characterisation of the erythrocyte-binding protein (PkβII) of Plasmodium knowlesi isolates from Malaysia. J. Genet. 2019, 98, 64. [Google Scholar] [CrossRef] [PubMed]
- Fong, M.Y.; Rashdi, S.A.A.; Yusof, R.; Lau, Y.L. Genetic diversity, natural selection and haplotype grouping of Plasmodium knowlesi gamma protein region II (PkγRII): Comparison with the Duffy binding protein (PkDBPαRII). PLoS ONE 2016, 11, e0155627. [Google Scholar] [CrossRef]
- Fong, M.Y.; Lau, Y.L.; Chang, P.Y.; Anthony, C.N. Genetic diversity, haplotypes and allele groups of Duffy binding protein (PkDBPαII) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia. Parasites Vectors 2014, 7, 161. [Google Scholar] [CrossRef]
- Ng, Y.L.; Lau, Y.L.; Hamid, M.H.A.; Jelip, J.; Ooi, C.H.; Mudin, R.N.; Jaimin, J.J.; Fong, M.Y. Genetic polymorphism of the thrombospondin-related apical merozoite protein (TRAMP) of Plasmodium knowlesi in Malaysia. Parasitol Res. 2023, 122, 195–200. [Google Scholar] [CrossRef]
- Azlan, U.W.; Lau, Y.L.; Hamid, M.H.A.; Jelip, J.; Ooi, C.H.; Mudin, R.N.; Jaimin, J.J.; Fong, M.Y. Genetic diversity of secreted protein with an altered thrombospondin repeat (SPATR) of Plasmodium knowlesi clinical isolates from Malaysia. Trop. Biomed. 2022, 39, 504–510. [Google Scholar]
- Azlan, U.W.; Lau, Y.L.; Fong, M.Y. Genetic diversity and clustering of the rhoptry associated protein-1 of Plasmodium knowlesi from Peninsular Malaysia and Malaysian Borneo. Korean J. Parasitol. 2022, 60, 393–400. [Google Scholar] [CrossRef]
- Pongvongsa, T.; Culleton, R.; Ha, H.; Thanh, L.; Phongmany, P.; Marchand, R.P.; Kawai, S.; Moji, K.; Nakazawa, S.; Maeno, Y. Human infection with Plasmodium knowlesi on the Laos-Vietnam border. Trop. Med. Health 2018, 46, 33. [Google Scholar] [CrossRef]
- Divis, P.C.; Lin, L.C.; Rovie-Ryan, J.J.; Kadir, K.A.; Anderios, F.; Hisam, S.; Sharma, R.S.; Singh, B.; Conway, D.J. Three divergent subpopulations of the malaria parasite Plasmodium knowlesi. Emerg. Infect. Dis. 2017, 23, 616–624. [Google Scholar] [CrossRef]
- Ahmed, M.A.; Fong, M.Y.; Lau, Y.L.; Yusof, R. Clustering and genetic differentiation of the normocyte binding protein (nbpxa) of Plasmodium knowlesi clinical isolates from Peninsular Malaysia and Malaysia Borneo. Malar. J. 2016, 15, 241. [Google Scholar] [CrossRef]
- Ng, Y.L.; Fong, M.Y.; Lau, Y.L. Genetic diversity of the full length apical membrane antigen-1 of Plasmodium knowlesi clinical isolates from peninsular Malaysia. Trop. Biomed. 2021, 38, 159–164. [Google Scholar]
- Ng, Y.L.; Lee, W.C.; Lau, Y.L.; Fong, M.Y. The impact of geographical variation in Plasmodium knowlesi apical membrane protein 1 (PkAMA-1) on invasion dynamics of P. knowlesi. Trop. Med. Infect. Dis. 2023, 8, 56. [Google Scholar] [CrossRef]
- Muh, F.; Lee, S.K.; Hoque, M.R.; Han, J.H.; Park, J.H.; Firdaus, E.R.; Moon, R.W.; Lau, Y.L.; Han, E.T. In vitro invasion inhibition assay using antibodies against Plasmodium knowlesi Duffy binding protein alpha and apical membrane antigen protein 1 in human erythrocyte-adapted P. knowlesi A1-H.1 strain. Malar. J. 2018, 17, 272. [Google Scholar] [CrossRef]
- WHO. Malaria vaccine: WHO position paper-March 2022. Wkly. Epidemiol. Rec. 2022, 9, 76. [Google Scholar]
- Rogers, W.O.; Baird, J.K.; Kumar, A.; Tine, J.A.; Weiss, W.; Aguiar, J.C.; Gowda, K.; Gwadz, R.; Kumar, S.; Gold, M.; et al. Multistage multiantigen heterologous prime boost vaccine for Plasmodium knowlesi malaria provides partial protection in rhesus macaques. Infect. Immun. 2001, 69, 5565–5572. [Google Scholar] [CrossRef]
- Nageswari, G.; Gnanadesigan, M.; Kiruthika, R. Polymeric particles as a delivery agent for malarial vaccines. In Applications of Nanobiotechnology for Neglected Tropical Diseases; Formiga, F.R., Inamudin, Severino, P., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 47–67. [Google Scholar]
- Kale, S.; Yadav, C.P.; Rao, P.N.; Shalini, S.; Eapen, A.; Srivasatava, H.C.; Sharma, S.K.; Pande, V.; Carlton, J.M.; Singh, O.P.; et al. Antibody responses within two leading Plasmodium vivax vaccine candidate antigens in three geographically diverse malaria-endemic regions of India. Malar. J. 2019, 18, 425. [Google Scholar] [CrossRef]
- Anand, G.; Biswas, S.; Yadav, N.; Mukherjee, P.; Chauhan, V.S. Production and immunogenicity of a tag-free recombinant chimera based on pfmsp-1 and pfmsp-3 using alhydrogel and dipeptide-based hydrogels. Vaccines 2021, 9, 782. [Google Scholar] [CrossRef]
- Cheong, F.W.; Fong, M.Y.; Lau, Y.L. Identification and characterization of epitopes on Plasmodium knowlesi merozoite surface protein-142 (MSP-142) using synthetic peptide library and phage display library. Acta Trop. 2016, 154, 89–94. [Google Scholar] [CrossRef]
- Goh, X.T.; Chua, K.H.; Kee, B.P.; Lim, Y.A.L. Identification of Plasmodium knowlesi Merozoite Surface Protein-119 (PkMSP-119) novel binding peptides from a phage display library. Trop. Med. Int. Health 2020, 25, 172–185. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence 5′ to 3′ | Annealing Temperature (°C) |
|---|---|---|
| PkMSP-1_F1 | CGTTGGCCACTTTTAAG | 56 |
| PkMSP-1_R1 | GCTTCCAACAAGGGTGTTGTCT | |
| PkMSP-1_F2 | CCGGATAATGGAAGGCAACCAA | 58 |
| PkMSP-1_R2 | AGGTCGTTGATCATGGTGTC | |
| PkMSP-1_F3 | CTACTGAAGCAGTACGCAC | 58 |
| PkMSP-1_R3 | CGTCTTCATCATTGCCGAACAG | |
| PkMSP-1_F4 | TGAAGCAGAACCAGCAAC | 56 |
| PkMSP-1_R4 | AATGTGCAGCCAAAGCC |
| Block | SNPs | Indels | No. of Haplotypes | Diversity | dN − dS | Tajima’s D | Fu and Li’s D * | Fu and Li’s F * | |
|---|---|---|---|---|---|---|---|---|---|
| Nucleotide (π) | Haplotype (Hd) | ||||||||
| I | 10 | 0 | 44 | 0.014 | 0.969 | −1.46 ** | −0.083 | −0.173 | −0.166 |
| II | 46 | 74 | 53 | 0.162 | 1.000 | 0.34 | 1.656 | 1.708 * | 2.019 * |
| III | 37 | 5 | 49 | 0.014 | 0.988 | −4.17 ** | −0.793 | −1.535 | −1.489 |
| IV | 231 | 326 | 55 | 0.174 | 1.000 | 0.98 | 0.545 | 1.105 | 1.057 |
| V | 12 | 0 | 36 | 0.012 | 0.971 | −0.56 | −1.190 | −1.778 | −1.861 |
| VI | 112 | 147 | 49 | 0.051 | 0.942 | 0.86 | 0.636 | 0.826 | 0.389 |
| VII | 25 | 9 | 49 | 0.018 | 0.987 | −3.82 ** | −0.250 | −1.114 | −0.930 |
| VIII | 138 | 244 | 51 | 0.100 | 1.000 | 3.19 ** | 0.495 | 1.462 | 1.314 |
| IX | 29 | 0 | 48 | 0.009 | 0.988 | −4.81 ** | −1.085 | −1.468 | −1.580 |
| Full length | 640 | 805 | 57 | 0.026 | 0.996 | −5.87 ** | −0.093 | −0.193 | −0.183 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noordin, N.R.; Lau, Y.L.; Cheong, F.W.; Fong, M.Y. Inter-Population Genetic Diversity and Clustering of Merozoite Surface Protein-1 (pkmsp-1) of Plasmodium knowlesi Isolates from Malaysia and Thailand. Trop. Med. Infect. Dis. 2023, 8, 285. https://doi.org/10.3390/tropicalmed8050285
Noordin NR, Lau YL, Cheong FW, Fong MY. Inter-Population Genetic Diversity and Clustering of Merozoite Surface Protein-1 (pkmsp-1) of Plasmodium knowlesi Isolates from Malaysia and Thailand. Tropical Medicine and Infectious Disease. 2023; 8(5):285. https://doi.org/10.3390/tropicalmed8050285
Chicago/Turabian StyleNoordin, Naqib Rafieqin, Yee Ling Lau, Fei Wen Cheong, and Mun Yik Fong. 2023. "Inter-Population Genetic Diversity and Clustering of Merozoite Surface Protein-1 (pkmsp-1) of Plasmodium knowlesi Isolates from Malaysia and Thailand" Tropical Medicine and Infectious Disease 8, no. 5: 285. https://doi.org/10.3390/tropicalmed8050285