
Ground States of Heisenberg Spin Clusters from a Cluster-Based Projected Hartree–Fock Approach
1
Department of Chemistry, Wesleyan University, Middletown, CT 06459, USA
2
Department of Chemistry, University of Potsdam, D-14476 Potsdam, Germany
*
Authors to whom correspondence should be addressed.
Condens. Matter 2023, 8(1), 18; https://doi.org/10.3390/condmat8010018
Submission received: 6 January 2023
/
Accepted: 26 January 2023
/
Published: 3 February 2023
(This article belongs to the Section Magnetism)
Abstract
:Recent work on approximating ground states of Heisenberg spin clusters by projected Hartree–Fock theory (PHF) is extended to a cluster-based ansatz (cPHF). Whereas PHF variationally optimizes a site–spin product state for the restoration of spin- and point-group symmetry, cPHF groups sites into discrete clusters and uses a cluster-product state as the broken-symmetry reference. Intracluster correlation is thus already included at the mean-field level, and intercluster correlation is introduced through symmetry projection. Variants of cPHF differing in the broken and restored symmetries are evaluated for ground states and singlet-triplet gaps of antiferromagnetic spin rings for various cluster sizes, where cPHF in general affords a significant improvement over ordinary PHF, although the division into clusters lowers the cyclical symmetry. In contrast, certain two- or three-dimensional spin arrangements permit cluster groupings compatible with the full spatial symmetry. We accordingly demonstrate that cPHF yields approximate ground states with correct spin- and point-group quantum numbers for honeycomb lattice fragments and symmetric polyhedra.
1. Introduction
The calculation of magnetic properties of exchange-coupled spin clusters, e.g., molecules with multiple open-shell transition-metal centers bridged by diamagnetic ligands [1,2], from the Heisenberg model, , usually relies on approximations, because exact diagonalization (ED) is only feasible for small systems. The ground state and perhaps a few excited states are needed to interpret electron-paramagnetic resonance (EPR) or inelastic neutron scattering (INS) spectra or to assess other low-temperature properties [3]. The density matrix renormalization group (DMRG [4]) is the most important variational method for ground states of one-dimensional (1D) systems (rings or chains) but is less suitable for 2D coupling topologies. The scarcity of computationally affordable and easily applicable alternatives motivated our recent exploration of projected Hartree–Fock theory (PHF [5]) for ground states of Heisenberg spin clusters [6]. PHF can be used in a black-box manner and has a mean-field (HF) scaling, with a prefactor depending on the size of the symmetry-projection grid. In finite spin systems, PHF restores spin (S) and point-group (PG) symmetry from a general product state. For a collection of sites, this broken-symmetry reference state is simply a three-dimensional spin configuration [6]. PHF yields rather accurate ground-state wave functions for symmetric rings with a moderate number of sites N and large local spin s and predicts reasonably accurate singlet-triplet gaps. Limitations become evident for larger rings, where the accuracy decreases [6]. PHF indeed recovers zero correlation energy per site in the thermodynamic limit . In other words, the method is not size extensive [7]. To ameliorate this problem and enable a more accurate treatment of larger systems by variational symmetry-projection methods, one could either adopt a multicomponent ansatz, where the broken-symmetry reference is a linear combination of nonorthogonal mean-field states [8], or work with a cluster basis that grants more flexibility than ordinary PHF, while still optimizing just a single reference. We pursue the latter option, which we call cPHF. Note, however, that both strategies could be combined into a multicomponent cPHF ansatz, which may be pursued in future work. For other correlated spin-cluster approaches (coupled-cluster and many-body perturbation theory) and for further literature on related methods, see, e.g., [9]. Note that the cluster mean-field is the same mean-field used in cluster extensions of dynamical mean-field theory [10] (DMFT). In this work, however, rather than attempting to obtain the interacting Green’s function, we use a variational ansatz to develop correlations in the ground state wave function using symmetry breaking and restoration. While symmetry breaking and restoration are somewhat limited in the flexibility they incorporate into the ansatz, it may be enough to obtain high-quality ground state wavefunctions for small systems with high symmetry, such as the ones considered in this work.
2. Theory and Computations
PHF optimizes a broken-symmetry mean-field state for the application of a symmetry projector [5]. In cPHF, is a product of individual cluster states :
where Q is the total number of clusters. As an example, for a cluster comprising two sites, the structure of is given in Equation (2).
The is independently optimized to minimize the variational energy, Equation (3), of the projected state :
The site-permutation invariance [11] of spin Hamiltonians representing systems with spatial symmetry (rings, symmetric polyhedra, etc.) is here referred to as point-group (PG) symmetry. Each level is thus characterized by its total spin S and its PG-label . To recover a substantial fraction of the correlation energy for all but the smallest systems, it is mandatory to combine S- with PG-projection in PHF [6,12]. In the PG-projector of Equation (3) (we consider only one-dimensional irreducible representations ), h is the order of the group, is the character of group element g, and is the respective symmetry operation [13].
Multidimensional irreducible representations become relevant for projection onto sectors. The projector for spin S and magnetic quantum number m (the eigenvalue) is expanded in terms of transfer operators ,
which are conveniently parameterized by Euler angles [14],
For a given , the coefficients (Equation (5)) correspond to the lowest-energy solution of the generalized eigenvalue problem for the Hamiltonian in the nonorthogonal basis spanned by , [12]. For combined S- and PG-projection, the projector is a product, (spin rotations commute with site permutations). In the trivial case where the projector is the identity, , that is, if no symmetry projection is performed, cPHF is equivalent to cHF, also called cluster mean-field theory [9,15]. If each cluster comprises just one site, cPHF becomes equivalent to PHF, specifically, the “single fermion” variety of PHF presented in [6]. Finally, note that cPHF trivially yields the exact ground state in the chosen symmetry sector if all sites are contained in a single cluster or if there are no couplings between clusters.
In quantum-chemical terminology, is of generalized HF type (GHF [16]) if it completely breaks spin symmetry. An unrestricted HF (UHF) state also breaks total spin symmetry (that is, is not an eigenfunction of ) but conserves . In a UHF-type reference, each cluster has a defined z-projection . Different clusters may have different , which add up to the total eigenvalue, . Compared to complete spin-symmetry breaking in GHF, the number of variational parameters is reduced in UHF. As an example, a general state of an dimer is a superposition of only two basis states, as shown in Equation (7):
PHF variants that restore S- or PG-symmetry from a GHF- or UHF-type reference are called SGHF, PGSUHF, etc. In cPHF, the cluster size q may be appended, e.g., SGHF(2) denotes a cluster-based SGHF calculation with dimers. For a given grouping, the lowest variational energy is obtained when working with the largest symmetry group (PGSGHF). We do not include complex-conjugation symmetry [5,17] in the cPHF scheme, because this involves a more complicated formalism [6,18,19] and has comparatively small effects for Heisenberg systems [6].
Self-consistent field (SCF, [5,6,19,20]) and gradient-based optimization ([12,21] and references cited therein) are two different strategies for the optimization of . In the SCF approach, the local cluster states result from successively building and diagonalizing an effective Fock matrix for each cluster. We found that reaching SCF convergence is often challenging in cPHF and therefore prefer gradient-based optimization, where each is parameterized in terms of a Thouless rotation from an initial guess . Details are provided in Appendix A. With q sites of spin s, the number of real variational parameters that define a general Thouless rotation for a single cluster is , leading to a total of for Q clusters (not counting the coefficients for S > 0, cf. Equation (5)). Note, however, that the Thouless parameterization, though convenient, is slightly redundant [6] with respect to S-projection from a GHF-type reference , because global spin rotations as well as certain gauge transformations of leave the spin-projected state unchanged [22].
The local cluster basis of dimension would have to be truncated for large clusters, e.g., by considering a limited number of lowest levels of the intracluster Hamiltonian. As an example, such a scheme could make a treatment of the Mn70 or Mn84 single-molecule magnets (with N = 70 or N = 84 s = 2 sites) feasible in terms of a division [23] but is beyond the scope of this work.
As recommended previously [20], we discretized transfer operators, Equation (6), with a combined Lebedev-Laikov [24] and Trapezoid integration grid. For S-projection from a UHF-type reference, the evaluation of integrals over Euler angles and is trivial [25], and integration over employs a Gauss–Legendre grid. A computational parallelization of the summation over the grid is trivial [20]. The quality of S-projection can be checked by computing from a sum of spin-pair correlation functions (SPCFs), as shown in Equation (8):
We ensured that deviates by from the ideal value of . Details on the calculation of SPCFs are provided in Appendix B.
Figure 1 illustrates that cluster formations are in general not fully compatible with spatial symmetry, meaning that working with the cyclic group (or the dihedral group that additionally includes vertical axes) of spin rings with N sites would involve complicated transformations between different cluster bases; on a similar note, complicated transformations between different coupling schemes prevent the combined use of the full spin- and point-group symmetry in ED [26]. A division in terms of q neighbors thus reduces the cyclical symmetry of rings according to or . For example, for , sectors and of group (the crystal momentum k indicates the eigenvalue of the cyclic permutation ) belong to the same sector of group . Thus, a state in is generally a mixture of (Mulliken label ) and in . This, however, does not imply that cPHF wave functions will significantly break symmetry with respect to the full point group (see Results section).
From the perspective of molecular magnetism, symmetry reduction through cluster formation may not be a major concern, because the cyclic-symmetry order of existing ring-like molecules is often lower than the number of open-shell ions. For example, the mere six-fold rotational symmetry of an Fe18 ring (studied by INS in [27]) suggests a treatment in terms of six equivalent clusters, each hosting three neighboring sites, which represent three chemically inequivalent FeIII ions. Furthermore, Mn70 and Mn84 tori, the largest single-molecule magnets known to date, have repeat units of 14 MnIII ions (s = 2), but the pattern of isotropic couplings in the Heisenberg model suggests a partitioning into two inequivalent types of clusters with seven sites each [23].
In contrast, it is straightforward to use all spatial symmetries in cPHF that keep clusters intact, meaning that a given operation transforms all sites of one cluster into sites of a specific second cluster. Operations associated with internal site permutations do not pose a problem. For example, vertical rotations in the dihedral group of rings involve such internal permutations; see Appendix A for details. The Results section additionally presents selected systems (honeycomb lattice fragments and symmetric polyhedra) permitting cluster groupings that are fully symmetry-compatible if operations associated with internal permutations are considered.
Unless noted otherwise, all clusters (total number ) are equivalent by symmetry and contain the same number of sites q with a uniform local spin s, although these are not requirements for the application of cPHF. It is usually reasonable to construct clusters from neighboring (interacting) sites to recover a substantial amount of correlation energy already at the mean-field level. In the absence of intracluster interactions, cHF recovers the ordinary HF (classical spin, ) solution. With respect to variational symmetry projection in cPHF, the cluster ansatz offers more flexibility than (ordinary PHF), even when there are no intracluster interactions (see Results section).
The relative correlation energy, , defined in Equation (9),
is an accuracy measure of cPHF for ground states, where is the exact result (ED), and refers to the classical solution, e.g., for the Néel configuration in a ring. All benchmark systems treated here have a nondegenerate ground state. Energies are reported in units of the uniform nearest-neighbor coupling constant (antiferromagnetic coupling).
In cases where the system size prohibits a comparison with exact energies, we compare cPHF against a cluster-variant of second-order perturbation theory (cPT2). The (nonvariational) cPT2 corrected energy is given by Equation (10),
where , , and is obtained from through excitations a and b in two neighboring (directly interacting) clusters. In other words, is a doubly excited cluster mean-field state. The denominator in the second term of Equation (10) is of Epstein–Nesbet type [28]. For cluster size and , such PT2 corrections were considered for the truncated icosahedron in [29]. Alternatively, one could apply Rayleigh–Schrödinger perturbation theory in a cluster basis, using differences of Fock-like orbital energies in the denominator, as seen in [9] (for the square lattice) and [30] (for the single-band Hubbard model).
3. Results and Discussion
For antiferromagnetic spin rings, honeycomb lattice fragments, and four tetrahedral or icosahedral polyhedra, we compare ground-state energies and SPCFs from different variants of cPHF against exact results (where available). In addition, we briefly consider singlet-triplet gaps in spin rings and larger local spin (up to ) in polyhedra and explore how the quality of predictions depends on the cluster size or shape. Rather than deriving specific new insights on any of these systems, our aim is to investigate the potential of cPHF as a variational black-box method with a cluster mean-field scaling.
3.1. Symmetric Rings
As in our previous work [6], we choose rings with sites as benchmark systems to compare against ED but restrict attention to the least classical case which had proven to be most problematic for ordinary PHF (q = 1) [6].
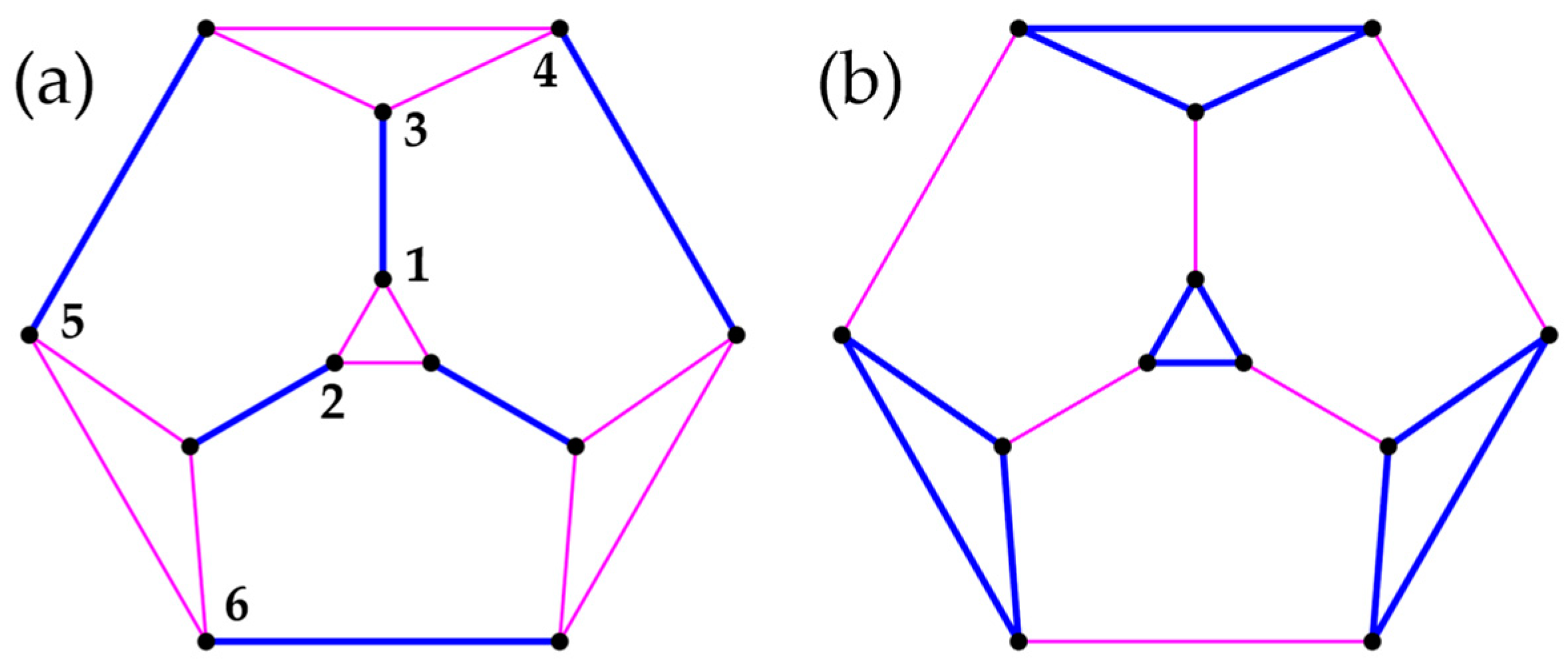
We note in passing that, in contrast to dimers formed from neighboring sites (see Theory section), clusters composed of diametrically opposite sites are indeed compatible with the full symmetry. Figure 2 illustrates that the cyclic operation leaves all clusters intact and exchanges sites in one dimer. For , with such a partitioning yields the exact ground state (exact within numerical double precision) in each k sector (k = 0, 1, …, N − 1), whereas turned out to be exact in sectors only [6]. This shows that the cluster ansatz is somewhat more flexible with respect to variational symmetry projection, even though HF(2) is equivalent to HF(1) due to the lack of intracluster interactions. For larger rings, the described pairing of the most distant sites does not provide a sizable improvement over PHF(1) and shall not be discussed further.
We construct clusters comprising neighbors (Figure 3), corresponding to the common divisors of N, thus reducing the point group accessible to cPHF, that is, , where . Rings with even N lack frustration and adopt UHF-type solutions in cHF. In contrast, for odd N (not studied here), frustration gives rise to genuine GHF solutions breaking symmetry.
It is simple to show that HF(2) produces a product of singlets, . We observed that local singlets are also formed in HF(6). Consequently, is a sum of the ground-state energies of the individual clusters: , and (for In contrast, for , all clusters assume z-projections with alternating signs, leading to a Néel-like spin-density pattern and nonvanishing interactions between clusters, . We note in passing that for and even q, all clusters have the same state in HF, but , and the intercluster interaction is nonzero; for odd q, states alternate between and .
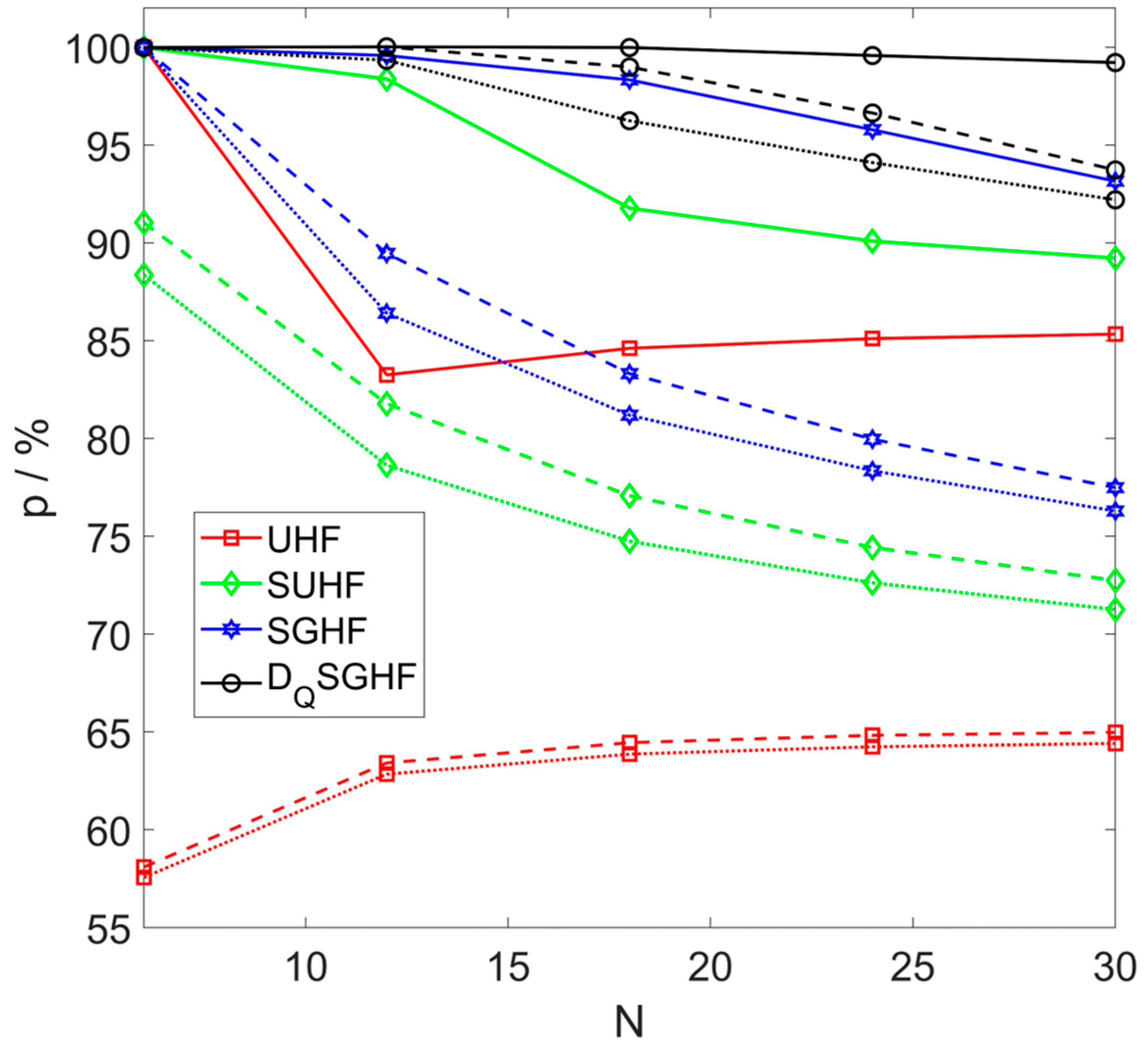
Relative correlation energies p for cluster variants of UHF, SUHF, SGHF, and are plotted in Figure 4. Relative singlet-triplet gaps from SUHF, SGHF, and ED are plotted in Figure 5. Numerical energy values are given for reference in Appendix C. The labels for the ground states in sectors S = 0 and S = 1 depend on q, S, and N, as detailed in Table 1. (This information can be derived straightforwardly from the respective labels in , which were given in Table III of [6]).
For or , all clusters are in the SUHF references for or projection. The fact that energies are lower than UHF shows that SUHF does not yield a singlet-product. For a given state, system, and q, the ordering reflects the breaking and restoration of additional symmetries. (In contrast, in the absence of a group/subgroup relation between cPHF variants, such as in the set SGHF, PGGHF, or PGSUHF, it is not possible to determine the ordering a priori.) The equality holds only when the lower-level method is exact. This applies to , , where UHF (and any PHF variant) is trivially exact, as well as in a few other cases, e.g., SGHF(2) yields the numerically exact and ground states for .
For or , an SUHF or SGHF wave function can be trivially expressed in the basis. Thus, will yield the best variational energy. In contrast, if -projection is included or if the larger cluster size is not a multiple of the smaller size we cannot rigorously predict if a larger q is advantageous. However, Figure 4 shows that this is indeed true in all studied cases. Excluding the exact , point, the relative correlation p captured by UHF increases with N because rings with even N approach the Bethe–Hulthén limit from below [31]. With , the limits for cHF in Equations (11)–(13),
also hold for cPHF due to the lack of size extensivity (see Introduction). This limitation of cPHF is clearly apparent from the fact that p decreases with increasing N (Figure 4), but the improvement over ordinary PHF (where for ) is still significant. For example, for , yields [6] versus with . Finally, from ; not included in Figure 1) is very close to [32].
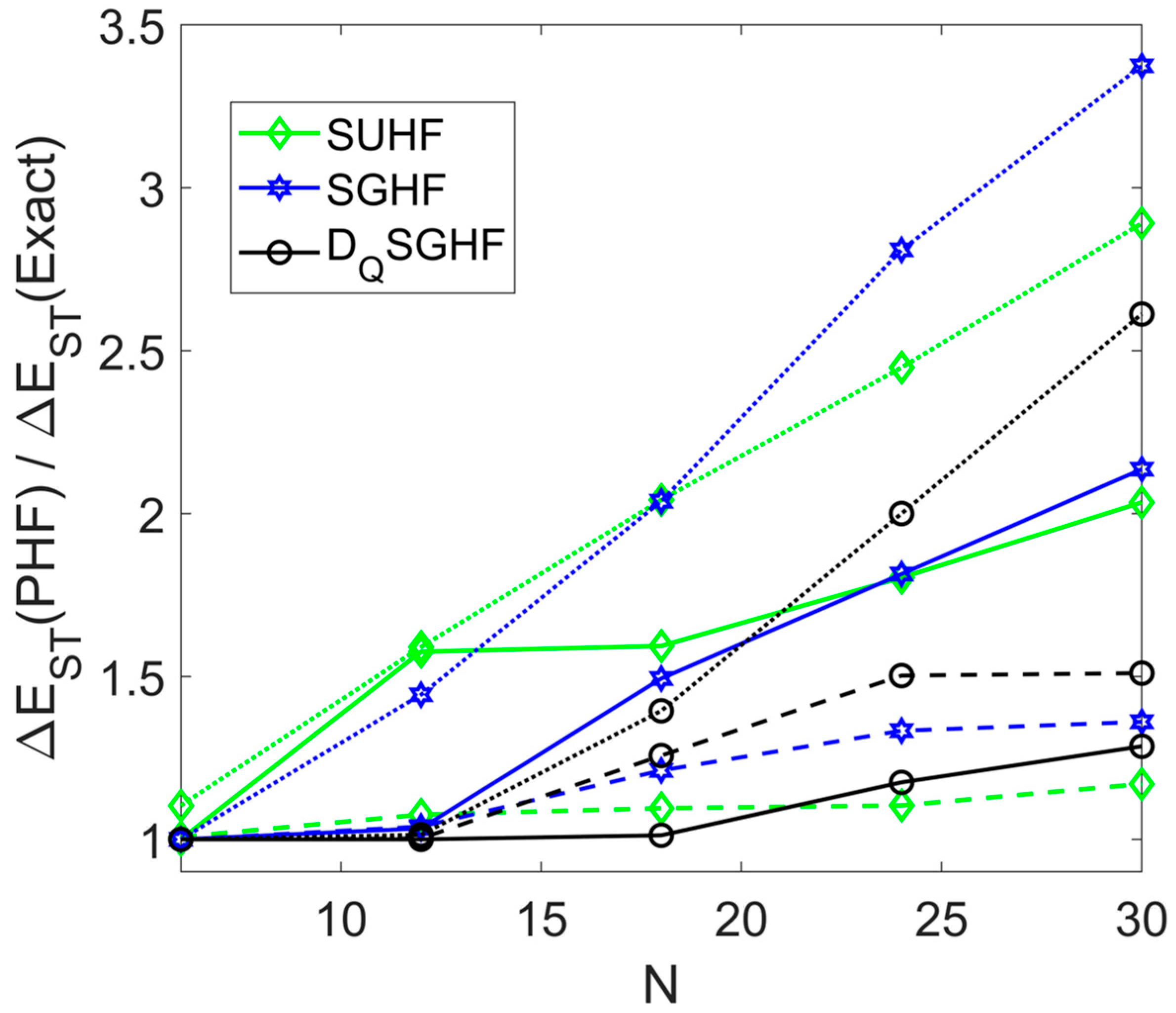
Triplet energies improve along the same hierarchy as singlet energies, but this does not guarantee an improvement in the gap . Somewhat unfortunately, is severely overestimated, except for small N and large q. The cluster approach can still afford better results than ordinary PHF. For example, for yields [6], compared to from and an exact value of [32].
3.2. Honeycomb Lattice Fragments
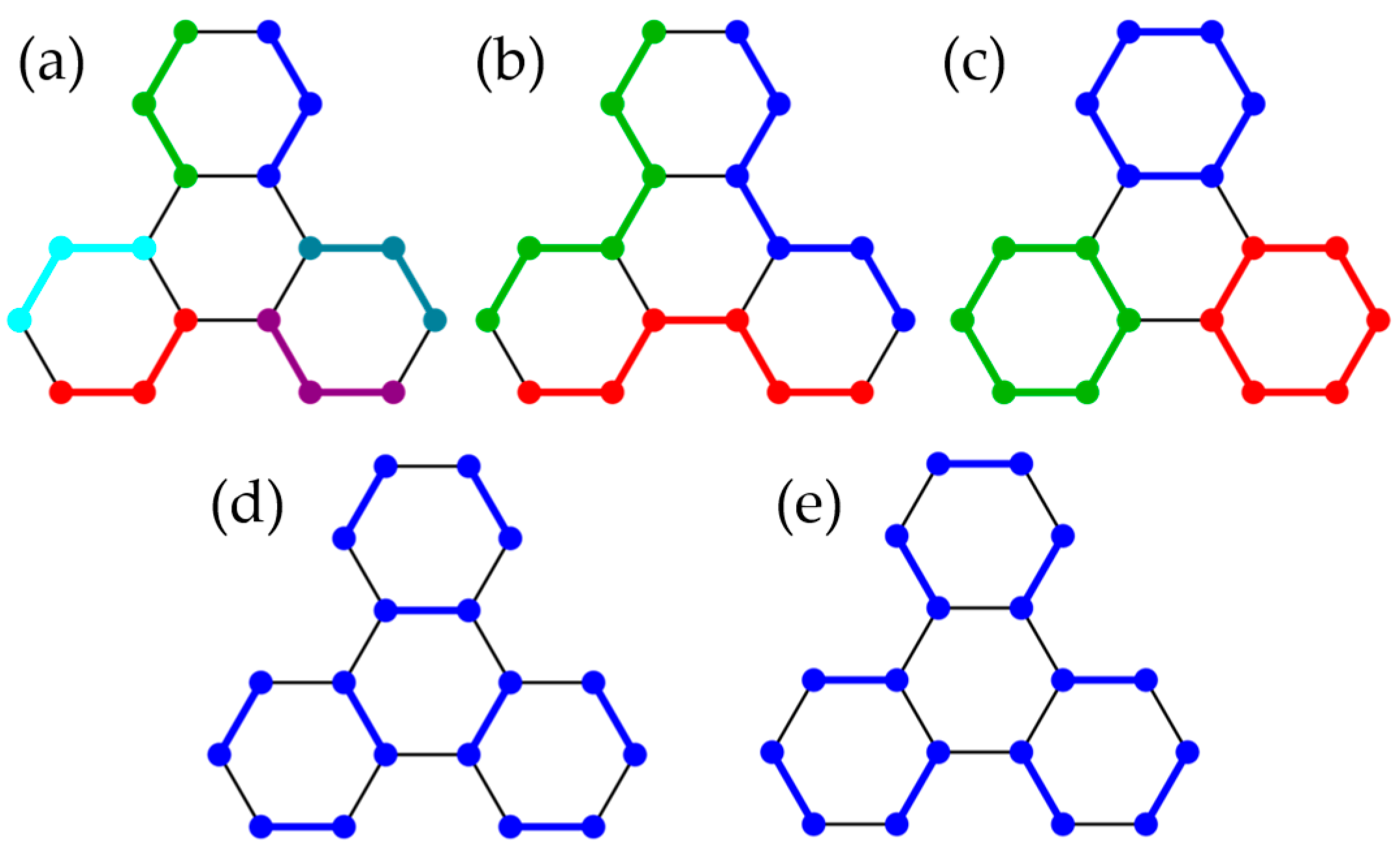
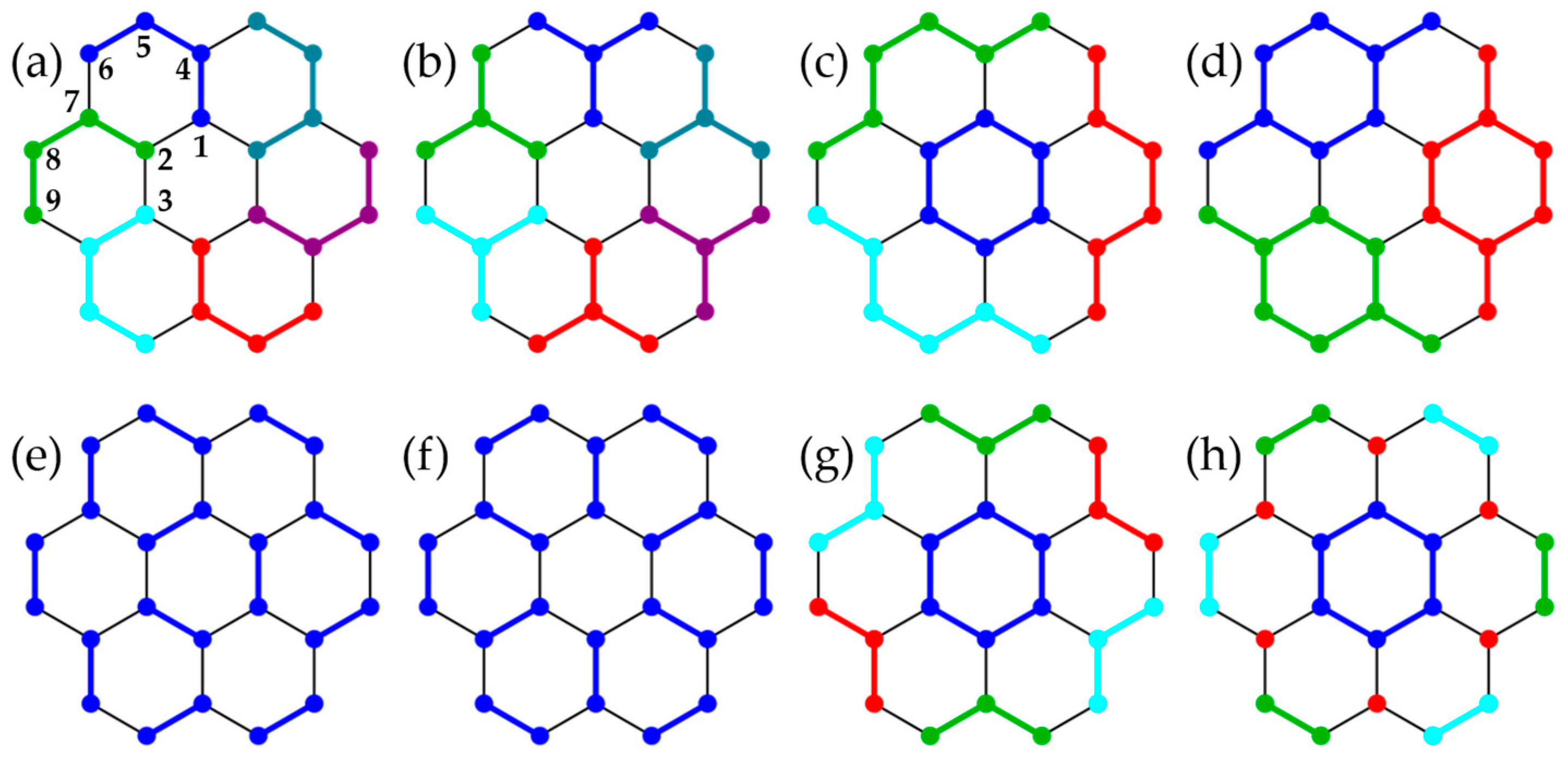
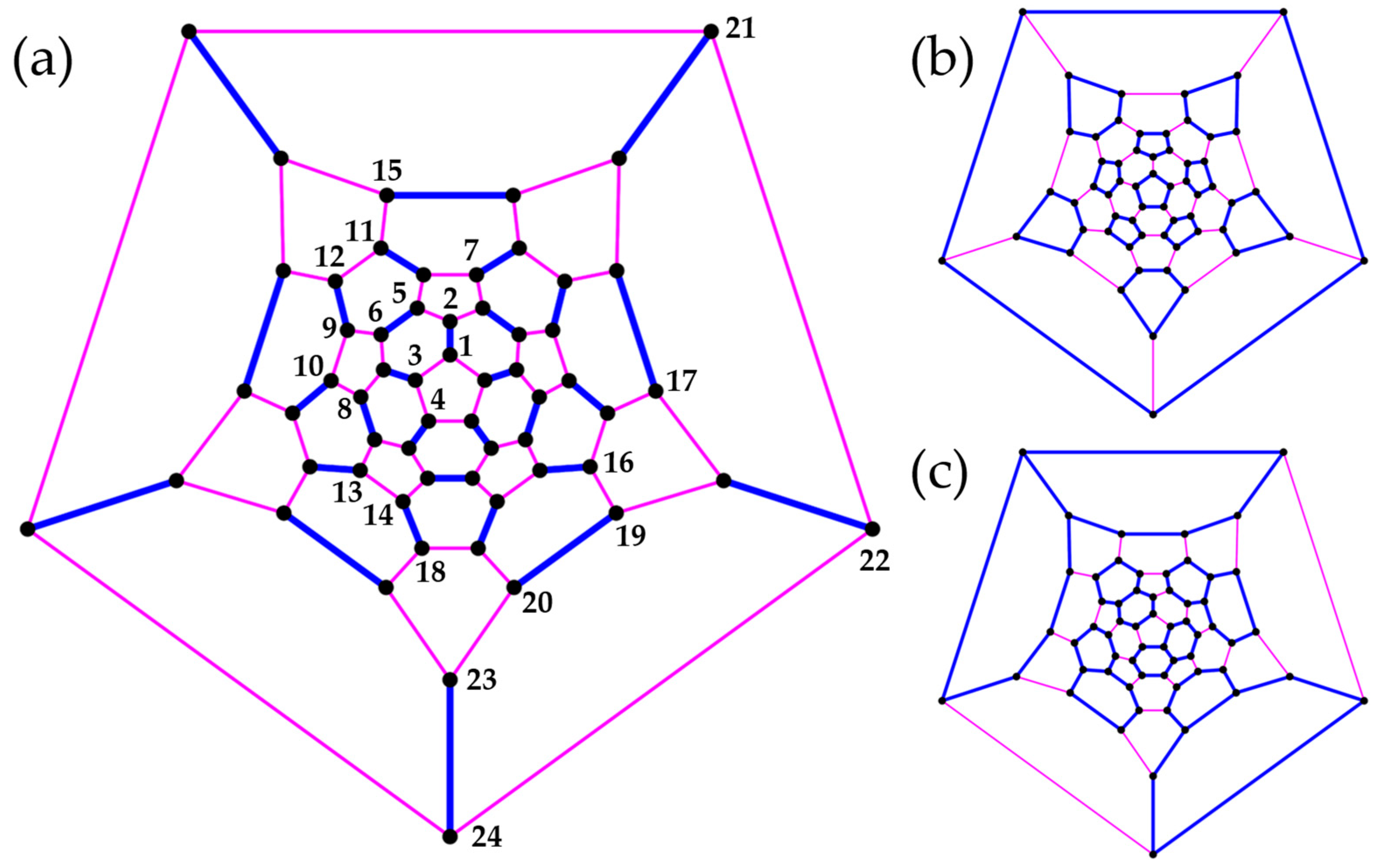
Systems with the connectivity of polycyclic aromatic hydrocarbons—triphenylene , coronene (N = 24), hexabenzocoronene (N = 42), hexa-cata-hexabenzocoronene (N = 48), and kekulene (N = 48)—allow us to briefly explore options for cluster formation (the chemical nomenclature for these systems is not meant to imply that the Heisenberg model describes properties of the respective organic molecules). The lattices are bipartite [33] and thus have S = 0 ground states. For and up to eight different groupings (Figure 6, Figure 7, Figure 8, Figure 9 and Figure 10; in some cases, the clusters are obviously not all equivalent), Table 2 compares energies from cUHF, cPHF, and cPT2 to exact results (available only for N = 18, 24).
In triphenylene, grouping (b) is obtained from (a) by merging clusters, while conserving the full PG-symmetry, and (b) is thus guaranteed to yield lower cPHF energies than (a). The same is true for (c) with respect to the two complementary dimer formations (d) and (e). In fact, based on (c) with three benzene units, each hosting a Clar sextet in the most favorable resonance structure [34], is very close to the exact ground state.
In coronene, grouping (d), including 24 of 30 bonds in the clusters, is obtained from (b) by merging neighboring clusters pairwise and recovers 99.75% of with . For cluster types (a)–(d), Table 3 collects all distinct nearest-neighbor (NN) and a few next-nearest neighbor (NNN) SPCFs. Their magnitude is generally somewhat over- or underestimated (with minor exceptions), depending on whether sites reside in the same (bold type in Table 3) or in different clusters, respectively. It thus appears generally advantageous to include pairs of strongly antiferromagnetically correlated sites in a cluster. The SPCFs evidence a moderate degree of spatial-symmetry breaking, in (a), in (c), and in (d), but not in (b), because this grouping allows full -projection.
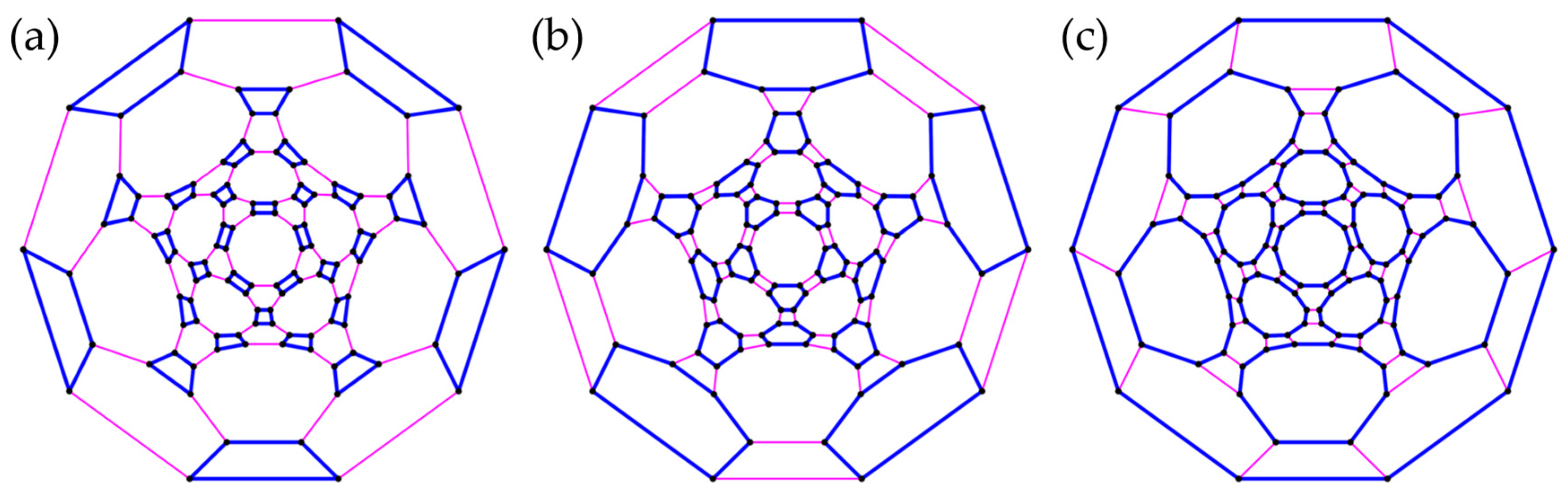
Due to computational limitations, we cannot carry out ED on the N = 42 and N = 48 lattices, but a comparison with cPT2 suggests that cPHF is still fairly accurate in these larger systems. In hexabenzocoronene, both options include a fraction of 42/54 bonds in the clusters. Although (Figure 8a) has seven intact rings, and thus loosely corresponds to the most favorable resonance structure with seven Clar sextets [34], (Figure 8b) with six sextets is energetically very slightly favored in , though not in UHF (Table 2). In hexa-cata-hexabenzocoronene, with 42/60 bonds in seven rings (Figure 9a) yields a significantly lower energy than with the same number of intracluster bonds but no intact rings (Figure 9c) and is even slightly better than with 48 intracluster bonds but only six rings (Figure 9b). Finally, in kekulene, grouping (d) predicts the lowest energy, despite symmetry breaking. In contrast to (a), (c), and (d), the six Clar sextets are broken up in (b), which features only 36/60 bonds and yields higher energies in cHF, cPHF, and cPT2.
3.3. Polyhedra
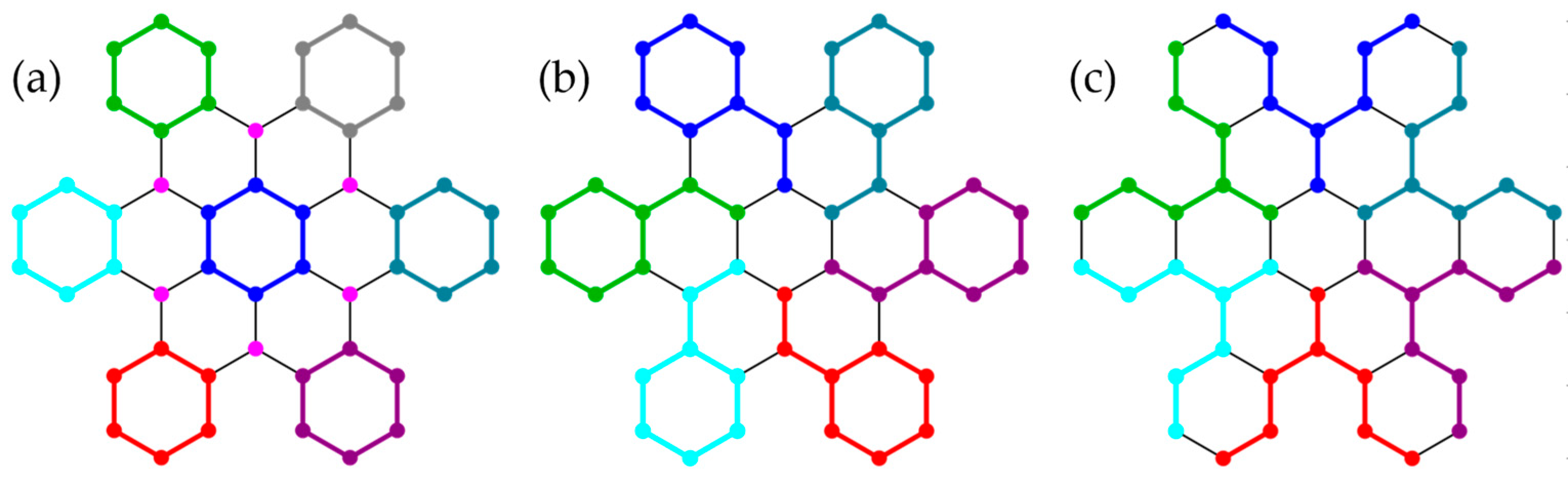
We lastly consider four polyhedra (Figure 11) with or symmetry, which allow cluster groupings that fully respect spatial symmetry.
- (a)
- Icosahedron
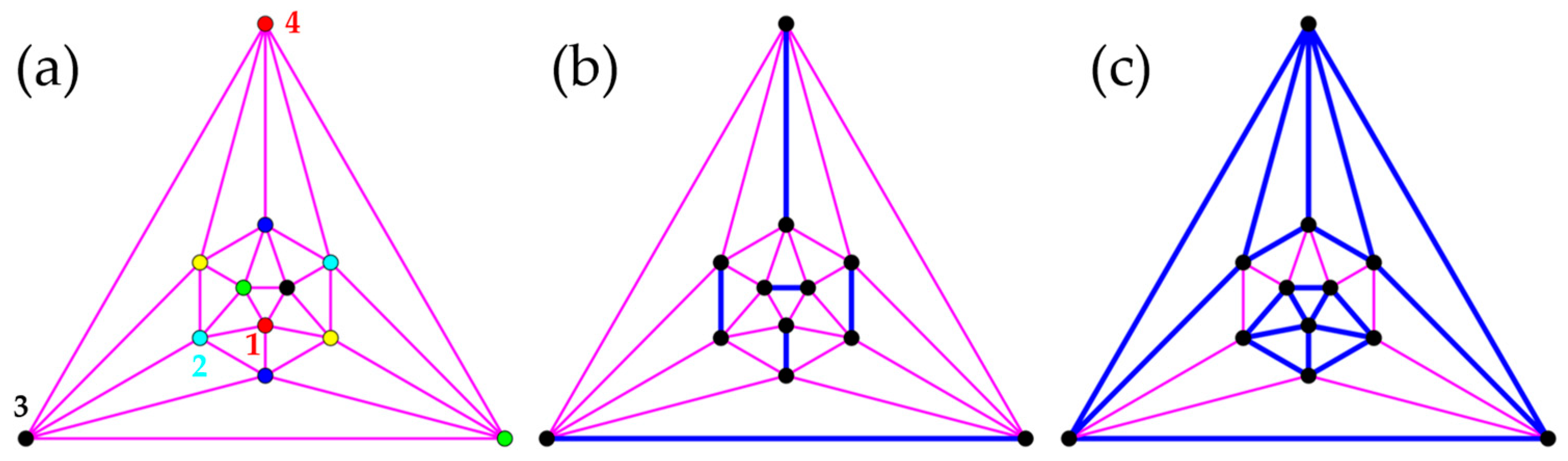
Dimers of the sites related by spatial inversion conserve symmetry (Figure 12a), while nearest-neighbor pairs break symmetry (, Figure 12b), as do hexamers (, Figure 12c). For , the antiferromagnetic spin-pair correlation between the most distant sites (numbers defined in Figure 12a) exceeds the NN correlation in the exact wave functions, see Table 4.
Interestingly, the grouping (a) is advantageous over (b), see Table 5, except for GHF, where (a) yields the classical solution [35], while (b) profits energetically from correlation in the dimers. For , GHF yields local singlets in (b), which are broken up in the SGHF reference, as apparent from the energy lowering, . For , yields 100%, 99.999%, 99.996%, and 99.995%, respectively, of the exact ground-state energy and thereby recovers most of the correlation energy missing from [6]. Not surprisingly, SPCFs are very close to the exact results (Table 4), corroborating the high quality of the wave functions.
Incidentally, (without explicit S-projection) converges onto the exact ground state of the system (the numerical deviation from is ). In contrast, in , the additional use of complex-conjugation symmetry (K) was required to reach the exact solution [6]. The fact that the exact ground state for is also obtained with (Figure 12c) shows that a cluster grouping that breaks symmetry does not necessarily lead to a symmetry-broken cPHF wave function. SGHF(2) wave functions are totally symmetric in group but have mixed symmetry under spatial inversion . For , the respective weights in the SGHF(2) wave function are and .
- (b)
- Truncated Tetrahedron
The classical solution for the truncated tetrahedron [36] minimizes frustration on the four triangles (with angles of 120 between spins) and aligns spins on the bonds between triangles antiparallel. Two complementary cluster groupings defined by either six classically unfrustrated bonds (u-bonds, , Figure 13a) or twelve frustrated bonds (f-bonds, , Figure 13b) maintain tetrahedral symmetry.
Table 6 shows that u-bonds yield lower GHF and SGHF energies than f-bonds , despite a smaller number of variational parameters, e.g., for , for , and for . However, the larger variational freedom provided by f-bonds leads to slightly better energies for compared to . The somewhat larger deviations between PGSGHF and exact results than in the icosahedron may be attributed to the lower order of the group ( in versus in ), although a direct comparison between these two polyhedra is not meaningful. For [6] and , though not for , and for all tested values of s, the SGHF reference can be revealed to have the classical spin-density structure by appropriate gauge transformations [22]. (For additional comments on this issue, see the following section on the truncated icosahedron.) SGHF(1) [6] and SGHF(2) wave functions transform like the exact ground state (A2 or A1 for half-integer or integer s, respectively).
- (c)
- Truncated Icosahedron
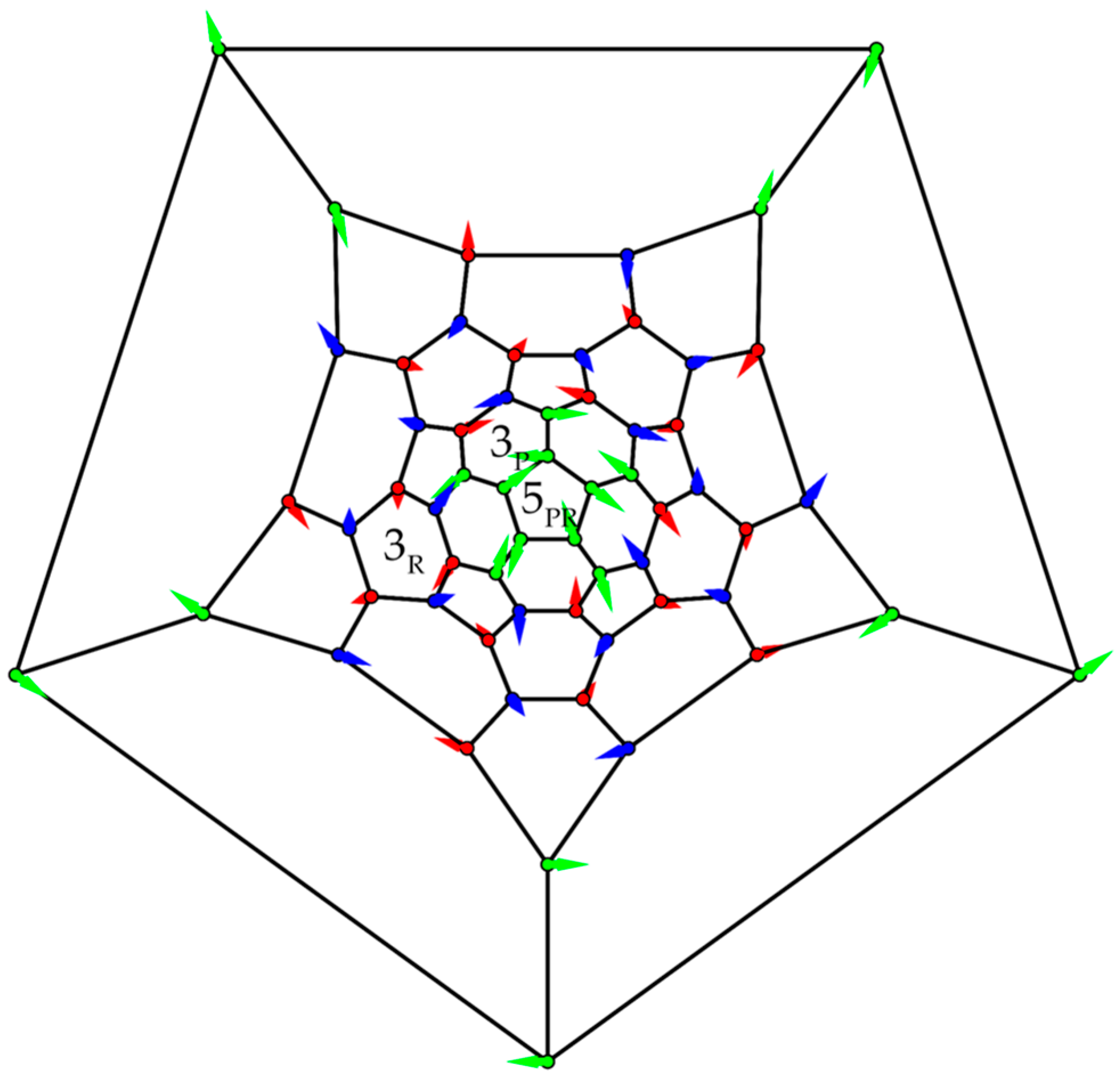
In contrast to icosidodecahedral arrangements of spin centers in Keplerate molecules [37,38], [39], [40], and [41,42], with for V3+, Cr3+, Fe3+, truncated icosahedra were not yet synthetically realized as magnetic molecules. The respective Heisenberg model was nevertheless addressed in a few works [29,43,44,45,46,47,48,49], some of which were motivated by gaining an understanding of the properties of buckminsterfullerene C60. However, electronic-structure calculations have shown that the Heisenberg model is at best of qualitative value for C60 because this molecule possesses unpaired electrons [50] (but note that there is no unique measure for this number). It was eventually concluded that C60 displays no significant strong correlation [51]. Ab initio GHF calculations [50] on C60 still reproduce the exotic three-dimensional spin-density pattern of the classical Heisenberg ground state [29]. Geometry optimization on the GHF level-of-theory yields a perfect structure for C60 [50], but the spatial inversion is the only obvious self-consistent symmetry of the GHF solution. We specify the hidden icosahedral symmetry I in Figure 14, where classical spin vectors are plotted in a Schlegel diagram. A uniform rotation was applied such that spins in the central pentagon in Figure 14 lie in the geometrical plane of that pentagon, with the spin on the first site pointing in the negative x-direction. This spin configuration is left unchanged by a combined spin rotation (R) by 144° and a five-fold site permutation (P). In the electronic-structure problem, the site permutation corresponds to a spatial rotation. These operations are performed about an axis through the coordinate origin (the center of the truncated icosahedron) and the center of the central pentagon (marked in Figure 14).
A spin rotation by 120 about an axis through the midpoint of a hexagon (, Figure 14), combined with a threefold site permutation about another hexagon is a second symmetry , and and are generators of a group isomorphic to group I. The full symmetry group of the classical (or GHF) solution is . This overall explains why GHF orbitals of C60 are up to six-fold degenerate: molecular orbitals span irreducible representations [52] of the double group .
Two complementary cluster formations preserve spatial symmetry by including either the 30 u-bonds between pentagons (q = 2, Figure 15a) or the 60 f-bonds in the pentagons (q = 5, Figure 15b). Classical spins are antiparallel on u-bonds (see Figure 15a) and span angles of 144° on the f-bonds. We additionally consider fused hexagons (, Figure 15c), thereby including 36 f-bonds and all 30 u-bonds in the clusters and reducing symmetry, .
Interestingly, can be solved analytically for . Starting from the classical solution [29], , we define a u-bond wave function in Equation (14),
that depends on a real parameter α. The local quantization axis for projections and in a cluster i are given by the classical solution, that is, recovers GHF(1). Thus, all spins retain their classical orientation in GHF(2), but Equation (14) allows the local singlet to acquire a higher weight than the triplet. Each dimer contributes an energy , as shown in Equation (15):
Compared to GHF(1), the local magnetization is reduced by a factor , Equation (16), in the GHF(2) wave function.
Interactions along the intercluster bonds (f-bonds) are evaluated classically,
with (144°). Minimization of the total GHF(2) energy E, Equation (18),
with respect to , affords in Equation (19),
corresponding to in Equation (20),
and overall results in Equation (21),
Numerical calculations indeed converge onto this solution. (An analogous analysis for the truncated tetrahedron yields , that is, singlets are formed on the six u-bonds.) GHF(2) significantly improves over GHF(1) but still falls short of an accurate description. Other works derived the following variational estimates for the ground state: [45] (resonating valence-bond, RVB), [44], or [43] (Variational Monte Carlo with a Gutzwiller projector, VMC), and [47] (DMRG). The latter most recent result (DMRG) may be regarded as a quasiexact benchmark value. The GHF and PHF energies are collected in Table 7. Our best estimate of , obtained with , is very close to the RVB result [45] and corresponds to p = 90%. In view of the Hilbert-space dimension of , which is presently still infeasible for ED, even if symmetries were employed to reduce the matrix size, we would like to emphasize the very significant state-space reduction afforded by cPHF in terms of for q = 2, for q = 5, and for q = 10.
As explained, GHF(2) spin densities (local magnetizations) maintain the classical structure. Our numerical calculations showed that this also holds true for GHF(5). Although SGHF(2) or SGHF(5) will generally converge onto a reference that does not display the classical spin-density pattern, we found that the latter can be restituted by gauge rotations on that leave the S-projected state unchanged (redundancies in the definition of with respect to gauge transformations related to spin symmetry were discussed in [22]): assumes the classical magnetization pattern when it is ensured through appropriate gauge transformations that the expectation value of the total-spin vector vanishes, . Irrespective of whether such transformations are applied to , SGHF(2) and SGHF(5) wave functions (S = 0) have pure symmetry in . For , is the optimal reference for projection in SGHF , which appears to be a rather general feature of highly symmetric polyhedra [6] but is not true for spin rings [6] or for the presently studied polyhedra if . In other words, for , the variation-after-projection (VAP) approach of SGHF is not equivalent to a far simpler projection-after-variation (PAV) “single-shot” S-projection of .
In Table 8, PHF predictions for SPCFs from SGHF and are compared to VMC [43]. Note that the energy of a variational trial function that respects icosahedral symmetry is . The magnitude of the antiferromagnetic u-bond correlation is underestimated by , while the magnitude of in the f-bonds is overestimated. The reverse is true for , which yields and values that match the VMC estimates more closely. This further illustrates the trend observed in the hexagonal lattices of over- or underestimating correlations in intra- or intercluster bonds, respectively. Except for , cPHF overestimates long-range correlation rather dramatically.
We are not aware of any previous works addressing ground states in the truncated icosahedron with . For , energies from GHF, SGHF, and (q = 2) are collected in Table 9, where they are compared with local singlets on fused benzene rings (q = 10) and with PT2(1) and PT2(2). Except for , where the singlet-product represents the GHF(10) solution, yields the best variational energy. For the system, the PT2(1) energy, [29], is incidentally rather close to the DMRG result, [47], but the PT2(2) prediction deviates significantly, .
The relative deviation between PT2 energies and is smallest for . The Hilbert-space of dimension for is completely out of reach of ED. The problem size is again drastically reduced by cPHF(2), which operates with merely variational parameters. Somewhat unfortunately, we cannot offer a reliable assessment of the accuracy of cPHF in this rather large system, where obtaining an accurate reference value with DMRG would be very challenging.
- (d)
- Truncated icosidodecahedron
Lastly, as another demonstration of the applicability of cPHF to systems that are far too large for ED, we consider a truncated icosidodecahedron with 120 vertices, Figure 16 presents three symmetry-compatible cluster groupings .
Being bipartite, the system has a nondegenerate ground state, which should transform as (like the classical Néel state, ). With 120/180 bonds included in the clusters, none of the three groupings has an obvious advantage: , , . For a given q, all clusters assume the same state, but no local singlets are formed , leading to spin densities of Néel-type. We selected for cPHF calculations , , . With respect to , q = 6 is still inferior to q = 4, yielding . The same trend is true for PT2: , .
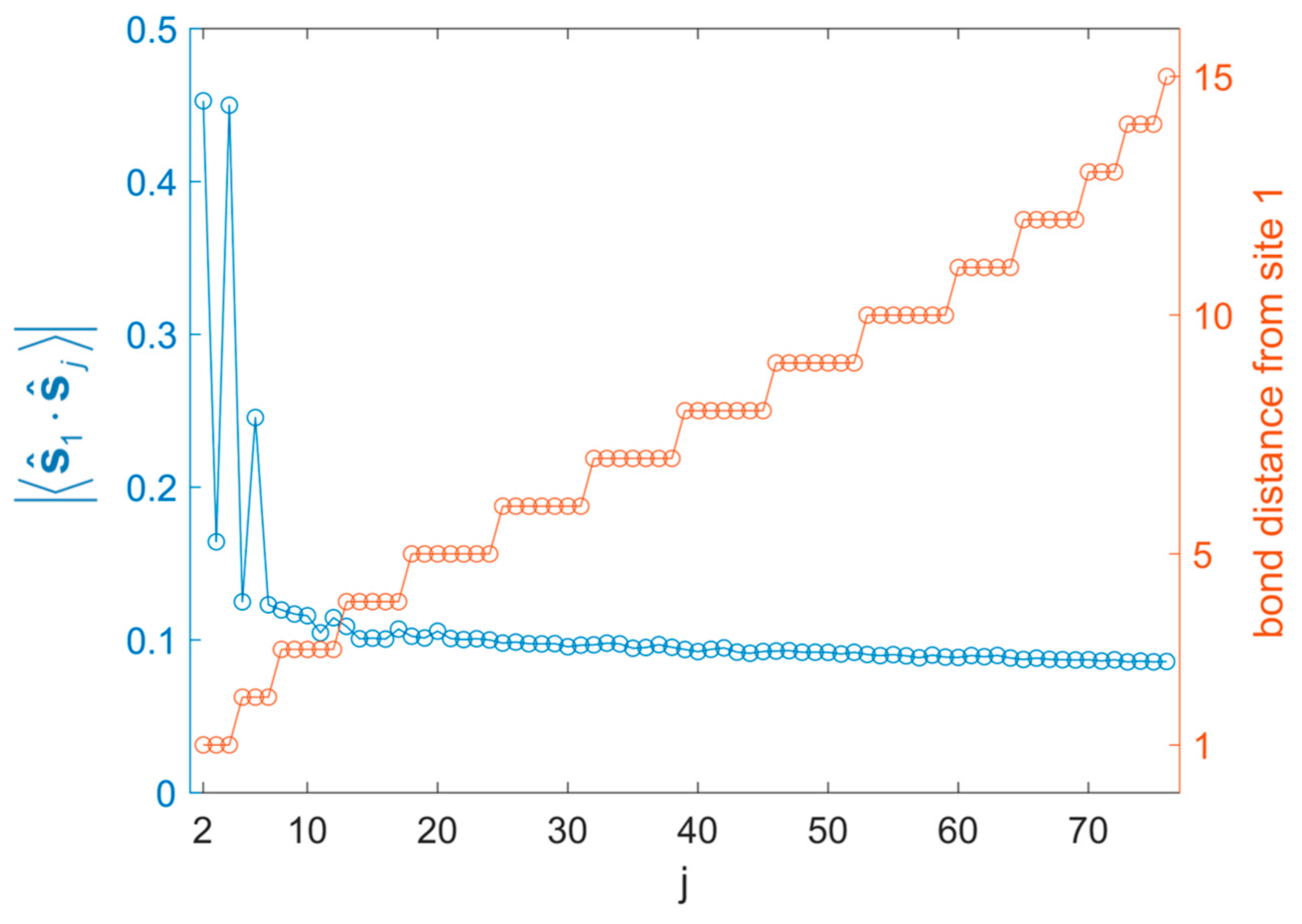
SPCFs (referring to the site numbering of Figure 17) from are plotted in Figure 18. All correlations within the same sublattice are positive; all correlations between different sublattices are negative. The strong correlation across the whole range of the molecule is most likely artifactual, because PHF (or cPHF) reverts to HF (or cHF) in the thermodynamic limit [5,6]. Therefore, long-range order for large systems is generally exaggerated.
4. Conclusions
By partitioning spin sites into clusters, the cPHF method extends the variational flexibility of PHF for a simple approximation of ground states of finite Heisenberg systems. The optimization of a cluster-product state for the restoration of good quantum numbers (spin and point group) comes at a mean-field cost, with a prefactor depending on the projection-grid size. The compact representation of the cPHF wave function in terms of a projector acting on a cluster mean-field state is suitable for the calculation of various properties.
We considered only energies and spin-pair correlations, but other quantities needed for modeling EPR or INS spectra, such as spin densities or expectation values of higher-rank local spin operators, as well as transition-density matrices, could also be obtained straightforwardly, opening a perspective for the application of cPHF to moderately large magnetic molecules. For antiferromagnetic spin rings, which are more challenging for PHF than systems [6], cPHF significantly improves over ordinary PHF by predicting rather accurate ground states for larger ring sizes. Although the cluster ansatz cannot access the full cyclic symmetry, the accuracy of cPHF improves with cluster size, where a smaller number of bonds is left to be correlated through symmetry projection.
We additionally studied hexagonal lattice fragments and symmetric polyhedra where cluster groupings can maintain the full spatial symmetry. In these systems, it is generally advantageous to include strongly antiferromagnetic bonds in the clusters. This may be accomplished by maximizing the number of intact rings (Clar sextets) in hexagonal lattices or by defining, if possible, clusters in terms of classically unfrustrated bonds in polyhedra. Applications to the truncated icosahedron and the truncated icosidodecahedron demonstrate that cPHF can be applied to systems whose size is prohibitive for exact diagonalization and challenging for other methods.
More advanced symmetry-projected methods have been under active development in various fields of many-body physics and appear worth pursuing for Heisenberg systems. Specifically, a linear combination of cluster-product states (configurations) is systematically improvable by increasing the number of configurations and would thus ameliorate problems associated with the lack of size-extensivity. Such a multiconfiguration variant of cPHF would also give access to excited states in the respective symmetry sectors that are needed for modeling spectra and low-temperature properties of molecular magnets.
Author Contributions
Conceptualization, S.G.T.; Methodology, S.G.T.; Formal analysis, S.G.T. and C.A.J.-H.; Writing—original draft, S.G.T.; Writing—review & editing, S.G.T. and C.A.J.-H.; Supervision, C.A.J.-H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
S.G.T. thanks the German Academic Exchange Service (DAAD) for support in the early stages of this project.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A. Optimization of the Broken-Symmetry Reference
The self-consistent field (SCF) optimization of the cluster-product state , Equation (A1),
In Equation (A2), a Thouless rotation relates to an initial guess :
A is a normalization constant. This rotation separates into rotations for all individual clusters,
The quantum-chemical terminology used in Equation (A4), occ = “occupied”, virt = “virtual”, refers to a fermionic formulation (cf. [6]), where defines a single occupied molecular orbital at the respective cluster in terms of a fermionic creation operator,
As indicated, we find it helpful to draw a conceptual connection to electronic-structure theory by associating the state of a spin cluster with a molecular orbital occupied by a single fermion. This is analogous to our approach to PHF [6], which can be regarded as a special case of cPHF with cluster size . A fermionic formulation leads to a second-quantized Hamiltonian,
Integrals in Equation (A6) have a simple block structure. The matrix t comprising matrix elements is of dimension but consists of Q blocks , where is the Hamiltonian of the i-th isolated cluster. We similarly define reduced two-body integrals (for ) for the interaction of site p of an arbitrary cluster with site of another arbitrary cluster. These integrals are generally zero for most combinations because only specific site pairs interact. Local spin matrices are of dimension . The number Z of nonzero entries in is independent of p (for , for all ). For a given , there are thus combinations of nonzero entries in and , each combination yielding a nonvanishing integral . The value of is the product of the respective nonzero entries in and (all couplings are assumed to have the same strength, ); (k, n) and (l, m) are the (row, column) indices of the nonzero entries in and , respectively.
The initial guess , that is, an initial set of molecular orbitals (MOs), is provided in terms of expansion coefficients in the uncoupled basis, where the latter corresponds to an orthonormal atomic-orbital (AO) basis in electronic-structure calculations. is an vector, and is . We generate through small random mixing of occupied and virtual cHF orbitals. At each iteration of the optimization process, a pure-state density matrix for each cluster can be formed:
In the first iteration, we set and and calculate (as described below) the energy E of the projected state as well as the global gradient with respect Z.
Equations (A8) and (A9) describe the updating of through a Thouless rotation from in each iteration. The number and the lower triangular matrix are obtained from Equations (A8) and (A9) by Cholesky decomposition:
Following Equations (3.45) and (3.47) in [21], we form the intermediate :
In the following, we explain the calculation of the energy E of the projected state (similar to the PHF algorithm described in [6]; see also [20]) and the calculation of the global gradient vector G with elements , defined in Equation (A14):
As noted above, mediates a transformation from the AO to the MO basis. Matrices defined in the MO basis carry a tilde, where assumes a simple form:
In Equation (A15), the unit matrix 1 is because there is only one occupied MO per site. For spin projection, the grid weights (an Euler-angle triplet defines a grid point) are combined with Wigner D-matrix elements for all combinations of magnetic quantum numbers m and k [20]:
For combined S- and PG-projection, denotes a combination of a spin-rotation and a site permutation . The loop over grid points thus comprises two nested loops, for and g. In the spin-rotation matrix (the matrix representation of ),
is the total-spin vector of an isolated cluster, .
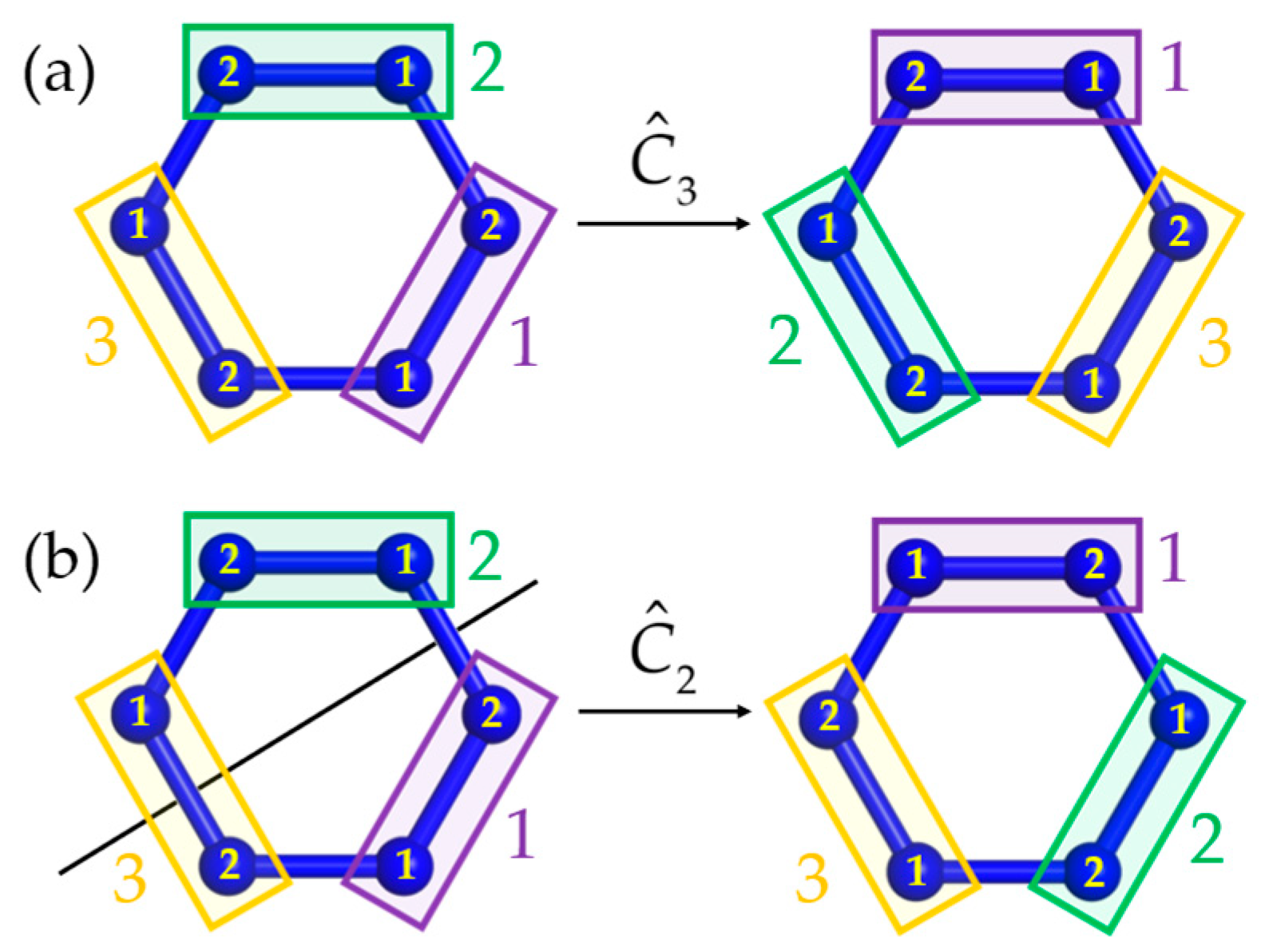
The PG-operation converts cluster i into cluster , which may be associated with a permutation among sites within the clusters (internal spin permutations). Take a dimerized symmetric spin ring as an example (Figure A1). The cyclic operation carries center of cluster into of , etc., and thus does not cause internal permutations; see Figure A1a. In contrast, the vertical two-fold rotation illustrated in Figure A1b exchanges site pairs in all three clusters. In other words, the operation is associated with internal permutations.
Figure A1.
The cyclic point-group operation keeps the numbering of spins in the clusters intact (a), but a vertical rotation causes internal permutations (b).
Figure A1.
The cyclic point-group operation keeps the numbering of spins in the clusters intact (a), but a vertical rotation causes internal permutations (b).

With causing an internal spin permutation in cluster , the single-cluster block of the combined spin-rotation/PG-operation becomes:
is the internal permutation operator. As an example, for , can have only two values (exchange or no exchange). For , the exchange operator has the simple representation [53]. Permutation operators for were derived in [54].
has four blocks,
The latter are transformed back to the AO basis:
The perturbation tensor is the sum of all increments, as summarized in Scheme A1:
Scheme A1.
Incremental calculation of the perturbation tensor.

We define as the sum of the local cluster Hamiltonian and the perturbation tensor:
The quantity is the trace of the sum , taken over all blocks:
Back in the MO basis, . At each grid point , the following quantities are incremented (for Equations (A28) and (A29), see Equations (3.37) and (3.38) in [21]):
The generalized eigenvalue-problem is solved for the lowest energy E:
The for all Q clusters is concatenated into , which is . The energy E of the projected state, the initial guess , and the global gradient G are passed to the fminunc function in Matlab. The separation into real and imaginary parts (Equation (A14)) is required for fminunc, which optimizes with respect to a set of real variables.
Note that for cPHF with a UHF (instead of a GHF reference) we first run a few iterations with an SCF algorithm to determine the local spin-projections on the z-axis, . With the initial guess thus established, we switch to gradient-based optimization, setting those elements of the gradient to zero which would change . In other words, in gradient-based optimization, all numbers are frozen at their values in the initial guess .
Appendix B. Spin-Pair Correlation Functions (SPCFs)
The calculation of SPCFs is analogous to the evaluation of the energy. A double-integration over the spin-projection grid can be avoided because is a spin scalar, which commutes with the (Hermitian and idempotent) spin-projection operator. For PG-projection, we consider only one-dimensional representations . Then only the totally symmetric part , Equation (A34):
The evaluation of the expectation value for each term occurring in the symmetrized operator depends on whether sites l and m are in the same cluster (corresponding to a single-particle term) or in different clusters (two-particle term).
Appendix C. Reference Energies for Spin Rings
Singlet and triplet energies for spin rings from SUHF, SGHF and PGSGHF are collected for reference in Table A1, Table A2 and Table A3, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Singlet and triplet energies and the gap in rings with N sites and cluster size q. SUHF predictions are compared to exact results (from Table V.1 in [32]).
Table A1.
Singlet and triplet energies and the gap in rings with N sites and cluster size q. SUHF predictions are compared to exact results (from Table V.1 in [32]).
| N | ||||||
|---|---|---|---|---|---|---|
| q | 6 | 12 | 18 | 24 | 30 | |
| 2 | −2.6514 | −4.8770 | −7.1335 | −9.3914 | −11.6485 | |
| −1.8956 | −4.3114 | −6.6414 | −8.9434 | −11.2237 | ||
| 0.756 | 0.566 | 0.492 | 0.448 | 0.425 | ||
| 6 | −2.8028 | −5.3482 | −7.7332 | −10.2071 | −12.6941 | |
| −2.1180 | −4.7874 | −7.3492 | −9.8772 | −12.3954 | ||
| 0.685 | 0.561 | 0.384 | 0.330 | 0.299 | ||
| Exact | −2.803 | −5.387 | −8.023 | −10.670 | −13.322 | |
| Exact | 0.685 | 0.356 | 0.241 | 0.183 | 0.147 | |
Table A2.
Singlet and triplet energies and the gap in rings with N sites and cluster size q. SGHF predictions are compared to exact results (from Table V.1 in [32]).
Table A2.
Singlet and triplet energies and the gap in rings with N sites and cluster size q. SGHF predictions are compared to exact results (from Table V.1 in [32]).
| N | ||||||
|---|---|---|---|---|---|---|
| q | 6 | 12 | 18 | 24 | 30 | |
| 2 | −2.8028 | −5.0625 | −7.3603 | −9.6589 | −11.9416 | |
| −2.1180 | −4.5485 | −6.8696 | −9.1446 | −11.4453 | ||
| 0.685 | 0.514 | 0.491 | 0.514 | 0.496 | ||
| 6 | −2.8028 | −5.3768 | −7.9641 | −10.4728 | −12.9231 | |
| −2.1180 | −5.0090 | −7.6042 | −10.1411 | −12.6090 | ||
| 0.685 | 0.368 | 0.360 | 0.332 | 0.314 | ||
| Exact | −2.803 | −5.387 | −8.023 | −10.670 | −13.322 | |
| Exact | 0.685 | 0.356 | 0.241 | 0.183 | 0.147 | |
Table A3.
Singlet and triplet energies and the gap in rings with N sites and cluster size q. predictions are compared to exact results (from Table V.1 in [32]).
Table A3.
Singlet and triplet energies and the gap in rings with N sites and cluster size q. predictions are compared to exact results (from Table V.1 in [32]).
| N | ||||||
|---|---|---|---|---|---|---|
| q | 6 | 12 | 18 | 24 | 30 | |
| 2 | −2.8028 | −5.3710 | −7.8905 | −10.3945 | −12.8677 | |
| −2.1180 | −5.0104 | −7.5544 | −10.0287 | −12.4834 | ||
| 0.685 | 0.361 | 0.336 | 0.366 | 0.384 | ||
| 6 | −2.8028 | −5.3874 | −8.0224 | −10.6501 | −13.2762 | |
| −2.1180 | −5.0315 | −7.7782 | −10.4356 | −13.0870 | ||
| 0.685 | 0.356 | 0.244 | 0.215 | 0.189 | ||
| Exact | −2.803 | −5.387 | −8.023 | −10.670 | −13.322 | |
| Exact | 0.685 | 0.356 | 0.241 | 0.183 | 0.147 | |
References
- Bencini, A.; Gatteschi, D. Electron Paramagnetic Resonance of Exchange Coupled Systems; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar]
- Schnack, J. Large Magnetic Molecules and What We Learn from Them. Contemp. Phys. 2019, 60, 127. [Google Scholar] [CrossRef]
- Ghassemi Tabrizi, S.; Arbuznikov, A.; Kaupp, M. Understanding Thermodynamic and Spectroscopic Properties of Tetragonal Mn12 Single-Molecule Magnets from Combined Density Functional Theory/Spin-Hamiltonian Calculations. J. Phys. Chem. A 2016, 120, 6864. [Google Scholar] [CrossRef] [PubMed]
- White, S.R. Density Matrix Formulation for Quantum Renormalization Groups. Phys. Rev. Lett. 1992, 69, 2863. [Google Scholar] [CrossRef]
- Jiménez-Hoyos, C.A.; Henderson, T.M.; Tsuchimochi, T.; Scuseria, G.E. Projected Hartree–Fock Theory. J. Chem. Phys. 2012, 136, 164109. [Google Scholar] [CrossRef]
- Ghassemi Tabrizi, S.; Jiménez-Hoyos, C.A. Ground States of Heisenberg Spin Clusters from Projected Hartree–Fock Theory. Phys. Rev. B 2022, 105, 35147. [Google Scholar] [CrossRef]
- Mayer, I. The Spin-Projected Extended Hartree–Fock Method. Adv. Quant. Chem. 1980, 12, 189. [Google Scholar]
- Jiménez-Hoyos, C.A.; Rodríguez-Guzmán, R.; Scuseria, G.E. Multi-Component Symmetry-Projected Approach for Molecular Ground State Correlations. J. Chem. Phys. 2013, 139, 204102. [Google Scholar] [CrossRef]
- Papastathopoulos-Katsaros, A.; Jiménez-Hoyos, C.A.; Henderson, T.M.; Scuseria, G.E. Coupled Cluster and Perturbation Theories Based on a Cluster Mean-Field Reference Applied to Strongly Correlated Spin Systems. J. Chem. Theory Comput. 2022, 18, 4293. [Google Scholar] [CrossRef]
- Potthoff, M. Cluster Extensions of Dynamical Mean-Field Theory. In DMFT: From Infinite Dimensions to Real Materials; Pavarini, E., Kock, E., Lichtenstein, A., Vollhardt, D., Eds.; Forschungszentrum Jülich GmbH: Jülich, Germany, 2018. [Google Scholar]
- Waldmann, O. Symmetry and Energy Spectrum of High-Nuclearity Spin Clusters. Phys. Rev. B 2000, 61, 6138. [Google Scholar] [CrossRef]
- Schmid, K.W.; Dahm, T.; Margueron, J.; Müther, H. Symmetry-Projected Variational Approach to the One-Dimensional Hubbard Model. Phys. Rev. B 2005, 72, 85116. [Google Scholar] [CrossRef]
- Tinkham, M. Group Theory and Quantum Mechanics; McGraw-Hill: New York, NY, USA, 1964. [Google Scholar]
- Percus, J.K.; Rotenberg, A. Exact Eigenfunctions of Angular Momentum by Rotational Projection. J. Math. Phys. 1962, 3, 928. [Google Scholar] [CrossRef]
- Ren, Y.-Z.; Tong, N.-H.; Xie, X.-C. Cluster Mean-Field Theory Study of J1 − J2 Heisenberg Model on a Square Lattice. J. Phys. Condens. Matter 2014, 26, 115601. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Hoyos, C.A.; Henderson, T.M.; Scuseria, G.E. Generalized Hartree–Fock Description of Molecular Dissociation. J. Chem. Theory Comput. 2011, 7, 2667. [Google Scholar] [CrossRef] [PubMed]
- Hendeković, J. Method of Complex Molecular Orbitals. Int. J. Quantum Chem. 1974, 8, 799. [Google Scholar] [CrossRef]
- Jiménez-Hoyos, C.A.; Rodríguez-Guzmán, R.; Scuseria, G.E. N-Electron Slater Determinants from Nonunitary Canonical Transformations of Fermion Operators. Phys. Rev. A 2012, 86, 52102. [Google Scholar] [CrossRef]
- Ghassemi Tabrizi, S.; Arbuznikov, A.; Jiménez-Hoyos, C.A.; Kaupp, M. Hyperfine-Coupling Tensors from Projected Hartree–Fock Theory. J. Chem. Theory Comput. 2020, 16, 6222. [Google Scholar] [CrossRef] [PubMed]
- Lestrange, P.J.; Williams-Young, D.B.; Petrone, A.; Jiménez-Hoyos, C.A.; Li, X. Efficient Implementation of Variation after Projection Generalized Hartree–Fock. J. Chem. Theory Comput. 2018, 14, 588. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Hoyos, C.A. Variational Approaches to the Molecular Electronic Structure Problem Based on Symmetry-Projected Hartree–Fock Configurations. Ph.D. Thesis, Rice University, Houston, TX, USA, 2013. [Google Scholar]
- Jiménez-Hoyos, C.A.; Rodríguez-Guzmán, R.R.; Henderson, T.M.; Scuseria, G.E. On a Dual Representation of the Goldstone Manifold. arXiv 2020, arXiv:2004.05047. [Google Scholar]
- Schurkus, H.F.; Chen, D.; O’Rourke, M.J.; Cheng, H.-P.; Chan, G.K.-L. Exploring the Magnetic Properties of the Largest Single-Molecule Magnets. J. Phys. Chem. Lett. 2020, 11, 3789. [Google Scholar] [CrossRef]
- Lebedev, V.I.; Laikov, D.N. A Quadrature Formula for the Sphere of the 131st Algebraic Order of Accuracy. Dokl. Math. 1999, 59, 477. [Google Scholar]
- Rivero, P.; Jiménez-Hoyos, C.A.; Scuseria, G.E. Entanglement and Polyradical Character of Polycyclic Aromatic Hydrocarbons Predicted by Projected Hartree–Fock Theory. J. Phys. Chem. B 2013, 117, 12750. [Google Scholar] [CrossRef] [PubMed]
- Heitmann, T.; Schnack, J. Combined Use of Translational and Spin-Rotational Invariance for Spin Systems. Phys. Rev. B 2019, 99, 134405. [Google Scholar] [CrossRef]
- Ummethum, J.; Nehrkorn, J.; Mukherjee, S.; Ivanov, N.B.; Stuiber, S.; Strässle, T.; Tregenna-Piggott, P.L.W.; Mutka, H.; Christou, G.; Waldmann, O.; et al. Discrete Antiferromagnetic Spin-Wave Excitations in the Giant Ferric Wheel Fe18. Phys. Rev. B 2012, 86, 104403. [Google Scholar] [CrossRef] [Green Version]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry; Dover Publications, Courier Corporation: Chelmsford, MA, USA, 1996. [Google Scholar]
- Coffey, D.; Trugman, S.A. Magnetic Properties of Undoped. C60 Phys. Rev. Lett. 1992, 69, 176. [Google Scholar] [CrossRef]
- Jiménez-Hoyos, C.A.; Scuseria, G.E. Cluster-Based Mean-Field and Perturbative Description of Strongly Correlated Fermion Systems: Application to the One-and Two-Dimensional Hubbard Model. Phys. Rev. B 2015, 92, 85101. [Google Scholar] [CrossRef]
- Schnack, J. Properties of the First Excited State of Nonbipartite Heisenberg Spin Rings. Phys. Rev. B 2000, 62, 14855. [Google Scholar] [CrossRef]
- Ummethum, J. Calculation of Static and Dynamical Properties of Giant Magnetic Molecules Using DMRG. Ph.D. Thesis, Bielefeld University, Bielefeld, Germany, 2012. [Google Scholar]
- Schnack, J. Effects of Frustration on Magnetic Molecules: A Survey from Olivier Kahn until Today. Dalt. Trans. 2010, 39, 4677. [Google Scholar] [CrossRef]
- Clar, E. The Aromatic Sextet and its Significance in Relation to the Stability of Aromatic Systems. In Polycyclic Hydrocarbons; Springer: Berlin/Heidelberg, Germany, 1964; pp. 32–39. [Google Scholar]
- Schmidt, H.-J.; Luban, M. Classical Ground States of Symmetric Heisenberg Spin Systems. J. Phys. A. Math. Gen. 2003, 36, 6351. [Google Scholar] [CrossRef]
- Coffey, D.; Trugman, S.A. Correlations for the S = 1/2 Antiferromagnet on a Truncated Tetrahedron. Phys. Rev. B 1992, 46, 12717. [Google Scholar] [CrossRef]
- Müller, A.; Todea, A.M.; van Slageren, J.; Dressel, M.; Bögge, H.; Schmidtmann, M.; Luban, M.; Engelhardt, L.; Rusu, M. Triangular Geometrical and Magnetic Motifs Uniquely Linked on a Spherical Capsule Surface. Angew. Chem. 2005, 117, 3925. [Google Scholar] [CrossRef]
- Botar, B.; Kögerler, P.; Hill, C.L. [{(Mo)Mo5O21(H2O)3(SO4)}12(VO)30(H2O)20]36−: A Molecular Quantum Spin Icosidodecahedron. Chem. Commun. 2005, 3138–3140. [Google Scholar] [CrossRef] [PubMed]
- Todea, A.M.; Merca, A.; Bögge, H.; Glaser, T.; Engelhardt, L.; Prozorov, R.; Luban, M.; Müller, A. Polyoxotungstates Now Also with Pentagonal Units: Supramolecular Chemistry and Tuning of Magnetic Exchange in {(M)M5}12V30 Keplerates (M = Mo, W). Chem. Commun. 2009, 3351–3353. [Google Scholar] [CrossRef]
- Todea, A.M.; Merca, A.; Boegge, H.; van Slageren, J.; Dressel, M.; Engelhardt, L.; Luban, M.; Glaser, T.; Henry, M.; Mueller, A. Extending the {(Mo)Mo5}12M30 Capsule Keplerate Sequence: A {Cr30} Cluster of S = 3/2 Metal Centers with a {Na(H2O)12} Encapsulate. Angew. Chemie Int. Ed. 2007, 46, 6106. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Sarkar, S.; Shah, S.Q.N.; Bögge, H.; Schmidtmann, M.; Sarkar, S.; Kögerler, P.; Hauptfleisch, B.; Trautwein, A.X.; Schünemann, V. Archimedean Synthesis and Magic Numbers: “Sizing” Giant Molybdenum-Oxide-Based Molecular Spheres of the Keplerate Type. Angew. Chemie Int. Ed. 1999, 38, 3238. [Google Scholar] [CrossRef]
- Müller, A.; Luban, M.; Schröder, C.; Modler, R.; Kögerler, P.; Axenovich, M.; Schnack, J.; Canfield, P.; Bud’ko, S.; Harrison, N. Classical and Quantum Magnetism in Giant Keplerate Magnetic Molecules. ChemPhysChem 2001, 2, 517. [Google Scholar] [CrossRef]
- Krivnov, V.Y.; Shamovsky, I.L.; Tornau, E.E.; Rosengren, A. Electronic Correlation Effects in a Fullerene Molecule Studied by the Variational Monte Carlo Method. Phys. Rev. B 1994, 50, 12144. [Google Scholar] [CrossRef]
- Sheng, D.N.; Weng, Z.Y.; Ting, C.S.; Dong, J.M. Magnetism and Pairing in a C60 Molecule: A Variational Monte Carlo Study. Phys. Rev. B 1994, 49, 4279. [Google Scholar] [CrossRef]
- Flocke, N.; Schmalz, T.G.; Klein, D.J. Variational Resonance Valence Bond Study on the Ground State of C60 Using the Heisenberg Model. J. Chem. Phys. 1998, 109, 873. [Google Scholar] [CrossRef]
- Konstantinidis, N.P. Unconventional Magnetic Properties of the Icosahedral Symmetry Antiferromagnetic Heisenberg Model. Phys. Rev. B 2007, 76, 104434. [Google Scholar] [CrossRef]
- Rausch, R.; Plorin, C.; Peschke, M. The Antiferromagnetic S = 1/2 Heisenberg Model on the C60 Fullerene Geometry. SciPost Phys. 2021, 10, 87. [Google Scholar] [CrossRef]
- Ghassemi Tabrizi, S. Point-Group Selection Rules and Universal Momentum-Transfer Dependencies for Inelastic Neutron Scattering on Molecular Spin Clusters. Phys. Rev. B 2021, 103, 214422. [Google Scholar] [CrossRef]
- Ghassemi Tabrizi, S. Symmetry-Induced Universal Momentum-Transfer Dependencies for Inelastic Neutron Scattering on Anisotropic Spin Clusters. Phys. Rev. B 2021, 104, 14416. [Google Scholar] [CrossRef]
- Jiménez-Hoyos, C.A.; Rodríguez-Guzmán, R.; Scuseria, G.E. Polyradical Character and Spin Frustration in Fullerene Molecules: An Ab Initio Non-Collinear Hartree–Fock Study. J. Phys. Chem. A 2014, 118, 9925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Head-Gordon, M. Distinguishing Artificial and Essential Symmetry Breaking in a Single Determinant: Approach and Application to the C60, C36, and C20 Fullerenes. Phys. Chem. Chem. Phys. 2019, 21, 4763. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.L.; Herzig, P. Point-Group Theory Tables; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Sakurai, J.J. Modern Quantum Mechanics, 2nd ed.; Tuan, S.F., Ed.; Addison Wesley: Reading, MA, USA, 1993. [Google Scholar]
- Brown, H.A. A Simple Derivation of the Spin-Exchange Operator. Am. J. Phys. 1972, 40, 1696. [Google Scholar] [CrossRef]
Figure 1.
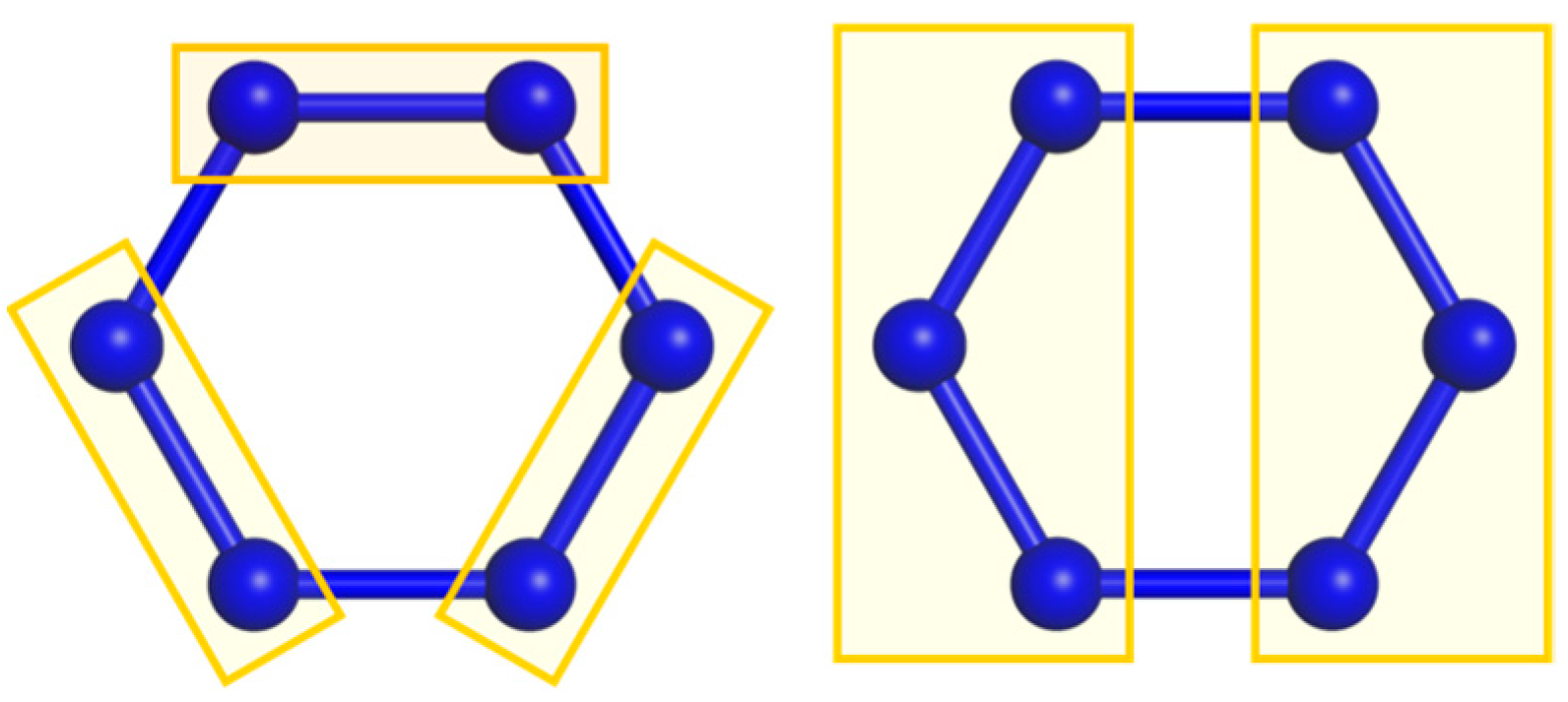
Two cluster groupings in an ring. Dihedral symmetry is broken by dimerization or trimerization . The respective reduced symmetry groups can be employed in cPHF.
Figure 1.
Two cluster groupings in an ring. Dihedral symmetry is broken by dimerization or trimerization . The respective reduced symmetry groups can be employed in cPHF.

Figure 2.
Dimers of diametrically opposite sites conserve the symmetry of rings (group in this example). A cyclic permutation interchanges sites in the last cluster (cluster 3, yellow box).
Figure 2.
Dimers of diametrically opposite sites conserve the symmetry of rings (group in this example). A cyclic permutation interchanges sites in the last cluster (cluster 3, yellow box).

Figure 3.
Clusters of size in an ring.

Figure 4.
Relative correlation energies p (Equation (9)) from cluster variants of UHF, SUHF, SGHF and in antiferromagnetic spin rings with sites. Data points are connected by dotted (q = 2), dashed (q = 3) or solid lines (q = 6).
Figure 4.
Relative correlation energies p (Equation (9)) from cluster variants of UHF, SUHF, SGHF and in antiferromagnetic spin rings with sites. Data points are connected by dotted (q = 2), dashed (q = 3) or solid lines (q = 6).

Figure 5.
Relative singlet-triplet gaps from cluster variants of SUHF, SGHF and in antiferromagnetic spin rings with sites. Data points are connected by dotted (cluster size q = 2), dashed (q = 3) or solid lines (q = 6).
Figure 5.
Relative singlet-triplet gaps from cluster variants of SUHF, SGHF and in antiferromagnetic spin rings with sites. Data points are connected by dotted (cluster size q = 2), dashed (q = 3) or solid lines (q = 6).

Figure 6.
Cluster groupings in triphenylene conserving the full symmetry. In (a–c), clusters are shown in different colors; in (d,e), all dimer clusters are shown in the same color for simplicity.
Figure 6.
Cluster groupings in triphenylene conserving the full symmetry. In (a–c), clusters are shown in different colors; in (d,e), all dimer clusters are shown in the same color for simplicity.

Figure 7.
Cluster groupings in triphenylene (N = 24) with symmetries (a), (b,f–h), and (c–e). In (e,f), all dimers (q = 2) are shown in the same color for simplicity. In (g), three clusters comprise two separate trimers each. In (h), two clusters comprise three separate dimers, and six disconnected sites (red) constitute another cluster.
Figure 7.
Cluster groupings in triphenylene (N = 24) with symmetries (a), (b,f–h), and (c–e). In (e,f), all dimers (q = 2) are shown in the same color for simplicity. In (g), three clusters comprise two separate trimers each. In (h), two clusters comprise three separate dimers, and six disconnected sites (red) constitute another cluster.

Figure 8.
Cluster groupings ((a), and ((b), in hexabenzocoronene (N = 42) conserving the full symmetry.
Figure 8.
Cluster groupings ((a), and ((b), in hexabenzocoronene (N = 42) conserving the full symmetry.

Figure 9.
Cluster groupings in hexa-cata-hexabenzocoronene (N = 48) have symmetries (a,c) or (b). In (a), six disconnected sites (pink) constitute one cluster.
Figure 9.
Cluster groupings in hexa-cata-hexabenzocoronene (N = 48) have symmetries (a,c) or (b). In (a), six disconnected sites (pink) constitute one cluster.

Figure 10.
Cluster groupings in kekulene (N = 48) with symmetries (a), (b,c), and (d). In (a,b), two clusters (grey and pink) each comprise three separate dimers (a) or six separate sites (b).
Figure 10.
Cluster groupings in kekulene (N = 48) with symmetries (a), (b,c), and (d). In (a,b), two clusters (grey and pink) each comprise three separate dimers (a) or six separate sites (b).

Figure 11.
Icosahedron (a), truncated tetrahedron (b), truncated icosahedron (c), and truncated icosidodecahedron (d).
Figure 11.
Icosahedron (a), truncated tetrahedron (b), truncated icosahedron (c), and truncated icosidodecahedron (d).

Figure 12.
Three cluster groupings in the icosahedron conserve ((a), ), ((b), ), or symmetry ((c), ). In the planar projections (Schlegel diagrams), interacting sites belonging to the same or to different clusters are connected by blue or pink lines, respectively. In (a), the color of sites assigns them to one of six clusters. Sites forming symmetry-inequivalent pairs with site 1 are numbered in (a).
Figure 12.
Three cluster groupings in the icosahedron conserve ((a), ), ((b), ), or symmetry ((c), ). In the planar projections (Schlegel diagrams), interacting sites belonging to the same or to different clusters are connected by blue or pink lines, respectively. In (a), the color of sites assigns them to one of six clusters. Sites forming symmetry-inequivalent pairs with site 1 are numbered in (a).

Figure 13.
Dimers (a) and trimers (b) respect the full symmetry of the truncated tetrahedron. For more details, see caption to Figure 12.
Figure 13.
Dimers (a) and trimers (b) respect the full symmetry of the truncated tetrahedron. For more details, see caption to Figure 12.

Figure 14.
Classical three-dimensional spin configuration of the truncated icosahedron. Green spin vectors lie in the xy-plane (paper plane); red/blue vectors point in the negative/positive z-direction. The first vector on the central pentagon points in the (horizontal) negative x-direction. Symmetry elements (, and , see main text for details) are defined with respect to an axis through the coordinate origin and the center of the respective pentagon or hexagon.
Figure 14.
Classical three-dimensional spin configuration of the truncated icosahedron. Green spin vectors lie in the xy-plane (paper plane); red/blue vectors point in the negative/positive z-direction. The first vector on the central pentagon points in the (horizontal) negative x-direction. Symmetry elements (, and , see main text for details) are defined with respect to an axis through the coordinate origin and the center of the respective pentagon or hexagon.

Figure 15.
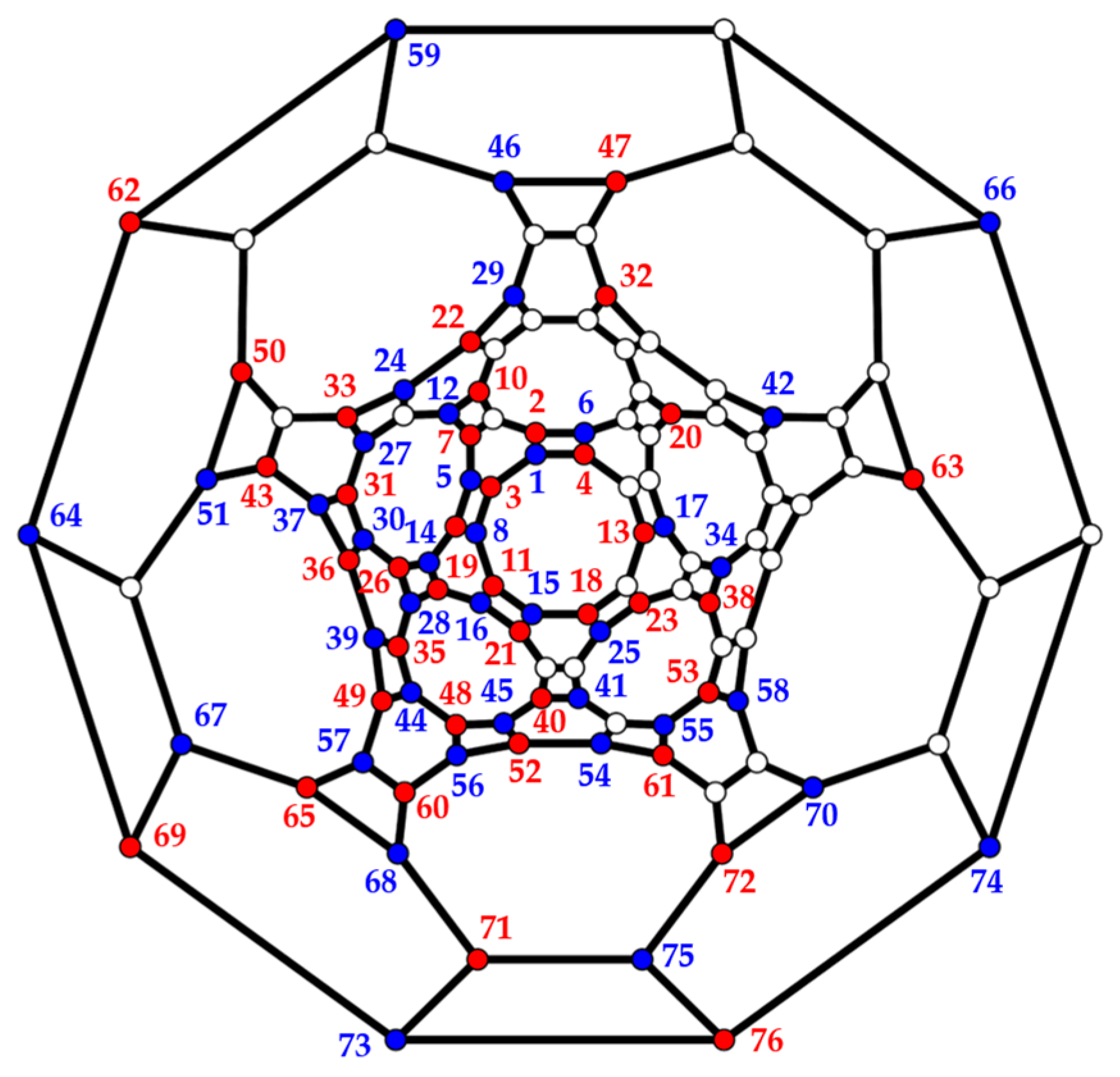
Interpentagon bonds ((a), q = 2) or pentagon clusters ((b), q = 5) maintain the symmetry of the truncated icosahedron. Fused hexagons ((c), q = 10) reduce symmetry to . The numbering (a) of centers forming inequivalent pairs with site 1 follows Figure 3 in [43].

Figure 16.
Cluster groupings compatible with symmetry in the truncated icosidodecahedron. The midpoints of the squares ((a), ), hexagons ((b), ), and decagons ((c), q = 10) form an icosidodecahedron (Q = 30), dodecahedron (Q = 20), or icosahedron (Q = 12), respectively.
Figure 16.
Cluster groupings compatible with symmetry in the truncated icosidodecahedron. The midpoints of the squares ((a), ), hexagons ((b), ), and decagons ((c), q = 10) form an icosidodecahedron (Q = 30), dodecahedron (Q = 20), or icosahedron (Q = 12), respectively.

Figure 17.
Numbering of centers forming inequivalent pairs with site 1 in the bicolorable (red, blue) truncated icosidodecahedron lattice. Sites without a number are white but still belong to one of the two sublattices.
Figure 17.
Numbering of centers forming inequivalent pairs with site 1 in the bicolorable (red, blue) truncated icosidodecahedron lattice. Sites without a number are white but still belong to one of the two sublattices.

Figure 18.
Magnitude of SPCFs (left y-axis; correlations are positive/negative for pairs in same/different lattices) and bond distances from reference site 1 (right y-axis) in the truncated icosidodecahedron. The site numbering follows Figure 17.
Figure 18.
Magnitude of SPCFs (left y-axis; correlations are positive/negative for pairs in same/different lattices) and bond distances from reference site 1 (right y-axis) in the truncated icosidodecahedron. The site numbering follows Figure 17.

Table 1.
Mulliken labels a of ground states in sectors and of even N antiferromagnetic rings in the reduced dihedral group .
Table 1.
Mulliken labels a of ground states in sectors and of even N antiferromagnetic rings in the reduced dihedral group .
| q even | q odd | q even | q odd | q even | q odd | q even | q odd |
| A2 | B1 | A1 | A1 | A1 | A1 | A2 | B1 |
a Label A(B) and subscript 1(2) respectively denote symmetry(antisymmetry) under the cyclic permutation or the vertical operation that exchanges all sites pairwise.
Table 2.
Ground-state energy estimates for honeycomb-lattice fragments from cluster variants of UHF, SGHF, PGSGHF, and PT2. The lowest energy for each grouping/method is given in bold type.
Table 2.
Ground-state energy estimates for honeycomb-lattice fragments from cluster variants of UHF, SGHF, PGSGHF, and PT2. The lowest energy for each grouping/method is given in bold type.
| System | Grouping (q, Bonds) a | UHF | SGHF | PT2 | Exact | |
|---|---|---|---|---|---|---|
| Triphenylene | a (3, 12) | −7.2753 | −8.2062 | −8.6556 (A2) | −8.6445 | −8.7697 |
| b (6, 15) | −7.5804 | −8.4865 | −8.7342 (A2) | −7.8205 | ||
| c (6, 18) | −8.4083 | −8.7556 | −8.7696 (A2) | −8.6640 | ||
| d (2, 9) | −7.0229 | −8.0255 | −8.5364 (A2) | −8.2068 | ||
| e (2, 9) | −6.9584 | −7.8672 | −8.4093 (A2) | −8.1914 | ||
| Coronene | a (4, 24) | −10.2764 | −11.2733 | −11.7966 (A) | −11.4628 | −11.9755 |
| b (4, 18) | −9.7660 | −10.7447 | −11.6399 (A1) | −11.3781 | ||
| c (6, 21) | −10.5736 | −11.3457 | −11.8103 (A1) | −11.3740 | ||
| d (8, 24) | −10.8304 | −11.6961 | −11.9459 (A1) | −11.4992 | ||
| e (2, 12) | −9.5702 | −10.6676 | −11.2997 (A1) | −11.3109 | ||
| f (2, 12) | −9.6055 | −10.7084 | −11.6190 (A1) | −11.3968 | ||
| g (6, 18) | −9.9867 | −10.9182 | −11.6693 (A1) | −11.1837 | ||
| h (6, 12) | −9.4117 | −11.1230 | −11.8635 (A1) | −11.1287 | ||
| Hexabenzo- coronene | a (6, 42) | −19.8044 | −20.4297 | −21.0044 (B1) | −20.7082 | – b |
| b (7, 42) | −19.6337 | −20.4824 | −21.0786 (B1) | −20.7724 | ||
| Hexa-cata-hexabenzo-coronene | a (6, 42) | −21.5999 | −23.0983 | −23.8296 (A1) | −23.1753 | – b |
| b (8, 48) | −22.4271 | −23.3169 | −23.7980 (A) | −23.4848 | ||
| c (8, 42) | −21.1057 | −21.9885 | −23.0109 (A1) | −23.0836 | ||
| Kekulene | a (6, 48) | −21.4983 | −22.2249 | −22.7387 (A1) | −22.9403 | – b |
| b (6, 36) | −19.8349 | −20.9791 | −22.4499 (A1) | −22.2943 | ||
| c (8, 48) | −21.4735 | −22.2083 | −23.0747 (A1) | −22.9187 | ||
| d (8, 48) | −22.1306 | −22.7459 | −23.3603 (A) | −23.2404 |
Table 3.
SPCFs in coronene from PGSGHF, compared against exact results. Letters (a, b, c, d) identifying cluster groupings, and the site numbers (first column) are defined in Figure 7 a.
Table 3.
SPCFs in coronene from PGSGHF, compared against exact results. Letters (a, b, c, d) identifying cluster groupings, and the site numbers (first column) are defined in Figure 7 a.
| i–j | Exact | ||||
|---|---|---|---|---|---|
| 1–2 | −0.33729 | −0.35059 | −0.39063 | −0.36850 | −0.35875 |
| 2–3 | −0.33729 | −0.35059 | −0.38706 | −0.33627 | −0.35875 |
| 1–4 | −0.40404 | −0.35322 | −0.30621 | −0.37926 | −0.37507 |
| 4–5 | −0.37034 | −0.40476 | −0.37196 | −0.37095 | −0.36665 |
| 5–6 | −0.54911 | −0.42665 | −0.52422 | −0.52685 | −0.52881 |
| 6–7 | −0.30533 | −0.40476 | −0.37196 | −0.37095 | −0.36665 |
| 7–8 | −0.37034 | −0.40476 | −0.41287 | −0.36483 | −0.36665 |
| 8–9 | −0.54911 | −0.42665 | −0.45278 | −0.52027 | −0.52881 |
| (1–3) | 0.17744 | 0.19472 | 0.19503 | 0.16792 | 0.16641 |
| (1–5) | 0.18265 | 0.20098 | 0.16410 | 0.17971 | 0.17661 |
| (2–6) | 0.18009 | 0.20098 | 0.16410 | 0.17971 | 0.17661 |
| (4–6) | 0.20777 | 0.18967 | 0.19643 | 0.19440 | 0.19291 |
| (5–7) | 0.17515 | 0.18967 | 0.19643 | 0.19440 | 0.19291 |
a SPCFs for all distinct NN pairs i–j (first column) are given, including pairs that are equivalent in the full symmetry group but inequivalent in some of the PGSGHF wave functions. The NNN set (pairs i–j in parentheses) is not complete, except for (b), which maintains the full symmetry. Bold type is used for SPCFs of pairs that belong to the same cluster.
Table 4.
Comparison of predictions of SPCFs in the ground state of the icosahedron with against exact values. Site numbers are defined in Figure 12a.
Table 4.
Comparison of predictions of SPCFs in the ground state of the icosahedron with against exact values. Site numbers are defined in Figure 12a.
| s | ||||
|---|---|---|---|---|
| 1/2 | Exact | −0.2063 | 0.0841 | −0.1397 |
| PHF | −0.2063 | 0.0841 | −0.1397 | |
| 1 | Exact | −0.6187 | 0.3680 | −0.7463 |
| PHF | −0.6187 | 0.3680 | −0.7464 | |
| 3/2 | Exact | −1.2580 | 0.9060 | −1.9899 |
| PHF | −1.2580 | 0.9062 | −1.9910 | |
| 2 | Exact | −2.1237 | 1.6616 | −3.6897 |
| PHF | −2.1236 | 1.6621 | −3.6926 |
Table 5.
GHF and PHF estimates of ground-state energies of the antiferromagnetic icosahedron with for two different groupings (Figure 12a,b) with or symmetry.
Table 5.
GHF and PHF estimates of ground-state energies of the antiferromagnetic icosahedron with for two different groupings (Figure 12a,b) with or symmetry.
| s | Grouping | GHF | SGHF | PGSGHF | |
|---|---|---|---|---|---|
| 1/2 | −4.5000 | −5.3224 | −6.1717 | −6.1879 (Au) | |
| −3.3541 | −5.7644 | −6.1879 a | |||
| 1 | −14.3025 | −17.4565 | −18.1678 | −18.5611 (Ag) | |
| −13.4164 | −18.2225 | −18.5609 | |||
| 3/2 | −31.4256 | −36.2633 | −37.3073 | −37.7412 (Au) | |
| −30.1869 | −37.3842 | −37.7396 | |||
| 2 | −55.2658 | −61.7751 | −63.1481 | −63.7104 (Ag) | |
| −53.6656 | −63.2529 | −63.7075 |
a Exact ground-state energy within numerical double precision.
Table 6.
GHF and PHF estimates of ground-state energies of the antiferromagnetic truncated tetrahedron for two different groupings (Figure 13).
Table 6.
GHF and PHF estimates of ground-state energies of the antiferromagnetic truncated tetrahedron for two different groupings (Figure 13).
| s | q | GHF | SGHF | TdSGHF | |
|---|---|---|---|---|---|
| 1/2 | 2 | −4.5000 | −5.2700 | −5.7009 a | −5.7009 (A2) |
| 3 | −3.8881 | −4.8147 | −5.7009 a | ||
| 1 | 2 | −14.0173 | −16.0342 | −17.1649 | −17.1955 (A1) |
| 3 | −13.8696 | −15.7195 | −17.1775 | ||
| 3/2 | 2 | −29.7756 | −32.8938 | −34.4456 | −34.6402 (A2) |
| 3 | −29.6977 | −32.5614 | −34.4796 | ||
| 2 | 2 | −51.5616 | −55.7815 | −57.7827 | −58.1140 (A1) |
| 3 | −51.5327 | −55.3924 | −57.8181 |
a Exact ground-state energy within numerical double precision.
Table 7.
cGHF and cPHF estimates of ground-state energies of the truncated icosahedron.
| q | GHF | SGHF | PGSGHF a |
|---|---|---|---|
| 2 | −24.2705 | −25.5486 | −27.8429 |
| 5 | −25.8525 | −26.6072 | −28.5653 |
| 10 | −28.6199 b | −29.2195 | −29.9842 |
a Projection onto , . b All clusters assume their local singlet ground state.
Table 8.
SPCFs, , for all distinct pair types (numbering defined in Figure 15) in the ground state of the truncated icosahedron. VMC results were taken from Table II in [43].
| q = 2 | q = 5 | ||||
|---|---|---|---|---|---|
| E | −25.5486 | −27.8429 | −26.6072 | −28.5653 | −30.69 |
| j | SGHF | SGHF | VMC | ||
| 2 | −0.562 | −0.610 | −0.186 | −0.277 | −0.529 |
| 3 | −0.145 | −0.159 | −0.351 | −0.337 | −0.247 |
| 4 | 0.051 | 0.051 | 0.076 | 0.073 | 0.030 |
| 5 | 0.136 | 0.137 | 0.142 | 0.154 | 0.141 |
| 6 | −0.145 | −0.154 | −0.151 | −0.154 | −0.142 |
| 7 | −0.056 | −0.054 | −0.059 | −0.061 | −0.023 |
| 8 | −0.090 | −0.080 | −0.094 | −0.093 | −0.038 |
| 9 | 0.084 | 0.070 | 0.087 | 0.083 | 0.031 |
| 10 | −0.002 | 0.001 | −0.003 | −0.001 | 0.001 |
| 11 | 0.051 | 0.049 | 0.052 | 0.051 | 0.027 |
| 12 | −0.090 | −0.072 | −0.094 | −0.088 | −0.026 |
| 13 | −0.090 | −0.042 | −0.094 | −0.084 | −0.002 |
| 14 | 0.051 | 0.017 | 0.052 | 0.046 | −0.001 |
| 15 | −0.002 | −0.002 | −0.003 | −0.003 | −0.004 |
| 16 | 0.084 | 0.037 | 0.087 | 0.078 | 0.001 |
| 17 | −0.090 | −0.036 | −0.094 | −0.081 | 0.002 |
| 18 | −0.056 | −0.018 | −0.059 | −0.051 | 0.013 |
| 19 | −0.145 | −0.042 | −0.151 | −0.129 | 0.000 |
| 20 | 0.136 | 0.039 | 0.142 | 0.124 | −0.002 |
| 21 | 0.051 | 0.016 | 0.053 | 0.046 | −0.030 |
| 22 | −0.145 | −0.040 | −0.150 | −0.128 | 0.007 |
| 23 | −0.179 | −0.046 | −0.186 | −0.158 | 0.016 |
| 24 | 0.168 | 0.044 | 0.176 | 0.152 | −0.008 |
Table 9.
Variational estimates from GHF; SGHF; (projection onto , ), a singlet-product on fused hexagons ; and PT2 for the ground state of the truncated icosahedron with .
Table 9.
Variational estimates from GHF; SGHF; (projection onto , ), a singlet-product on fused hexagons ; and PT2 for the ground state of the truncated icosahedron with .
| s | GHF (2) | SGHF (2) | IhSGHF (2) | PT2 (1) | PT2 (2) | |
|---|---|---|---|---|---|---|
| 1/2 | −24.2705 | −25.5486 | −27.8429 | −28.6199 | −31.0543 | −29.1216 |
| 1 | −85.6371 | −87.7764 | −90.1147 | −89.8943 | −96.7113 | −96.9910 |
| 3/2 | −186.6961 | −189.5706 | −192.4293 | −183.7630 | −202.2428 | −203.7112 |
| 2 | −327.0802 | −330.4438 | −343.1173 | −310.5941 | −347.1526 | −349.5852 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ghassemi Tabrizi, S.; Jiménez-Hoyos, C.A. Ground States of Heisenberg Spin Clusters from a Cluster-Based Projected Hartree–Fock Approach. Condens. Matter 2023, 8, 18. https://doi.org/10.3390/condmat8010018
AMA Style
Ghassemi Tabrizi S, Jiménez-Hoyos CA. Ground States of Heisenberg Spin Clusters from a Cluster-Based Projected Hartree–Fock Approach. Condensed Matter. 2023; 8(1):18. https://doi.org/10.3390/condmat8010018
Chicago/Turabian StyleGhassemi Tabrizi, Shadan, and Carlos A. Jiménez-Hoyos. 2023. "Ground States of Heisenberg Spin Clusters from a Cluster-Based Projected Hartree–Fock Approach" Condensed Matter 8, no. 1: 18. https://doi.org/10.3390/condmat8010018