

Transcriptional Profiling of Populations in the Clam Ruditapes decussatus Suggests Genetically Determined Differentiation in Gene Expression along Parallel Temperature Gradients and between Races of the Atlantic Ocean and West Mediterranean Sea
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
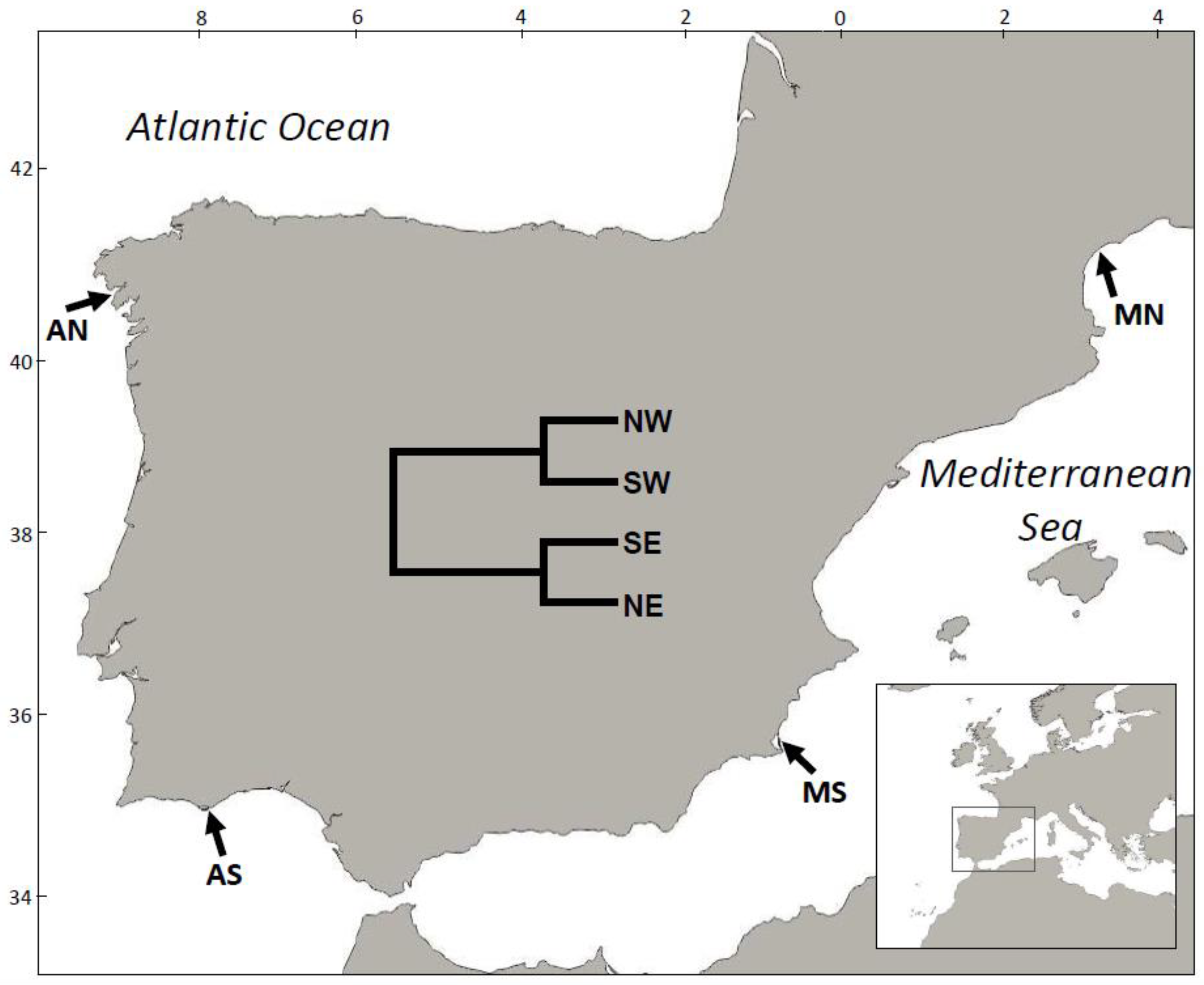
2.1. Clam Sampling and Experimental Setting
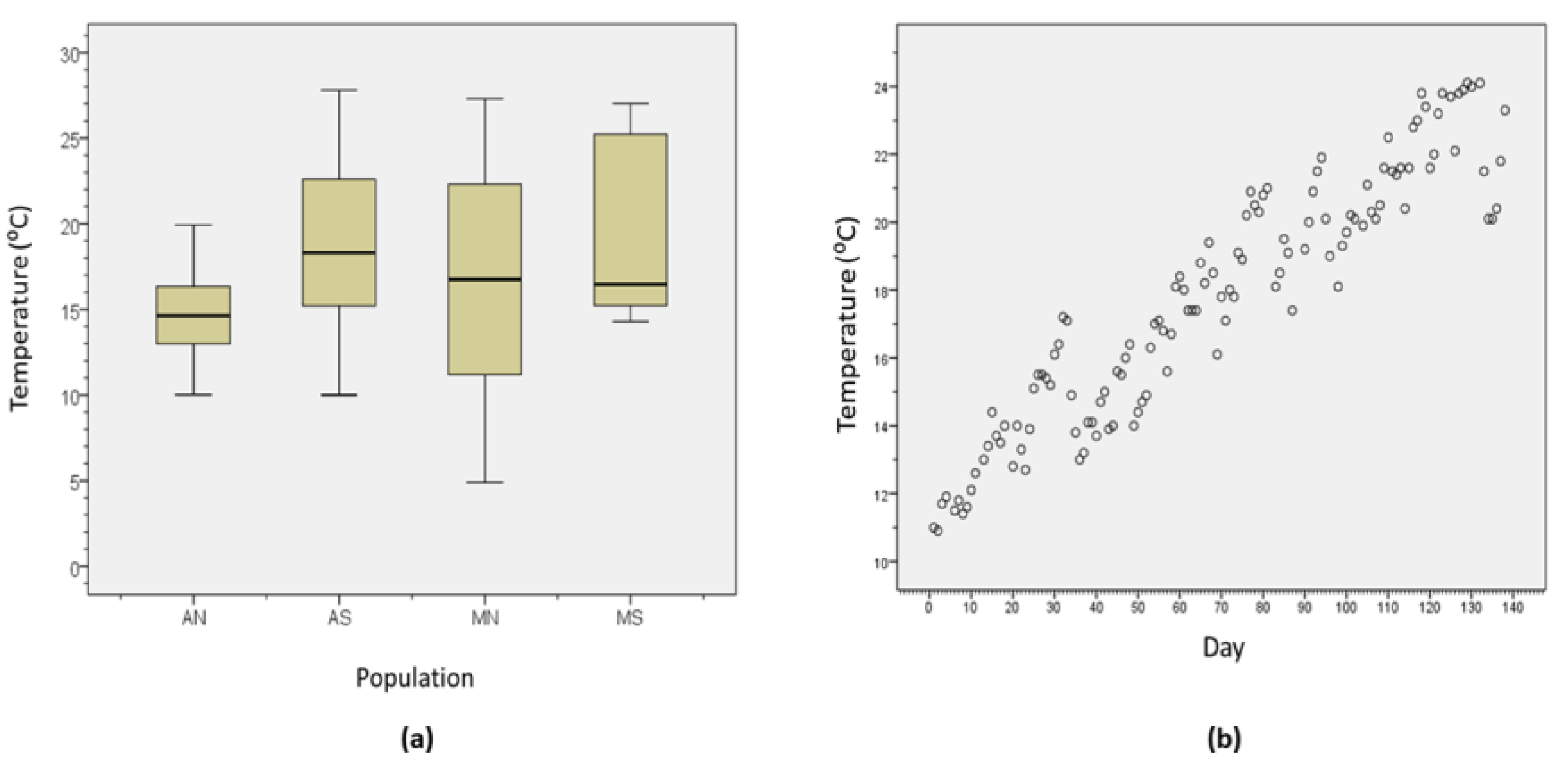
2.2. Environmental Variables
2.3. DNA and RNA Extraction
2.4. Intron Genotyping and Population Genetics
2.5. Microarray Hybridization
2.6. Microarray Data Treatment and Analysis
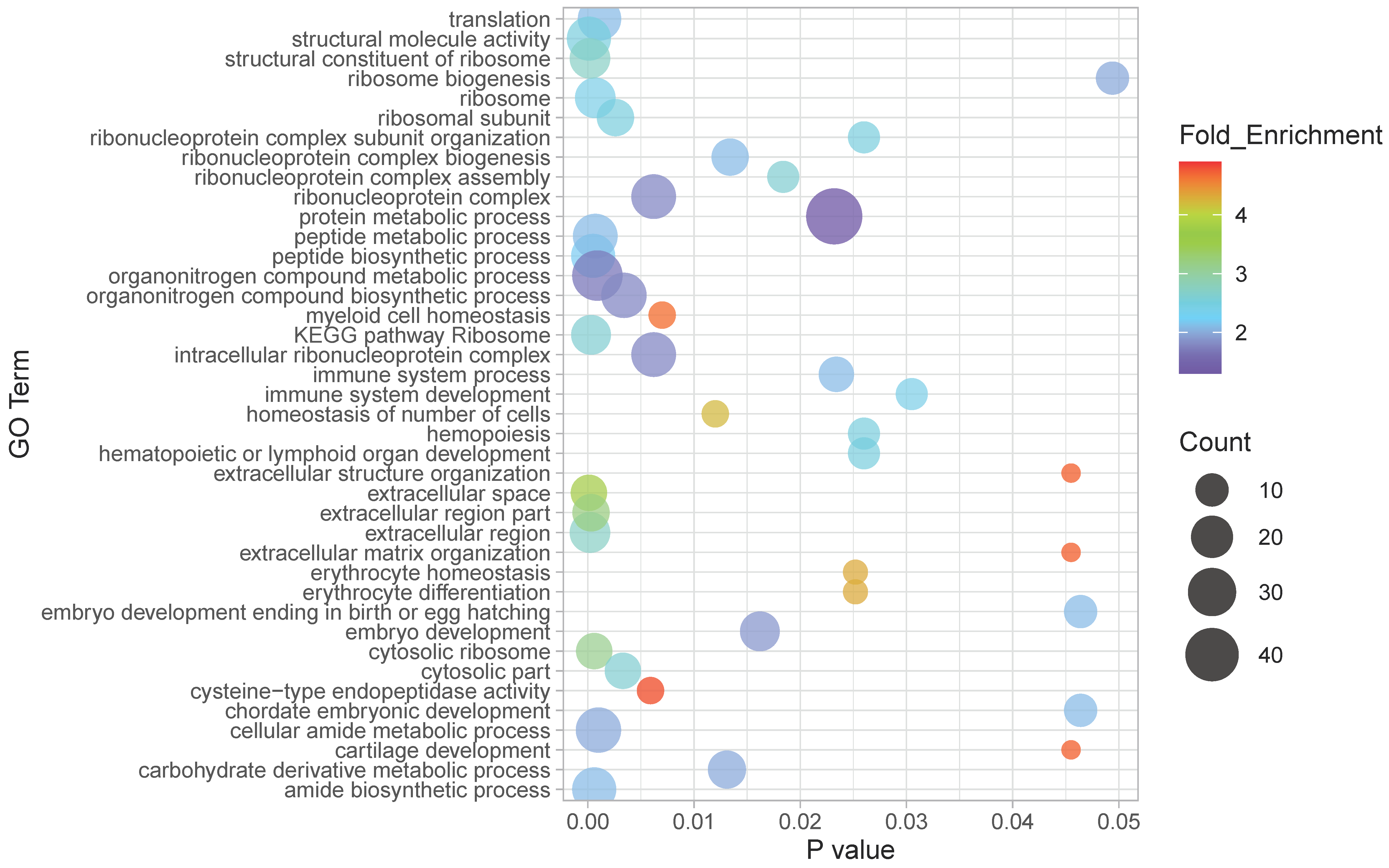
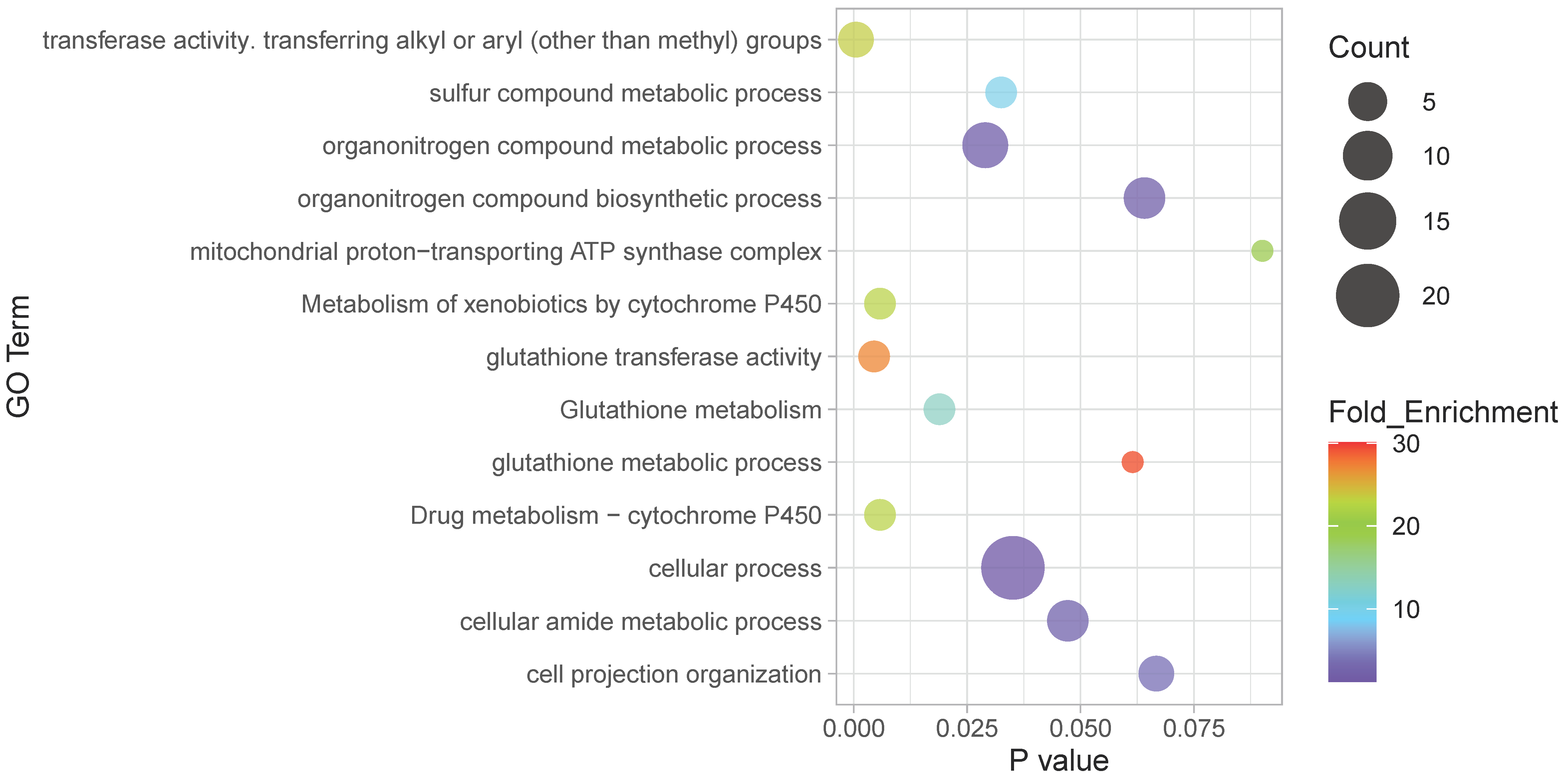
2.7. Functional Analysis
2.8. Neutrality Tests
3. Results
3.1. Environmental Variables
3.2. Population Genetics
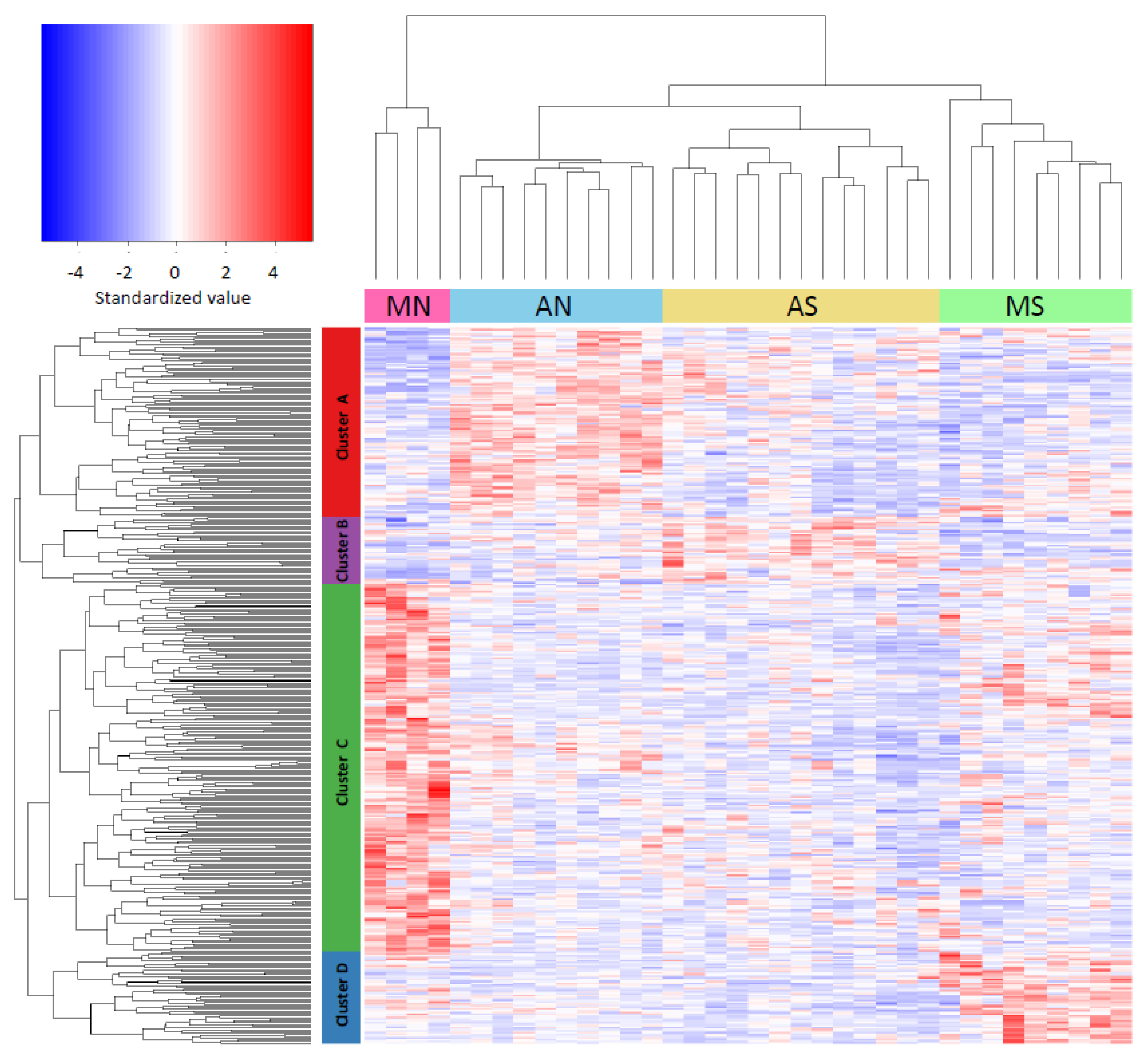
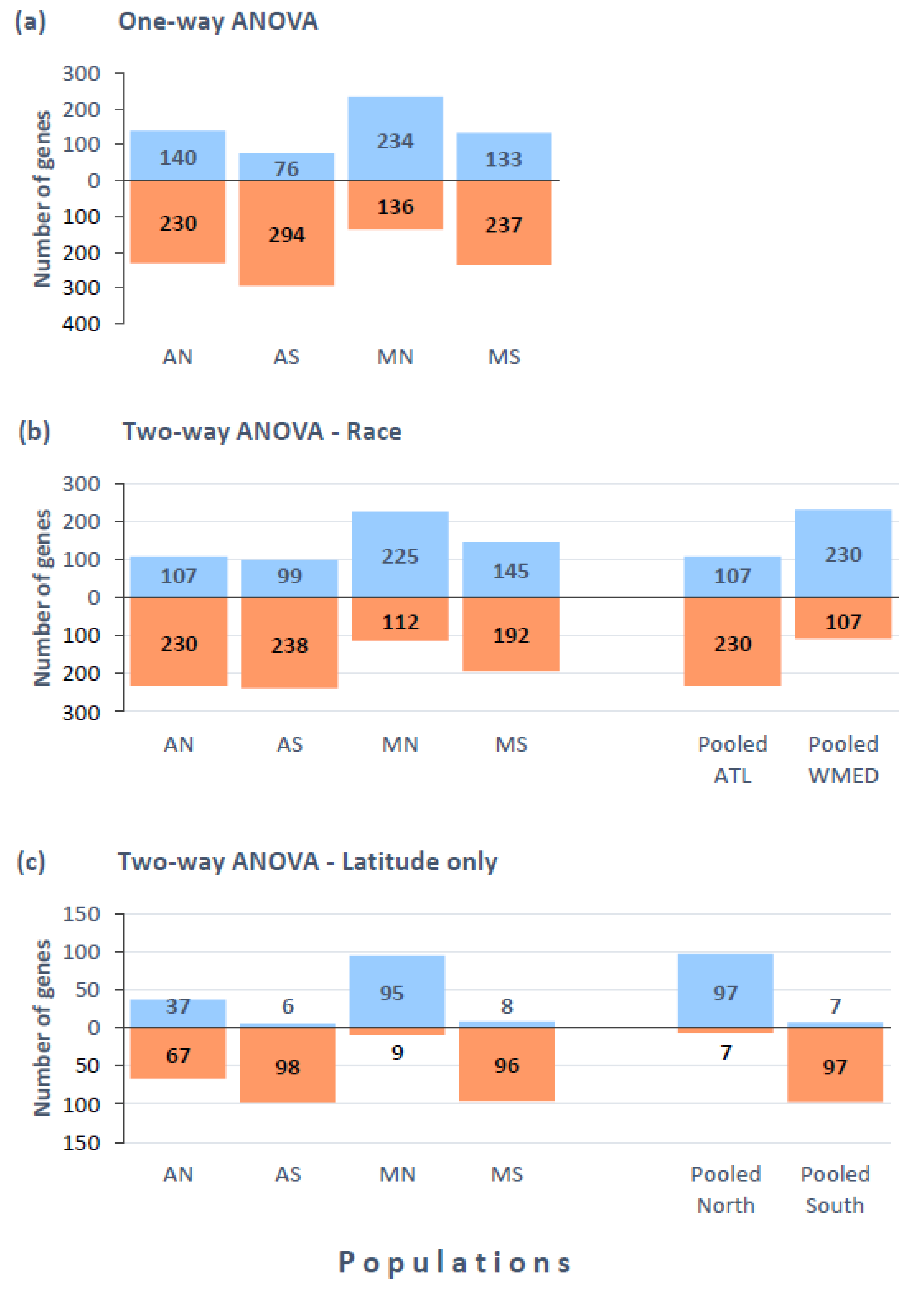
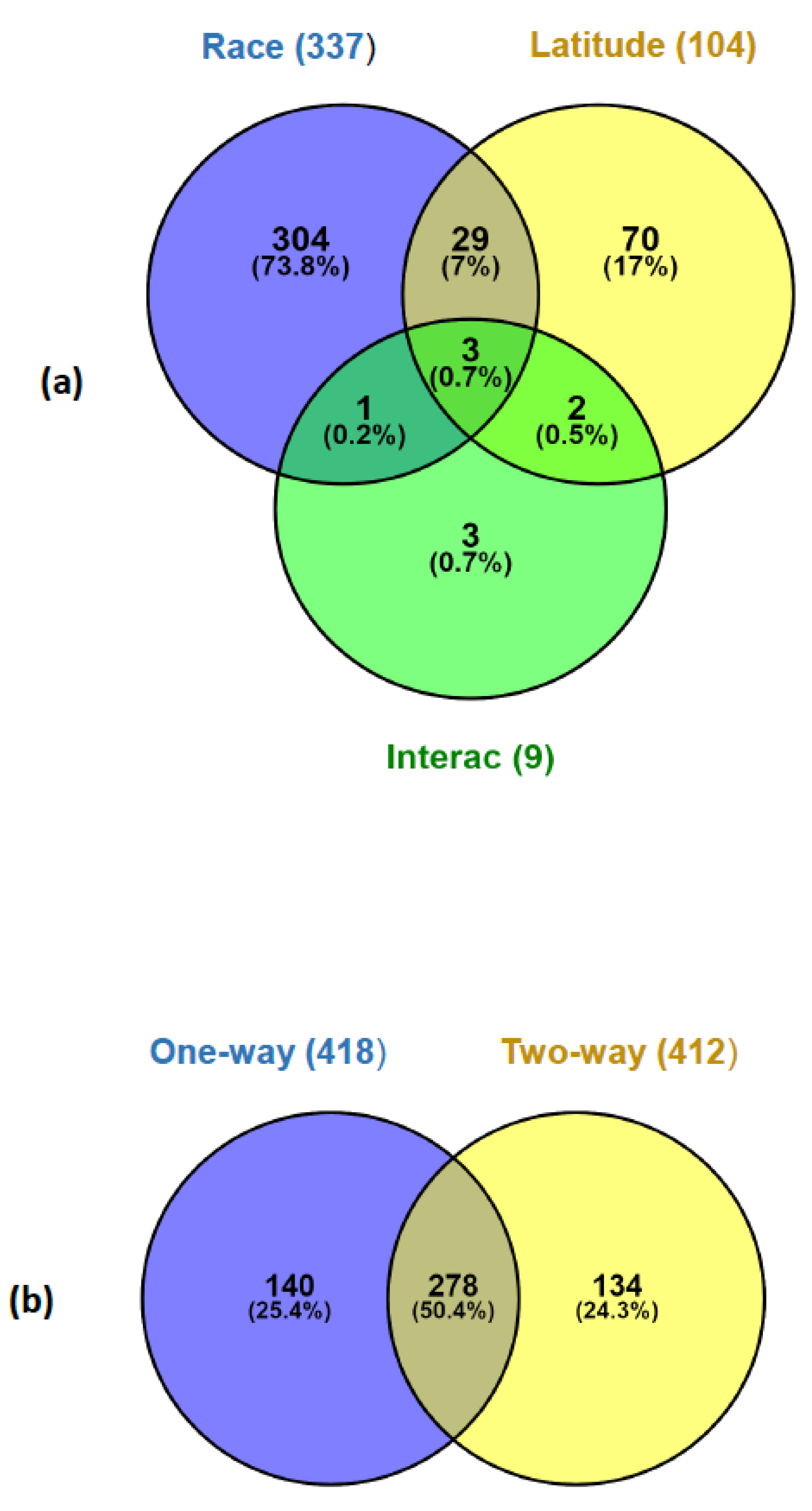
3.3. Gene Expression Variation across Populations (One-Way ANOVA)
3.4. Gene Expression Differences Associated to Race and Latitude (Two-Way ANOVA)
3.5. Test of Selection on Gene Expression Variation
4. Discussion
4.1. Transcriptional Differences between Races
4.2. Transcriptional Differences Associated to Latitude
4.3. Potential Adaptive Role of the Observed Differential Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franks, S.J.; Hoffmann, A.A. Genetics of Climate Change Adaptation. Annu. Rev. Genet. 2012, 46, 185–208. [Google Scholar] [CrossRef] [PubMed]
- Froehlich, H.E.; Gentry, R.R.; Halpern, B.S. Global Change in Marine Aquaculture Production Potential under Climate Change. Nat. Ecol. Evol. 2018, 2, 1745–1750. [Google Scholar] [CrossRef]
- Frölicher, T.L.; Fischer, E.M.; Gruber, N. Marine Heatwaves under Global Warming. Nature 2018, 560, 360–364. [Google Scholar] [CrossRef]
- Somero, G.N. The Physiology of Global Change: Linking Patterns to Mechanisms. Annu. Rev. Mar. Sci. 2012, 4, 39–61. [Google Scholar] [CrossRef]
- Deliberto, A.N.; Drown, M.K.; Ehrlich, M.A.; Oleksiak, M.F.; Crawford, D.L. Feeling the Heat: Variation in Thermal Sensitivity within and among Populations. J. Exp. Biol. 2022, 225, jeb.244831. [Google Scholar] [CrossRef] [PubMed]
- Batta-Lona, P.G.; Maas, A.E.; O’Neill, R.J.; Wiebe, P.H.; Bucklin, A. Transcriptomic Profiles of Spring and Summer Populations of the Southern Ocean Salp, Salpa thompsoni, in the Western Antarctic Peninsula Region. Polar Biol. 2017, 40, 1261–1276. [Google Scholar] [CrossRef]
- Whitehead, A.; Crawford, D.L. Neutral and Adaptive Variation in Gene Expression. Proc. Natl. Acad. Sci. USA 2007, 103, 5425–5430. [Google Scholar] [CrossRef] [PubMed]
- Coscia, I.; Vogiatzi, E.; Kotoulas, G.; Tsigenopoulos, C.; Mariani, S. Exploring Neutral and Adaptive Processes in Expanding Populations of Gilthead Sea Bream, Sparus aurata L., in the North-East Atlantic. Heredity 2011, 108, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.M. Expression of Interest: Transcriptomics and the Designation of Conservation Units. Mol. Ecol. 2010, 19, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.F.; Nielsen, E.E.; Williams, T.D.; Hemmer-Hansen, J.; Chipman, J.K.; Kruhøffer, M.; Grønkjær, P.; George, S.G.; Dyrskjøt, L.; Loeschcke, V. Adaptive Differences in Gene Expression in European Flounder (Platichthys flesus). Mol. Ecol. 2007, 16, 4674–4683. [Google Scholar] [CrossRef]
- Whitehead, A.; Crawford, D.L. Variation within and among Species in Gene Expression: Raw Material for Evolution. Mol. Ecol. 2006, 15, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Passow, C.N.; Henpita, C.; Shaw, J.H.; Quackenbush, C.R.; Warren, W.C.; Schartl, M.; Arias-Rodriguez, L.; Kelley, J.L.; Tobler, M. The Roles of Plasticity and Evolutionary Change in Shaping Gene Expression Variation in Natural Populations of Extremophile Fish. Mol. Ecol. 2017, 26, 6384–6399. [Google Scholar] [CrossRef]
- Etges, W.J.; de Oliveira, C.C.; Rajpurohit, S.; Gibbs, A.G. Effects of Temperature on Transcriptome and Cuticular Hydrocarbon Expression in Ecologically Differentiated Populations of Desert Drosophila. Ecol. Evol. 2017, 7, 619–637. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wilson, C.C.; Wellband, K.W.; Houde, A.L.S.; Neff, B.D.; Heath, D.D. Transcriptional Profiling of Two Atlantic Salmon Strains: Implications for Reintroduction into Lake Ontario. Conserv. Genet. 2015, 16, 277–287. [Google Scholar] [CrossRef]
- Tymchuk, W.; O’Reilly, P.; Bittman, J.; MacDonald, D.; Schulte, P. Conservation Genomics of Atlantic Salmon: Variation in Gene Expression between and within Regions of the Bay of Fundy. Mol. Ecol. 2010, 19, 1842–1859. [Google Scholar] [CrossRef] [PubMed]
- Quinn, N.L.; Mcgowan, C.R.; Cooper, G.A.; Koop, B.F.; Davidson, W.S. Identification of Genes Associated with Heat Tolerance in Arctic Charr Exposed to Acute Thermal Stress. Physiol. Genom. 2011, 43, 685–696. [Google Scholar] [CrossRef]
- Allen, S.L.; Bonduriansky, R.; Sgro, C.M.; Chenoweth, S.F. Sex-Biased Transcriptome Divergence along a Latitudinal Gradient. Mol. Ecol. 2017, 26, 1256–1272. [Google Scholar] [CrossRef]
- De Frenne, P.; Graae, B.J.; Rodríguez-Sánchez, F.; Kolb, A.; Chabrerie, O.; Decocq, G.; De Kort, H.; De Schrijver, A.; Diekmann, M.; Eriksson, O.; et al. Latitudinal Gradients as Natural Laboratories to Infer Species’ Responses to Temperature. J. Ecol. 2013, 101, 784–795. [Google Scholar] [CrossRef]
- Juneja, P.; Quinn, A.; Jiggins, F.M. Latitudinal Clines in Gene Expression and Cis-Regulatory Element Variation in Drosophila melanogaster. BMC Genom. 2016, 17, 981. [Google Scholar] [CrossRef]
- Pörtner, H.O.; Bock, C.; Knust, R.; Lannig, G.; Lucassen, M.; Mark, F.C.; Sartoris, F.J. Cod and Climate in a Latitudinal Cline: Physiological Analyses of Climate Effects in Marine Fishes. Clim. Res. 2008, 37, 253–270. [Google Scholar] [CrossRef]
- Schmidt, P.S.; Serrão, E.A.; Pearson, G.A.; Riginos, C.; Rawson, P.D.; Hilbish, T.J.; Brawley, S.H.; Trussell, G.C.; Carrington, E.; Wethey, D.S.; et al. Ecological Genetics in the North Atlantic: Environmental Gradients and Adaptation at Specific Loci. Ecology 2008, 89, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: Retrospect and Prospect. J. Biogeogr. 2009, 36, 3–15. [Google Scholar] [CrossRef]
- Moritz, C.; Langham, G.; Kearney, M.; Krockenberger, A.; vanDerWal, J.; Williams, S. Integrating Phylogeography and Physiology Reveals Divergence of Thermal Traits between Central and Peripheral Lineages of Tropical Rainforest Lizards. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1680–1687. [Google Scholar] [CrossRef]
- Jueterbock, A.; Franssen, S.U.; Bergmann, N.; Gu, J.; Coyer, J.A.; Reusch, T.B.H.; Bornberg-Bauer, E.; Olsen, J.L. Phylogeographic Differentiation versus Transcriptomic Adaptation to Warm Temperatures in Zostera marina, a Globally Important Seagrass. Mol. Ecol. 2016, 25, 5396–5411. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, F.; Cerviño-Otero, A.; Iglesias, Ó.; Cruz, A.; Guévélou, E. Hatchery Culture of European Clam Species (Family Veneridae). Aquac. Int. 2020, 28, 1675–1708. [Google Scholar] [CrossRef]
- Borsa, P.; Millet, B. Recruitment of the Clam Ruditapes decussatus in the Lagoon of Thau, Mediterranean. Estuar. Coast. Shelf Sci. 1992, 35, 289–300. [Google Scholar] [CrossRef]
- Gharbi, A.; Chatti, N.; Said, K.; Wormhoudt, A. Genetic Variation and Population Structure of the Carpet Shell Clam Ruditapes decussatus along the Tunisian Coast Inferred from MtDNA and ITS1 Sequence Analysis. Biologia 2010, 65, 688–696. [Google Scholar] [CrossRef]
- Habtemariam, B.T.; Arias, A.; García-Vázquez, E.; Borrell, Y.J. Impacts of Supplementation Aquaculture on the Genetic Diversity of Wild Ruditapes decussatus from Northern Spain. Aquac. Environ. Interact. 2015, 6, 241–254. [Google Scholar] [CrossRef]
- Leite, R.B.; Afonso, R.; Cancela, M.L. Perkinsus Sp. Infestation in Carpet-Shell Clams, Ruditapes decussatus (L), along the Portuguese Coast. Results from a 2-Year Survey. Aquaculture 2004, 240, 39–53. [Google Scholar] [CrossRef]
- Cordero, D.; Peña, J.B.; Saavedra, C. Phylogeographic Analysis of Introns and Mitochondrial DNA in the Clam Ruditapes decussatus Uncovers the Effects of Pleistocene Glaciations and Endogenous Barriers to Gene Flow. Mol. Phylogenet. Evol. 2014, 71, 274–287. [Google Scholar] [CrossRef]
- Arias-Pérez, A.; Cordero, D.; Borrell, Y.; Sánchez, J.A.; Blanco, G.; Freire, R.; Insua, A.; Saavedra, C. Assessing the Geographic Scale of Genetic Population Management with Microsatellites and Introns in the Clam Ruditapes decussatus. Ecol. Evol. 2016, 6, 3380–3404. [Google Scholar] [CrossRef] [PubMed]
- Markaide, P.; Gairín, I.; Cordero, D.; Ibarrola, I.; Saavedra, C. No Hybridization and Marked Interspecific Differences in Individual Growth Rate in Mixed Cultures of Manila Clam (Ruditapes philippinarum) and Grooved Carpet-Shell Clam (R. decussatus). Aquaculture 2021, 541, 736824. [Google Scholar] [CrossRef]
- Pascual, M.; Rives, B.; Schunter, C.; MaCpherson, E. Impact of Life History Traits on Gene Flow: A Multispecies Systematic Review across Oceanographic Barriers in the Mediterranean Sea. PLoS ONE 2017, 12, e0176419. [Google Scholar] [CrossRef]
- Patarnello, T.; Volckaert, F.A.M.J.; Castilho, R. Pillars of Hercules: Is the Atlantic-Mediterranean Transition a Phylogeographical Break? Mol. Ecol. 2007, 16, 4426–4444. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A Complete Re-Implementation of the GENEPOP Software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The Neighbor-Joining Method: A New Method for Reconstructing Phylogenetic Trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Leite, R.B.; Milan, M.; Coppe, A.; Bortoluzzi, S.; dos Anjos, A.; Reinhardt, R.; Saavedra, C.; Patarnello, T.; Cancela, M.L.; Bargelloni, L. MRNA-Seq and Microarray Development for the Grooved Carpet Shell Clam, Ruditapes decussatus: A Functional Approach to Unravel Host -Parasite Interaction. BMC Genom. 2013, 14, 741. [Google Scholar] [CrossRef]
- Milan, M.; Coppe, A.; Reinhardt, R.; Cancela, L.M.; Leite, R.B.; Saavedra, C.; Ciofi, C.; Chelazzi, G.; Patarnello, T.; Bortoluzzi, S.; et al. Transcriptome Sequencing and Microarray Development for the Manila Clam, Ruditapes philippinarum: Genomic Tools for Environmental Monitoring. BMC Genom. 2011, 12, 234. [Google Scholar] [CrossRef]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A Free, Open-Source System for Microarray Data Management and Analysis. Biotechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster Analysis and Display of Genome-Wide Expression Patterns. Genetics 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Lempicki, R.A.; Sherman, B.T. Systematic and Integrative Analysis of Large Gene Lists Using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Rohlfs, R.V.; Nielsen, R. Phylogenetic ANOVA: The Expression Variance and Evolution Model for Quantitative Trait Evolution. Syst. Biol. 2015, 64, 695–708. [Google Scholar] [CrossRef]
- Gleason, L.U.; Burton, R.S. RNA-Seq Reveals Regional Differences in Transcriptome Response to Heat Stress in the Marine Snail Chlorostoma funebralis. Mol. Ecol. 2015, 24, 610–627. [Google Scholar] [CrossRef]
- Hughes, D.A.; Kircher, M.; He, Z.; Guo, S.; Fairbrother, G.L.; Moreno, C.S.; Khaitovich, P.; Stoneking, M. Evaluating Intra- and Inter-Individual Variation in the Human Placental Transcriptome. Genome Biol. 2015, 16, 54. [Google Scholar] [CrossRef]
- Thiagalingam, S. Epigenetic Memory in Development and Disease: Unraveling the Mechanism. Biochim. Biophys. Acta-Rev. Cancer 2020, 1873, 188349. [Google Scholar] [CrossRef]
- Wang, X.; Li, A.; Wang, W.; Que, H.; Zhang, G.; Li, L. DNA Methylation Mediates Differentiation in Thermal Responses of Pacific Oyster (Crassostrea gigas) Derived from Different Tidal Levels. Heredity 2020, 126, 10–22. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Q.; Kong, L.; Yu, H. Epigenetic Variation of Wild Populations of the Pacific Oyster Crassostrea gigas Determined by Methylation-Sensitive Amplified Polymorphism Analysis. Fish. Sci. 2018, 84, 61–70. [Google Scholar] [CrossRef]
- Schunter, C.; Carreras-Carbonell, J.; MacPherson, E.; TintorÉ, J.; Vidal-Vijande, E.; Pascual, A.; Guidetti, P.; Pascual, M. Matching Genetics with Oceanography: Directional Gene Flow in a Mediterranean Fish Species. Mol. Ecol. 2011, 20, 5167–5181. [Google Scholar] [CrossRef] [PubMed]
- Lahtvee, P.-J.; Sá Nchez, B.J.; Smialowska, A.; Kasvandik, S.; Elsemman, I.E.; Gatto, F.; Nielsen, J. Absolute Quantification of Protein and MRNA Abundances Demonstrate Variability in Gene-Specific Translation Efficiency in Yeast. Cell Syst. 2017, 4, 495–504.e5. [Google Scholar] [CrossRef] [PubMed]
- Yon Rhee, S.; Wood, V.; Dolinski, K.; Draghici, S. Use and Misuse of the Gene Ontology Annotations. Nat. Rev. Genet. 2008, 9, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Petibon, C.; Malik Ghulam, M.; Catala, M.; Abou Elela, S. Regulation of Ribosomal Protein Genes: An Ordered Anarchy. Wiley Interdiscip. Rev. RNA 2021, 12, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, B.; Feng, X.; Singh, B.R. Marine Glutathione S-Transferases. Mar. Biotechnol. 2007, 9, 513–542. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Hellou, J.; Ross, N.W.; Moon, T.W. Glutathione, Glutathione S-Transferase, and Glutathione Conjugates, Complementary Markers of Oxidative Stress in Aquatic Biota. Environ. Sci. Pollut. Res. 2012, 19, 2007–2023. [Google Scholar] [CrossRef]
- Verlecar, X.N.; Jena, K.B.; Chainy, G.B.N. Seasonal Variation of Oxidative Biomarkers in Gills and Digestive Gland of Green-Lipped Mussel Perna viridis from Arabian Sea. Estuar. Coast. Shelf Sci. 2008, 76, 745–752. [Google Scholar] [CrossRef]
- Kim, M.; Ahn, I.Y.; Cheon, J.; Park, H. Molecular Cloning and Thermal Stress-Induced Expression of a Pi-Class Glutathione S-Transferase (GST) in the Antarctic Bivalve Laternula elliptica. Comp. Biochem. Physiol.-A Mol. Integr. Physiol. 2009, 152, 207–213. [Google Scholar] [CrossRef]
- Anacleto, P.; Maulvault, A.L.; Lopes, V.M.; Repolho, T.; Diniz, M.; Nunes, M.L.; Marques, A.; Rosa, R. Ecophysiology of Native and Alien-Invasive Clams in an Ocean Warming Context. Comp. Biochem. Physiol.-Part A Mol. Integr. Physiol. 2014, 175, 28–37. [Google Scholar] [CrossRef]
- Junge, W.; Nelson, N. ATP Synthase. Annu. Rev. Biochem. 2015, 84, 631–657. [Google Scholar] [CrossRef] [PubMed]
- Itoi, S.; Kinoshita, S.; Kikuchi, K.; Watabe, S. Changes of Carp FoF1-ATPase in Association with Temperature Acclimation. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2003, 284, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, K.; Itoi, S.; Watabe, S. Increased Levels of Mitochondrial ATP Synthase β-Subunit in Fast Skeletal Muscle of Carp Acclimated to Cold Temperature. Fish. Sci. 1999, 65, 629–636. [Google Scholar] [CrossRef]
- Chenghua Li Isolation of an ATP Synthase CDNA from Sinonovacula constricta and Its MRNA Expression by Thermal Stress. Afr. J. Biotechnol. 2012, 11, 1719–1723. [CrossRef]
- Fields, P.A.; Eraso, A. A Year in the Salt Marsh: Seasonal Changes in Gill Protein Expression in the Temperate Intertidal Mussel Geukensia demissa. Mar. Environ. Res. 2020, 161, 105088. [Google Scholar] [CrossRef]
- Wijers, S.L.J.; Smit, E.; Saris, W.H.M.; Mariman, E.C.M.; Van Marken Lichtenbelt, W.D. Cold- and Overfeeding-Induced Changes in the Human Skeletal Muscle Proteome. J. Proteome Res. 2010, 9, 2226–2235. [Google Scholar] [CrossRef]
- Coppe, A.; Agostini, C.; Marino, I.A.M.; Zane, L.; Bargelloni, L.; Bortoluzzi, S.; Patarnello, T. Genome Evolution in the Cold: Antarctic Icefish Muscle Transcriptome Reveals Selective Duplications Increasing Mitochondrial Function. Genome Biol. Evol. 2013, 5, 45–60. [Google Scholar] [CrossRef]
- Price, P.D.; Palmer Droguett, D.H.; Taylor, J.A.; Kim, D.W.; Place, E.S.; Rogers, T.F.; Mank, J.E.; Cooney, C.R.; Wright, A.E. Detecting Signatures of Selection on Gene Expression. Nat. Ecol. Evol. 2022, 6, 1035–1045. [Google Scholar] [CrossRef]
- Sussarellu, R.; Huvet, A.; Lapègue, S.; Quillen, V.; Lelong, C.; Cornette, F.; Jensen, L.F.; Bierne, N.; Boudry, P. Additive Transcriptomic Variation Associated with Reproductive Traits Suggest Local Adaptation in a Recently Settled Population of the Pacific Oyster, Crassostrea gigas. BMC Genom. 2015, 16, 808. [Google Scholar] [CrossRef]
- Brauer, C.J.; Unmack, P.J.; Beheregaray, L.B. Comparative Ecological Transcriptomics and the Contribution of Gene Expression to the Evolutionary Potential of a Threatened Fish. Mol. Ecol. 2017, 26, 6841–6856. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ANOVA Type | Factor (Levels) | No. of Significant Genes at p < 0.05 (FDR) | No. of Significant Genes at FDR = 0.05 |
|---|---|---|---|
| One-way | Population (MN, MS, AN, and AS) | 2287 (0.24) | 418 |
| Two-way (Race × Latitude) | 412 | ||
| Race (ATL; WMED) | 1897 (0.29) | 337 | |
| Latitude (North; South) | 1553 (0.36) | 104 | |
| Interaction | 1406 (0.39) | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saavedra, C.; Milan, M.; Leite, R.B.; Cordero, D.; Patarnello, T.; Cancela, M.L.; Bargelloni, L. Transcriptional Profiling of Populations in the Clam Ruditapes decussatus Suggests Genetically Determined Differentiation in Gene Expression along Parallel Temperature Gradients and between Races of the Atlantic Ocean and West Mediterranean Sea. Fishes 2023, 8, 203. https://doi.org/10.3390/fishes8040203
Saavedra C, Milan M, Leite RB, Cordero D, Patarnello T, Cancela ML, Bargelloni L. Transcriptional Profiling of Populations in the Clam Ruditapes decussatus Suggests Genetically Determined Differentiation in Gene Expression along Parallel Temperature Gradients and between Races of the Atlantic Ocean and West Mediterranean Sea. Fishes. 2023; 8(4):203. https://doi.org/10.3390/fishes8040203
Chicago/Turabian StyleSaavedra, Carlos, Massimo Milan, Ricardo B. Leite, David Cordero, Tomaso Patarnello, M. Leonor Cancela, and Luca Bargelloni. 2023. "Transcriptional Profiling of Populations in the Clam Ruditapes decussatus Suggests Genetically Determined Differentiation in Gene Expression along Parallel Temperature Gradients and between Races of the Atlantic Ocean and West Mediterranean Sea" Fishes 8, no. 4: 203. https://doi.org/10.3390/fishes8040203