
Labeling T Cells to Track Immune Response to Immunotherapy in Glioblastoma


Abstract
:1. Introduction
2. Immunotherapy for Gliomas
3. Importance of T Cells
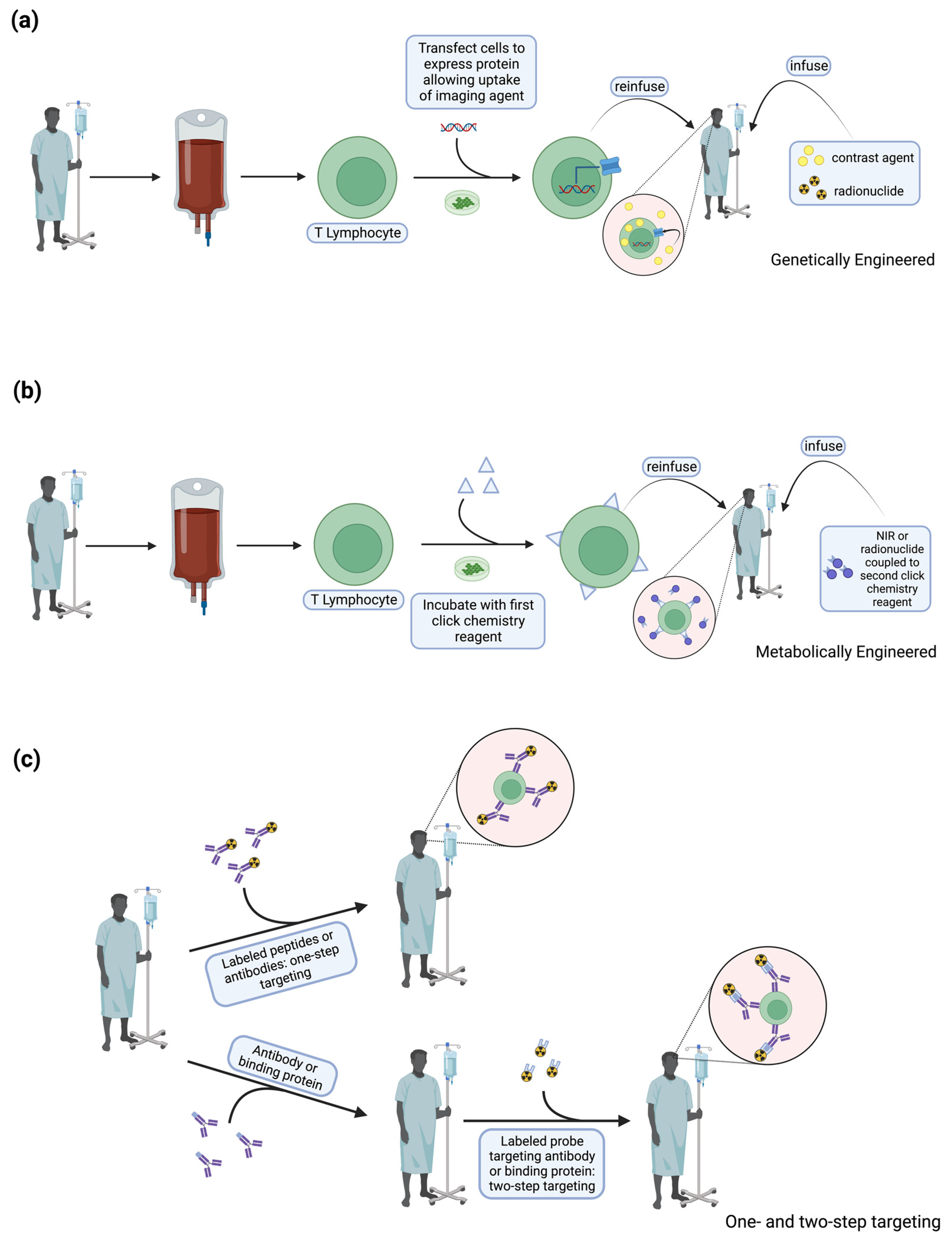
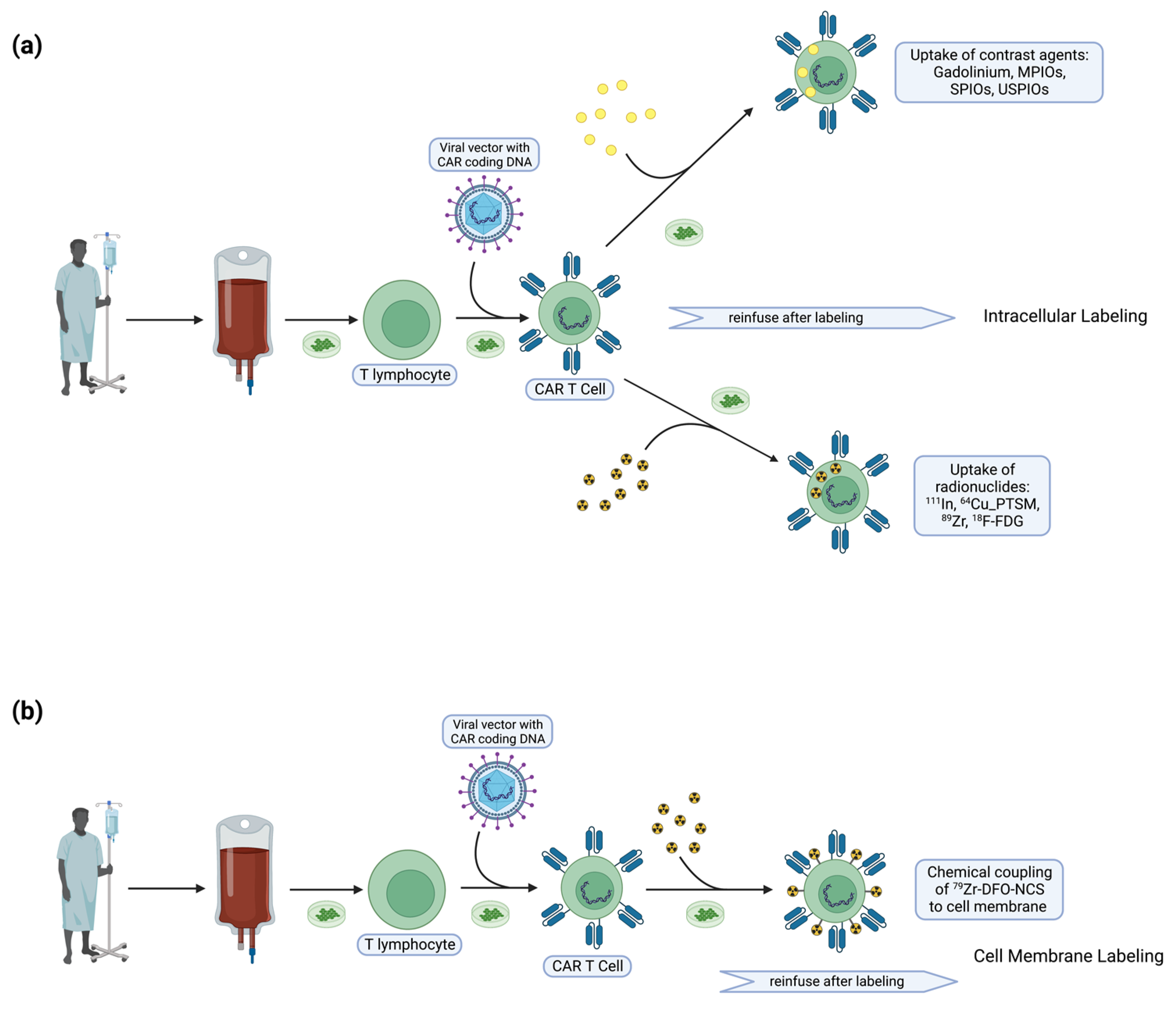
4. T Cell Labeling
5. Animal Studies
6. Human Studies
7. T Cell Tracking in Gliomas
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [PubMed] [Green Version]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar]
- Park, J.; Kim, C.G.; Shim, J.K.; Kim, J.H.; Lee, H.; Lee, J.E.; Kim, M.H.; Haam, K.; Jung, I.; Park, S.H.; et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. Oncoimmunology 2019, 8, e1525243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; Bent, M.V.D.; Bähr, O.; et al. Radiotherapy Combined with Nivolumab or Temozolomide for Newly Diagnosed Glioblastoma with Unmethylated MGMT Promoter: An International Randomized Phase 3 Trial. Neuro-Oncology 2022, 25, noac099. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Mashouf, L.A.; Lim, M. CAR T Cell Therapy in Primary Brain Tumors: Current Investigations and the Future. Front. Immunol. 2022, 13, 817296. [Google Scholar] [CrossRef]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T cell Strategies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2042–2048. [Google Scholar]
- Choi, B.D.; Curry, W.T.; Carter, B.S.; Maus, M.V. Chimeric antigen receptor T cell immunotherapy for glioblastoma: Practical insights for neurosurgeons. Neurosurg. Focus 2018, 44, E13. [Google Scholar]
- Choi, B.D.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; McLendon, R.E.; Bigner, D.D.; Sampson, J.H. EGFRvIII-Targeted Vaccination Therapy of Malignant Glioma. Brain Pathol. 2009, 19, 713–723. [Google Scholar] [PubMed] [Green Version]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar]
- Datsi, A.; Sorg, R.V. Dendritic Cell Vaccination of Glioblastoma: Road to Success or Dead End. Front. Immunol. 2021, 12, 770390. [Google Scholar] [PubMed]
- Wen, P.Y.; Reardon, D.A.; Armstrong, T.S.; Phuphanich, S.; Aiken, R.D.; Landolfi, J.C.; Curry, W.T.; Zhu, J.-J.; Glantz, M.; Peereboom, D.M.; et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5799–5807. [Google Scholar]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [PubMed] [Green Version]
- Perrin, J.; Capitao, M.; Mougin-Degraef, M.; Guérard, F.; Faivre-Chauvet, A.; Rbah-Vidal, L.; Gaschet, J.; Guilloux, Y.; Kraeber-Bodéré, F.; Chérel, M.; et al. Cell Tracking in Cancer Immunotherapy. Front. Med. 2020, 7, 34. [Google Scholar]
- Kircher, M.F.; Gambhir, S.S.; Grimm, J. Noninvasive cell-tracking methods. Nat. Rev. Clin. Oncol. 2011, 8, 677–688. [Google Scholar] [PubMed]
- Kang, S.-W.; Lee, S.; Na, J.H.; Yoon, H.I.; Lee, D.-E.; Koo, H.; Cho, Y.W.; Kim, S.H.; Jeong, S.Y.; Kwon, I.C.; et al. Cell labeling and tracking method without distorted signals by phagocytosis of macrophages. Theranostics 2014, 4, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, C.; Gouard, S.; Lacombe, M.; Saëc, P.R.-L.; Chalopin, B.; Bourgeois, M.; Chouin, N.; Tripier, R.; Halime, Z.; Haddad, F.; et al. Comparison of Immuno-PET of CD138 and PET imaging with 64CuCl2 and 18F-FDG in a preclinical syngeneic model of multiple myeloma. Oncotarget 2018, 9, 9061–9072. [Google Scholar]
- Hartimath, S.V.; Draghiciu, O.; Van De Wall, S.; Manuelli, V.; Dierckx, R.A.J.O.; Nijman, H.W.; Daemen, T.; De Vries, E.F.J. Noninvasive monitoring of cancer therapy induced activated T cells using [18F]FB-IL-2 PET imaging. Oncoimmunology 2016, 6, e1248014. [Google Scholar]
- Seo, J.W.; Tavaré, R.; Mahakian, L.M.; Silvestrini, M.T.; Tam, S.; Ingham, E.S.; Salazar, F.B.; Borowsky, A.D.; Wu, A.M.; Ferrara, K.W. CD8+ T cell density imaging with 64Cu-labeled cys-diabody informs immunotherapy protocols. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4976–4987. [Google Scholar]
- Vera, D.B.; Smith, C.C.; Bixby, L.M.; Glatt, D.M.; Dunn, S.S.; Saito, R.; Kim, W.Y.; Serody, J.S.; Vincent, B.G.; Parrott, M.C. Immuno-PET imaging of tumor-infiltrating lymphocytes using zirconium-89 radiolabeled anti-CD3 antibody in immune-competent mice bearing syngeneic tumors. PLoS ONE 2018, 13, e0193832. [Google Scholar]
- Larimer, B.M.; Wehrenberg-Klee, E.; Caraballo, A.; Mahmood, U. Quantitative CD3 PET Imaging Predicts Tumor Growth Response to Anti-CTLA-4 Therapy. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57, 1607–1611. [Google Scholar]
- Charoenphun, P.; Meszaros, L.K.; Chuamsaamarkkee, K.; Sharif-Paghaleh, E.; Ballinger, J.R.; Ferris, T.J.; Went, M.J.; Mullen, G.E.D.; Blower, P.J. [89Zr]oxinate4 for long-term in vivo cell tracking by positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; von See, M.P.; Yu, E.; Gunel, B.; Lu, K.; Vazin, T.; Schaffer, D.V.; Goodwill, P.W.; Conolly, S.M. Quantitative Magnetic Particle Imaging Monitors the Transplantation, Biodistribution, and Clearance of Stem Cells In Vivo. Theranostics 2016, 6, 291–301. [Google Scholar] [PubMed] [Green Version]
- Zheng, B.; Vazin, T.; Goodwill, P.W.; Conway, A.; Verma, A.; Saritas, E.U.; Schaffer, D.; Conolly, S.M. Magnetic Particle Imaging tracks the long-term fate of in vivo neural cell implants with high image contrast. Sci. Rep. 2015, 5, 14055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, I.S.; Mayer, A.T.; Sagiv-Barfi, I.; Wang, K.; Vermesh, O.; Czerwinski, D.K.; Johnson, E.; James, M.L.; Levy, R.; Gambhir, S.S. Imaging activated T cells predicts response to cancer vaccines. J. Clin. Investig. 2018, 128, 2569–2580. [Google Scholar] [CrossRef] [PubMed]
- Griessinger, C.M.; Maurer, A.; Kesenheimer, C.; Kehlbach, R.; Reischl, G.; Ehrlichmann, W.; Bukala, D.; Harant, M.; Cay, F.; Brück, J.; et al. 64Cu antibody-targeting of the T cell receptor and subsequent internalization enables in vivo tracking of lymphocytes by PET. Proc. Natl. Acad. Sci. USA 2015, 112, 1161–1166. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.; Pandey, M.K.; Demirhan, Y.E.; Nesbitt, J.J.; Crespo-Diaz, R.J.; Terzic, A.; Behfar, A.; DeGrado, T.R. Novel 89Zr cell labeling approach for PET-based cell trafficking studies. EJNMMI Res. 2015, 5, 19. [Google Scholar]
- Kiru, L.; Zlitni, A.; Tousley, A.M.; Dalton, G.N.; Wu, W.; Lafortune, F.; Liu, A.; Cunanan, K.M.; Nejadnik, H.; Sulchek, T.; et al. In vivo imaging of nanoparticle-labeled CAR T cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2102363119. [Google Scholar] [CrossRef]
- Srinivas, M.; Heerschap, A.; Ahrens, E.T.; Figdor, C.G.; de Vries, I.J.M. 19F MRI for quantitative in vivo cell tracking. Trends Biotechnol. 2010, 28, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Markovic, S.N.; Galli, F.; Suman, V.J.; Nevala, W.K.; Paulsen, A.M.; Hung, J.C.; Gansen, D.N.; Erickson, L.A.; Marchetti, P.; Wiseman, G.A.; et al. Non-invasive visualization of tumor infiltrating lymphocytes in patients with metastatic melanoma undergoing immune checkpoint inhibitor therapy: A pilot study. Oncotarget 2018, 9, 30268–30278. [Google Scholar] [CrossRef] [Green Version]
- Pharmaceuticals, R. Study of REGN3767 (Anti-LAG-3) with or without REGN2810 (Anti-PD1) in Advanced Cancers. Available online: https://clinicaltrials.gov/ct2/show/NCT03005782 (accessed on 10 January 2023).
- Bhargava, K.K.; Gupta, R.K.; Nichols, K.J.; Palestro, C.J. In vitro human leukocyte labeling with 64Cu: An intraindividual comparison with 111In-oxine and 18F-FDG. Nucl. Med. Biol. 2009, 36, 545–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronald, J.A.; Kim, B.-S.; Gowrishankar, G.; Namavari, M.; Alam, I.S.; D’Souza, A.; Nishikii, H.; Chuang, H.-Y.; Ilovich, O.; Lin, C.-F.; et al. A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant. Cancer Res. 2017, 77, 2893–2902. [Google Scholar] [CrossRef] [Green Version]
- Keu, K.V.; Witney, T.H.; Yaghoubi, S.; Rosenberg, J.; Kurien, A.; Magnusson, R.; Williams, J.; Habte, F.; Wagner, J.R.; Forman, S.; et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci. Transl. Med. 2017, 9, eaag2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, A.; Mayer, A.T.; Xu, L.; Reeves, R.E.; Gano, J.; Gambhir, S.S. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjug. Chem. 2015, 26, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 4664. [Google Scholar] [CrossRef] [PubMed]
- Hm, G.; McKnight, B.N.; Malysa, A.; Dyson, G.; Wiesend, W.N.; McCarthy, C.E.; Reyes, J.; Wei, W.Z.; Viola-Villegas, N.T. IFNγ PET Imaging as a Predictive Tool for Monitoring Response to Tumor Immunotherapy. Cancer Res. 2018, 78, 5706–5717. [Google Scholar]
- Martinez, O.; Sosabowski, J.K.; Maher, J.; Papa, S. New Developments in Imaging Cell-Based Therapy. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2019, 60, 730–735. [Google Scholar] [CrossRef]
- Weist, M.R.; Starr, R.; Aguilar, B.; Chea, J.; Miles, J.K.; Poku, E.; Gerdts, E.; Yang, X.; Priceman, S.J.; Forman, S.J.; et al. PET of Adoptively Transferred Chimeric Antigen Receptor T Cells with 89Zr-Oxine. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2018, 59, 1531–1537. [Google Scholar] [CrossRef] [Green Version]
- Rivera-Rodriguez, A.; Hoang-Minh, L.B.; Chiu-Lam, A.; Sarna, N.; Marrero-Morales, L.; Mitchell, D.A.; Rinaldi-Ramos, C.M. Tracking adoptive T cell immunotherapy using magnetic particle imaging. Nanotheranostics 2021, 5, 431–444. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Label | Target | Imaging Modality | Cancer |
|---|---|---|---|
| 64Copper-labeled diabody [19] | CD8 | PET | Her2 breast cancer |
| 89zirconium-labeled antibody [20,21] | CD3 | PET | Colon cancer, bladder cancer |
| 18fluorobenzoyl-interleukin-2 radiotracer | CD25 | PET | Cervical cancer |
| 64Copper-conjugated murine Ab specific for OX40 receptor [19] | OX40 receptor | PET | lymphoma |
| 89Zr-oxinate [22] | CART | SPECT | Breast cancer, myeloma, glioblastoma |
| Ferucarbotran [23,24] | CART | Magnetic particle imaging | Glioblastoma |
| Granzyme B (GZP—peptide in PET imaging) | Granzyme B | GZP PET signal | Colon carcinoma |
| Label | Target | Imaging Modality | Cancer |
|---|---|---|---|
| 99mTc [30] | IL-2 | SPECT/CT | Metastatic melanoma |
| 89Zr-DFO [31] | Lymphocyte-activating gene 3 | PET | Lymphoma |
| 64Copper-labeled antibody [32] | PD-1 | PET | Non-small-cell lung cancer |
| 89Zirconium [32] | PD-1 IFN-gamma, IL13Rα2-targeted CAR T cells | PET | Non-small-cell lung cancer, breast cancer, glioblastoma |
| 18Fluorobenzoyl-labeled clofarabine [33] | Enzyme in deoxycytidine kinase pathway | PET | Lymphoma |
| 2’-deoxy-2’-fluoro-9-B-arabinofuranosylguanine [33] | Enzyme in deoxyguanosine kinase pathway | PET | Acute graft versus host disease |
| 9-[4-[18F]fluoro-3-(hydroxymethyl)butyl]guanine ([18F]FHBG) [34] | CART engineered to express herpes simplex virus type-1 thymidine kinase (HSV1-TK) and interleukin-13 | PET | Glioma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rhee, J.Y.; Ghannam, J.Y.; Choi, B.D.; Gerstner, E.R. Labeling T Cells to Track Immune Response to Immunotherapy in Glioblastoma. Tomography 2023, 9, 274-284. https://doi.org/10.3390/tomography9010022
Rhee JY, Ghannam JY, Choi BD, Gerstner ER. Labeling T Cells to Track Immune Response to Immunotherapy in Glioblastoma. Tomography. 2023; 9(1):274-284. https://doi.org/10.3390/tomography9010022
Chicago/Turabian StyleRhee, John Y., Jack Y. Ghannam, Bryan D. Choi, and Elizabeth R. Gerstner. 2023. "Labeling T Cells to Track Immune Response to Immunotherapy in Glioblastoma" Tomography 9, no. 1: 274-284. https://doi.org/10.3390/tomography9010022