
Title-Inflammatory Signaling Pathways in Allergic and Infection-Associated Lung Diseases


1
Department of Ophthalmic Research, Cole Eye Institute, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195, USA
2
Department of Medicine, Division of Regenerative Medicine, Loma Linda University, Loma Linda, CA 92354, USA
*
Author to whom correspondence should be addressed.
Allergies 2022, 2(2), 57-74; https://doi.org/10.3390/allergies2020006
Submission received: 22 February 2022
/
Revised: 2 April 2022
/
Accepted: 27 May 2022
/
Published: 1 June 2022
(This article belongs to the Section Asthma/Respiratory)
Abstract
:Lung inflammation can be caused by pathogen infection alone or by allergic disease, leading to pneumonitis. Most of the allergens (antigens) that cause allergic lung diseases, including asthma and hypersensitivity pneumonitis (HP), are derived from microorganisms, such as bacteria, viruses, and fungi, but some inorganic materials, such as mercury, can also cause pneumonitis. Certain allergens, including food and pollen, can also cause acute allergic reactions and lead to lung inflammation in individuals predisposed to such reactions. Pattern recognition-associated and damage-associated signaling by these allergens can be critical in determining the type of hypersensitization and allergic disease, as well as the potential for fibrosis and irreversible lung damage. This review discusses the signs, symptoms, and etiology of allergic asthma, and HP. Furthermore, we review the immune response and signaling pathways involved in pneumonitis due to both microbial infection and allergic processes. We also discuss current and potential therapeutic interventions for infection-associated and allergic lung inflammation.
1. Introduction
Allergic lung disease is a term that includes many different conditions, such as allergic asthma, hypersensitivity pneumonitis (also known as extrinsic allergic alveolitis), granulomatosis with polyangiitis (Wegener granulomatosis), and eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) [1]. The former two conditions are caused by a combination of individual susceptibility and exposure to different allergy-inducing antigens, while the latter two are autoimmune vasculitides with idiopathic or unknown etiologies [1,2,3]. The conditions arise from a large diversity of etiologies and have different mechanisms of pathogenesis.
Of the various asthma phenotypes, allergic asthma is the most prevalent in the general population [4]. Asthma is physiologically characterized by the conducting airways’ tendency to spontaneously contract or close in response to various exogenous stimuli, producing type I hypersensitivity reactions and symptoms such as coughing, wheezing, or dyspnea [5,6]. This tendency to contract is accompanied by a chronic state of inflammation, which begins in the conducting airways but spreads deeper as the disease progresses [5]. In this regard, eosinophilia is almost always a standard feature of the airway inflammation that accompanies asthma and is present in most types of allergic inflammation [5,7,8,9]. Along with eosinophilia, the infiltrate includes other elements of an allergic inflammatory response, such as the excessive presence or infiltration of Th2 CD4+ lymphocytes, neutrophils, basophils, mast cells, and class switching of B cells to IgE [5,8]. Symptoms are thought to be related to mast cell degranulation and the release of biologically active mediators, such as histamine, serotonin, and leukotrienes [5]. This is one of the primary distinguishing factors from hypersensitivity pneumonitis (HP), which exhibits a Th1-mediated inflammatory response with IgG [8].
HP is another type of allergic lung disease characterized by a combined type III and IV hypersensitivity reaction due to repeated inhalation exposure to various antigens in susceptible people [2,10]. HP can be divided into acute and chronic forms depending on the amount and type of exposure to the causative antigen and the progression of the disease [2]. Acute allergic reactions, such as those observed in acute HP, are thought to be due to high levels of antigen exposure, with symptoms occurring within a day following exposure and presenting as a flu-like illness, while chronic HP is most often associated with occupational exposure over a long period of time with an insidious onset of symptoms including cough, dyspnea, and fatigue [2,10]. Occupations relating to agriculture, bird breeding, and certain occupations in industrial settings are at significantly higher risk for HP [5,11,12]. However, not everyone exposed to the same allergens will develop an allergic reaction. Individuals often need to be exposed to large amounts of allergens and have some susceptibility to allergy before a sensitivity reaction can be initiated [2,10]. Even among people who develop a response, few cases progress towards chronic, fibrotic allergic lung inflammation that can cause permanent lung damage [2]. Symptoms can begin even years after exposure to the antigen has ceased, making it hard to distinguish chronic HP from other interstitial lung diseases such as idiopathic pulmonary fibrosis, especially since the chronic form of HP can cause pulmonary fibrosis [2,13].
According to the WHO, about 262 million people worldwide were affected by asthma in 2019, of whom, 461,000 died [14]. The prevalence of asthma and other allergic lung diseases is increasing, especially in western countries [11,14,15]. In fact, asthma is the most common chronic disease affecting children [12,14]. According to the U.S. Department of Health and Human Services, asthma prevalence increased from 7.3% in 2001 to 8.4% in 2010, while a Swedish study found that the prevalence of asthma increased from 8.4% in 1996 to 10.9% in 2016 [6,11]. New allergic asthma diagnoses are highest among children in the 0–9 age group, while new non-allergic asthma diagnoses peak between 50 and 59 years of age [16].
Information on the epidemiology of HP is scant, but one United States-based study suggests a national prevalence between 1.67–2.71 per 100,000 population, with a yearly incidence between 1.28 and 1.94 per 100,000 [17]. Mortality rates are significantly higher among those with fibrotic disease from HP—between 54.7 and 80.4 deaths per 1000 person-years compared to between 38.8 and 51.5 deaths per 1000 person-years for those without fibrosis [17]. Age also plays a significant role, with a much higher mortality rate among 65 and older [17].
The pathogenesis of allergic lung disease can vary depending on which disease process is causing the patient’s illness. Allergic asthma is due to a type I hypersensitivity reaction from a combination of intrinsic susceptibility and exposure to extrinsic allergens [5,8]. As a result, chronic eosinophilia, elevated IgE levels, and Th2-mediated inflammation predominate, while mast cell degranulation and histamine release cause bronchoconstriction acutely and sometimes precipitate an “asthma attack,” warranting immediate treatment with inhaled bronchodilators [7,8,13,18]. In some cases, asthma can be so severe that there is irreversible damage to airway function, secondary to remodeling of tissue that often results from chronic inflammatory processes [19]. Inhaled corticosteroids (ICS) with short-acting β2 agonists as needed have long been the mainstay of asthma management. However, long-acting β2 agonists, muscarinic antagonists, and long-acting leukotriene antagonists are sometimes used in cases that cannot be controlled alone with ICS [18,20,21]. More recently, several biologics are being used in severe cases that do not respond to conventional treatment [22,23]. It has been suggested that adherence to asthma treatment is vital to prevent acute attacks since successful asthma management can prevent airway remodeling and irreversible damage and reduce mortality [24,25].
HP pathogenesis is thought to be due to a combined type III and type IV hypersensitivity reaction from inhalation of various antigens, leading to activation of fibrogenic growth factors in some people, causing fibrotic disease and a poorer prognosis [2,13,17,18,26]. Treatment of HP currently focuses on avoiding the causative antigen and blunting the immune response using corticosteroids [5,27,28]. However, this treatment is most helpful in hastening the resolution of acute allergic reactions and is ineffective in preventing disease progression in the long term [20]. Therefore, in some cases, antifibrotic agents that target fibrogenic pathways, such as pirfenidone and nintedanib, are used to slow the progression of the fibrotic disease [21,29].
2. Etiology of Allergic Lung Disease
Several factors play a role in the development of asthma. It appears that a combination of both intrinsic and environmental factors contributes to the etiology of asthma, and a strong predictor of asthma in children is atopy—a tendency for developing the allergic disease [3]. There are many possible triggers for those with asthma, which can include substances such as pollen, fungal spores, tobacco smoke, perfumes, and many other substances that can cause an allergic response in these individuals [3]. Asthma prevalence is higher among those of low socioeconomic status, which may be caused by a higher-than-usual exposure to the various allergens that can trigger asthma [6]. Like asthma, developing HP requires some amount of genetic or environmental susceptibility and sensitization and subsequent re-exposure to various antigens [5,12,30]. Specific major histocompatibility complex (MHC) II polymorphisms or haplotypes may contribute to a greater risk of disease [26]. The variety of antigens comes from various sources, including bacteria, fungi, proteins from animals and plants, and certain synthetic compounds and metals [26,31]. Some of the most common organisms that can cause HP are thermophilic actinomycetes inhaled from hay and grains stored in high humidity [20,31]. This type of exposure is associated with farm work, and HP resulting from this type of exposure is often termed “farmer’s lung” [31]. Fungi such as Aspergillus species may also be a common cause of farmer’s lung [10]. Another common source of exposure is birds, with a substantial proportion of pigeon breeders (5–20%) showing symptoms [2,32]. Along with farmer’s lung and avian-related HP, improperly cleaned humidifiers can also lead to the development of HP from inhalation of antigens from various fungi [2,27]. Identifying the causative antigen is an essential strategy in managing and treating HP since this can improve symptoms and prevent disease progression. Persistent exposure to antigens is associated with higher mortality [5,12,27,30]. Unfortunately, HP can occasionally be idiopathic, and a causative agent may not be found in some cases [2,27].
3. Signaling Pathways of Infection-Induced Lung Inflammation
Lung inflammation can happen in response to infection, injury, environmental stimuli, or allergic reaction and can be broadly classified into acute and chronic inflammation. The magnitude and delicate balance of molecular events and cellular interplay determine whether the outcome will be tissue homeostasis or disease [28]. Lung inflammation is a vast and complex topic; it is crucial to understand both allergic and non-allergic lung inflammation signaling events. The section below discusses lung inflammation and related signaling pathways in response to common bacterial, viral and fungal infections.
3.1. Lung Inflammation and Receptor Signaling Pathways in Response to Bacterial Infection
The most common disease caused by a bacterial infection in the lungs is pneumonia. It is classified as either community-acquired or hospital-acquired. Pneumonia is caused by typical bacteria (e.g., Streptococcus pneumoniae, Hemophilus influenzae, Klebsiella pneumoniae, or Staphylococcus aureus) or atypical bacteria (e.g., Mycoplasma pneumoniae, Chlamydophila pneumoniae, or Legionella pneumophila). Pathogen-associated pattern molecules (PAMPs), such as endotoxins, flagellin, peptidoglycan, lipopolysaccharide (LPS), and nucleic acids from pathogenic bacteria, are detected by pattern recognition receptors (PRRs) expressed on alveolar and interstitial macrophages and dendritic cells (DCs) of lungs [30,33,34]. These PRRs include Toll-like receptor 4 (TLR4), which recognizes LPS and endotoxins; Toll-like receptor 2 (TLR2), which binds to peptidoglycans and lipoproteins; and Toll-like receptor 9 (TLR9), which is activated by CpG islands on bacterial DNA. Downstream signaling through these receptors activates NFκB, the master regulator of the inflammatory response, leading to the expression or production of chemokines, adhesion molecules, and pro-inflammatory cytokines, such as tumor necrosis factor—α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and interleukin-8 (IL-8), of which IL-8 acts as a chemoattractant by recruiting neutrophils to the site of infection. Meanwhile, dendritic cells phagocytose bacteria and present antigens to T lymphocytes to initiate an adaptive immune response. TLR signaling in airway epithelium stimulates the production of chemokines, including Granulocyte macrophage-colony stimulating factor (GM-CSF), macrophage-inflammatory protein 2 (MIP-2), and keratinocyte chemoattractant (KC), and modulate the inflammatory response. Lung γδ T cells are another set of innate immune cells that secrete interleukin-2 (IL-2), interleukin-12 (IL-12), IFN-γ, and interleukin-17 (IL-17) to further enhance Th1 and Th17 responses. IgA and B-1 B cells have also been found to mount a response against early bacterial infection. Another set of PRRs, NOD-like receptors (NLRs), are cytosolic and bind to a specific component of bacteria leading to inflammasome formation [35,36,37]. TNF-α, IL-1β, IL-6, and other pro-inflammatory cytokines bind to their receptors to further amplify signaling and promote inflammation. Please refer Table 1 for various infectious agents-derived PAMPs and their PRRs.
3.2. Lung Inflammation and Receptor Signaling Pathways in Response to Virus Infection
Viral pneumonia is caused by influenza viruses A and B (Orthomyxoviridae), respiratory syncytial virus (RSV; most common in infants), adenoviruses, and paramyxoviruses. In immunocompromised subjects, cytomegalovirus (CMV), herpes simplex virus (HSV), and varicella-zoster virus (VZV) can also cause pneumonia [38,39,40]. Viral antigens are also recognized by cell surface and cytosolic TLRs and NLRs on innate immune cells. TLR2 recognizes hemagglutinin (HA) on the measles virus, TLR3 recognizes double-stranded RNA of the influenza virus, and TLR7 and 8 recognize single-stranded RNA. TLR9 recognizes the DNA of adenoviruses and other DNA viruses. Retinoic acid-inducible gene 1 (RIG-1), a member of the RIG-like receptor (RLR) family, is essential for sensing ssRNA of paramyxovirus and influenza A virus [41,42]. These activated immune cells produce Type-I interferon (IFNα, β) and pro-inflammatory cytokines IL-1β, IL-6, IL-12, monocyte chemoattractant protein 1 (MCP-1), and other cytokines. Type I interferon through interferon receptors (IFNR) on DCs directly enhance maturation, migration, and antigen presentation to CD4+ T cells and cross-presentation to CD8+ cells. IL-15 secreted by DCs directly supports the survival of memory T cells and natural killer (NK) cells. The relative timing of IFNR and T-cell receptor (TCR) signaling can also directly stimulate proliferation, survival, and differentiation of T cells. It also impacts B cells by modulating TLR7 expression, cell migration, cytokine production, and survival [43,44,45,46,47].
3.3. Lung Inflammation and Receptor Signaling Pathways in Response to Fungal Infection
Fungal infection in the lung mainly occurs in immunocompromised individuals as opportunistic infections. Aspergillus, Cryptosporidium, and Pneumocystis species are major pathogens causing aspergillosis, cryptococcosis, and Pneumocystis pneumonia, respectively [48,49,50]. Alveolar macrophages, neutrophils, and DCs are the first line of defense against these opportunistic pathogens. Fungus-derived PAMPs are complex carbohydrates consisting of mannoproteins, phospholipomannans, beta-glucans, and chitin. C-lectin receptors including Dectin1/2, mannose receptors, Clec4d, Clec4e, TLR2, and TLR4 on innate immune cells initially sense these PAMPs. Certain glycoantigens bind to DC-SIGN on DCs [51,52]. Upon stimulation by fungus-derived antigens, alveolar macrophages, and DCs mount inflammatory responses and secrete TNF-α, IL-1β, IL-6, IL-8, MCP-1, and GM-CSF. Pro-inflammatory cytokines recruit neutrophils at the site of infection, which kill conidia through NADPH oxidase activity and affect DC maturation. NK cells indirectly shape inflammation by secreting IFN-γ. Cytokines and chemokines secreted by innate immune cells further trigger the proliferation of adaptive immune cells. CD4+ T cells (Th1 type) and Th17 responses are associated with the adaptive immune response. It has also been reported that lung epithelial cells mount a protective response against fungal infection via indoleamine 2,3-dioxygenase signaling. Type I interferon signaling protects in some instances such as in C. neoformans infection by increasing Th2 and Th17 responses [50,53,54,55].
3.4. Transducers and Effectors of Receptor Signaling in Lung Inflammation
The signals through TLRs happen in either a MyD88-dependent or MyD88-independent manner. TLRs 2, 4, 6, 7, 8, and 9 utilize MyD88, whereas TLR3 and TLR4 transduce signals through TIR-domain-containing adapter-inducing interferon-β (TRIF). Myd88 activates TGF-β-activated kinase 1 (TAK1), which parallelly activates nuclear factor kappa B (NFκB) directly and activator protein 1 (AP-1) through mitogen-activated protein kinase (MAPKs).
Extracellular signal-regulated kinase (ERK1/2), p38, and jun N-terminal kinases (JNK) are three MAP kinases that act as effectors for the inflammatory response. In the TRIF pathway, TRIF interacts with TANK binding kinase 1 (TBK1) to activate interferon regulatory factor 3/7 (IRF3/7), transcription factors for Type I interferon cytokine generation, or interacts with RIP1 and TRAF6 to activate NFκB directly [32].
IL1-IL1R signal transduction is also classified as both a MyD88-dependent and independent pathway. Upon receptor activation, MyD88 binds to the TIR domain of the receptor, and subsequent interaction leads to IRAK1/2 phosphorylation, which is associated with TRAF6 and TAK1 recruitment. TAK1 parallelly activates NFκB directly and AP-1 through MAPKs as well as IRF3/7 [56]. IL-6 signals through two pathways: classical and trans-signaling. In classical IL-6 signaling, IL-6, membrane-bound IL-6R and gp130 make a complex, whereas in trans-signaling IL-6, soluble IL-6R and gp130 form the complex. The formation of this complex activates the janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway and transcription of STAT3 target genes. MAPK and PI3K/AKT pathways are also activated by the IL6-IL6R-gp130 complex [57]. TNF-α binds to TNFR1 and TNFR2 and recruits TNFRSF1A associated via death domain (TRADD), TNF receptor associated factor 2 (TRAF2), receptor interacting protein (RIP), and interleukin 1 receptor associated kinase 1 (IRAK1) to activate MAPK and NF-κB [58]. NOD1 and NOD2, upon ligand interaction, recruit RIP2 by caspase recruitment domain (CARD) interaction and becomes phosphorylated [59]. TRAF family members (TRAF1, 5, 6), cIAP1, cIAP2, and BID binds to RIP2 for it to get ubiquitinated. Ubiquitinated RIP2 recruits TAK1, activating MAPK and NF-κB [60]. In RLR signaling, viral RNA binds to retinoic acid-inducible gene I and melanoma differentiation-associated protein 5, which results in conformational changes to unmask their CARDs. Through its C-terminal CARD, mitochondrial antiviral-signaling protein binds to these receptors and activates NF-κB and IRF3 and 7 via TBK1 [61].
Type I IFNα/β signaling is initiated when IFNα/β binds to IFNR and activates JAK1 and tyrosine kinase 2 (TYK2). Receptors get phosphorylated and recruit STAT proteins. These STAT proteins are activated by phosphorylation and translocate to the nucleus. STAT1, STAT2, and IRF9 complex transcribe antiviral response genes, STAT1 homodimers induce pro-inflammatory gene transcription, and STAT3 activates genes for inflammation suppression [62].
4. Signaling Pathways of Allergic Lung Inflammation and Diseases
In the airway, delayed allergic reactions cause the progression of asthma as expressed by chronic airway inflammation that can cause airway hindrance, airway hyper-responsiveness, and lung tissue alteration [63]. Deferred allergic reactions are endorsed by cells within the innate and adaptive immune systems. The allergic airway diseases associated with eosinophilic inflammation develop due to the dominant differentiation of the Th2 cell subset, which preferentially produces interleukin-4 (IL-4), interleukin-5 (IL-5), and interleukin-13 (IL-13), rather than IFN-γ and TNF-α (Figure 1) [64].
As discussed in the previous section, developing HP requires exposure and sensitization to an antigen to develop the allergic immune response and subsequent inflammation [2,26]. Upon primary exposure to the antigen, antigen-presenting cells in the alveolar space, namely, alveolar macrophages and DCs, induce differentiation of T cells [5,66,67]. While some allergic pulmonary diseases such as asthma are Th2-mediated, HP is caused by a primarily Th1 CD4+ mediated disease process. Whereas asthma is predominantly a type I IgE-mediated hypersensitivity reaction, HP has features of both type III (immune complex) and type IV (cell-mediated) hypersensitivity reactions [13,66,67]. In addition to the Th1-mediated response, mouse models showed some Th17 differentiation, which may contribute to the fibrosis present in some instances of HP [68]. However, this is most likely not the only method by which fibrosis occurs. Although the exact mechanism for disease progression into the chronic form and development of pulmonary fibrosis is currently unknown, there is evidence that the immune system switches from a Th1 to Th2-mediated response through inhibition of Tregs and activation of NKT cells [68,69]. Fibrosis may be partially explained through this mechanism since Th2-mediated immune responses can lead to the development of fibrosis in tissue [70].
The section below describes the contributions of specific components of innate and adaptive immune responses, such as innate lymphoid cells type 2 (ILC2) mast cells, basophils, and CD4+ T cells, and the corresponding signaling pathways.
4.1. Innate Lymphoid Cells (ILCs) in Allergic Lung Inflammation
Innate lymphoid cells (ILCs) are categorized as a small population of non-T and non-B lymphoid cells. ILCs are tissue-resident cells that play essential roles in tissue homeostasis, repair, and regeneration. In addition, ILCs are essential regulators of innate immunity in response to pathogens and allergens. A specific subtype of ILC, ILC2, exists in the human lung parenchyma, expressing IL-7Rα and the ST2 receptor for IL-33 [66], and contributes to the pathophysiology of allergic inflammation [71]. ILC2s play a central role in lung tissue homeostasis by maintaining the respiratory tract epithelial barrier to limit pathogen exposure. The primary mediators of ILC2 that serve to maintain a functional epithelial layer are type 2 interleukins IL-4, IL-5, IL-9, and IL-13 [66]. In the case of allergic asthma, ILC2 responds to intranasal exposure of IL-25 and IL-33 by binding to their specific receptors IL-7Ra and ST2, respectively. The specific binding of IL-25 and IL33 to their receptors induces the production of IL-5 and IL-13, leading to the development of airway hyper-reactivity, eosinophilia, and airway inflammation. Although both IL-25 and IL-33 are involved in evoking type 2 immunity, it is IL-33 that shows a greater response to allergen challenge, as evident from the studies on mouse models induced with house dust and ovalbumin (OVA) [67].
Regarding the mechanism, it was described that exposure to an allergen with protease activity, such as papain, initially injures the airway epithelial cells, inducing the production of IL-33, which in turn activates ILC2 [72]. Similar results were obtained when Alternaria alternata, another allergen with protease activity, induced IL25 and IL-33 release by epithelial cells that activated lung ILC2 [73]. These studies suggested that ILC2s are a critical early source of IL-5, IL-13, and IL-9 in allergen-induced lung inflammation and that this occurs in a T cell-independent manner.
Regarding the specific signaling pathways, studies with glucocorticoid treatment of patients reveal that phosphorylation of STAT3, STAT5, and STAT6 primarily regulate in ILC2-mediated immune response. The OVA challenge model demonstrated that TSLP was responsible for inducing IL-33 that activated the STAT-5 signaling pathway. A recent study by the Yu group demonstrated that upon stimulation with IL-25 and IL-33, a significant increase was observed in phosphorylated STAT 3, -5, and -6 and the levels of phosphorylated JAK MEK1/2 in patient-derived and purified ILC2. Specific inhibitors for STAT3, STAT5, STAT6, and JAK3 phosphorylation significantly decreased IL-5 and IL-13 secretion (Figure 2). It suggests that MEK/JAK-STAT signaling pathways produced Th2 cytokines by ILC2s [74]. Apart from the major regulatory pathway discussed above, the p38 MAPK signaling has been shown to regulate IL-33 mediated cytokine production in ILC2 [75]
4.2. Mast Cells in Allergic Lung Inflammation
Mast cells are involved in the pathogenesis of allergic lung diseases. The cellular accumulation of mast cells in the lung correlates with the severity of the allergic response. A biomarker for allergic asthma is a high serum level of IgE [76]. Serum IgE levels are generally not high in HP; however, infiltration of lymphocytes and mast cells along with high serum IgE levels have been reported in summer type-HP patients [77].
The progenitor cells differentiate into functional mast cells responding to the allergen or other inflammatory stimuli. The lung mast cells then transfer through blood vessel endothelium and confine themselves to vascularized connective tissue underneath epithelial layers. The differentiated mast cells are capable of effector cell function, expressing the high-affinity IgE receptor FcεR1, a receptor that binds to IgE. Mast cell activation and the discharge of multiple inflammatory mediators occur due to interaction between receptor-bound IgE and antigen [78,79].
The IL-4-induced up-regulation of IgE causes increased binding to the FcεRI of the mast cell or basophil surface, leading to FcεRI cross-linking followed by the granulation and release of biologically active mediators such as histamine, serotonin, and leukotrienes within minutes. The antigen-IgE complex formation leads to an activation of a series of signaling pathways. First, activation of the Lyn-Src kinase pathway occurs. Activation of Lyn leads to intracellular calcium efflux and PKC activation. Lyn-Src pathways regulate the mast cell degranulation process; Lyn deficient mast cells exhibit defective degranulation phenotype. The Lyn phosphorylation and activation further induces positive regulation of the Syk pathway (another major mediator of IgE-FcεRI signaling), whereas it negatively regulates signal transduction of Cbp (Csk binding protein) [80] (Figure 3).
Contact between FcεR1-bound IgE and antigen prompts an intricate system of signaling paths that provokes cell activation. FcεR1 arrangement stimulates SFKs, Fyn, and Lyn by a vague process that causes immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation and consequently the production of high-affinity docking sites for proteins that have SH2 domains. These comprise Lyn, Fyn, and the protein tyrosine kinase Syk. All three proteins bind the phosphorylated ITAMs and thus stimulate trans- and auto-phosphorylation actions. These actions cause the tyrosine phosphorylation of many downstream substrates. The most important proteins are L-type amino acid transporter (LAT and LAT2) [LAB/NTAL], which are phosphorylated on numerous tyrosines and interlink with various SH2 domain-containing proteins. These include the adapter proteins growth factor receptor-bound protein 2 (Grb2), SH2 domain-containing leukocyte phosphoprotein of 76 kDa (SLP-76), GRB2-associated-binding protein 2 (Gab2), and GTP exchange factors of Vav and son of sevenless (SOS). The former two proteins trigger Ras, which activates MAPK signaling cascades. SOS, Vav, and Gab2 stimulate PI3-kinase activation, creating membrane-docking sites for proteins comprising pleckstrin homology domains. These include the main enzyme PLCγ, which links with LAT/LAT2 to form complexes and becomes activated through tyrosine phosphorylation. PLCγ generates second messenger molecules that promote calcium flux and protein kinase C activity. These proceedings, along with signals delivered by MAPK cascades, augment the various events related to basophil and mast cell activation [82]. FcεR1 signaling also stimulates PI3-kinase, producing phosphatidylinositol 1,4,5- triphosphate [PIP3] and, hence, membrane docking sites for other signaling proteins. PIP3 is transformed to PIP2 by the inositol phosphatases SH-2 containing inositol 5′ polyphosphatase 1 (SHIP-1), SH-2 containing inositol 5′ polyphosphatase 2 (SHIP-2), and phosphatase and tensin homolog (PTEN), resulting in downregulation of signaling [83].
4.3. Basophils in Allergic Inflammation
Basophils and mast cells arise from a common lineage and express the FcεR1 that is triggered by antigen-IgE induced aggregation of the receptor. In addition, basophils are key factors in immune reactions to protease allergens, which substantially contribute to human allergic disease. Furthermore, basophils are recruited to inflamed lungs and exacerbate memory Th2 responses in mice and humans [84]. Apart from the well-studied IgE response from mast cells and basophils, another mechanistic axis related to basophils is TSLP and its receptors (TSLP-TSLPR) interaction that has been reported in models of airway inflammation and hyper-responsiveness. Initial investigations revealed that TSLP induces activation of human DCs that promote CD4+ T cell differentiation to effector cells with pro-inflammatory and pro-allergic cytokine functional properties [85]. TSLP binds to IL17R-TSLPR complexes that induce activation of downstream signaling pathways in basophils such as JAKs, which, in turn, activate STAT factors including STAT1, STAT3, STAT4, STAT5a, STAT5b, and STAT6 [86]. Apart from the JAK-STAT pathway, the IL-7 ligand-induced 1L-17R and TSLPR dimerization leads to the activation of PI3-kinase and MAPK/Erk signaling cascades and their respective responses [87].
4.4. T Cells and T Cell Receptor Signaling Pathways in Allergic Inflammation
CD4+ T cells play essential roles in antigen-induced allergic lung inflammation. The simple CD4+ T cells differentiate into effector cells upon the interaction of TCR by peptide-MHCII complexes. These effector cells can be classified into four groups: Th1 cells that generate interferon-γ (IFNγ); Th2 cells which produce IL-4, IL-5, IL-13, and IL-25; Th17 cells that cause the production of IL-17A, IL-17F, IL-21, and IL-22; and regulatory T (Treg) cells, which secrete suppressive cytokines. In general, allergic lung disease is primarily mediated through the eosinophilic inflammation that occurs due to Th2 responses responsible for IL-4, IL-5, and IL-13 production, whereas the Th1 response plays a minor role and produces IFN-γ and TNF-α [63]. In airway inflammation, activated CD4+ T cells can be found in the mucosal membrane of the patients, and suppression of CD4+ T cells in murine models have been reported to improve the symptoms of allergic airway inflammation [88]. These data suggest that activation of CD4+ T cells may be a predisposing factor for the development of allergic inflammation in the lung.
The T cell receptor (TCR) consists of antigen-specific α and β chains linked with CD4 and the CD3 complex. Cross- and auto-phosphorylation causes upregulation zeta-chain-associated protein kinase 70 (Zap-70) kinase activity, which along with LCK proto-oncogene, Src family tyrosine kinase (Lck) and the IL2 inducible T cell kinase (Itk) facilitates phosphorylation of LAT and SLP-76. As in the FcεR1 signaling pathway, LAT and SLP-76 cause the production of a complex containing phospholipase C gamma 1 (PLCγ1). This increases calcium flux and MAPK activity tempted by PLCγ1, and other paths cause induction of several transcription factors and variations in cytoskeletal structure linked with T cell stimulation [89]. The precarious parts of protein tyrosine kinases in the TCR signaling pathway make these enzymes potential inhibitory targets that might be used to subdue chronic allergic inflammatory responses. Zap-70 holds tyrosine residues that have been involved in both negative regulation and connections with other proteins that stimulate TCR signaling. Reduced Zap-70 function results in thymic T cell development. It suggested that the autoimmune responses that had been observed could be due to faults in the negative selection or Treg development. Itk signifies a captivating target for inhibitors that might be used to treat chronic allergic inflammation [90].
5. Therapeutic Strategies for Lung Inflammation Associated with Infection
Therapeutics strategies for lung inflammation, such as pneumonia due to infections, include antibiotics and immunomodulators focusing on host-immune responses. The immunomodulatory therapeutics are attractive because they are driven on host-microbe immune response and designed based on the nature of infection-causing agents. Several immunomodulatory therapies control pneumonia and appropriate antibiotics, antiviral, and antifungal treatments [91]. Here, we will briefly discuss corticosteroids, statins, cytokines-targeted therapies, and immunomodulatory antibiotics.
5.1. Corticosteroids
Corticosteroids, such as prednisone, cortisone, and dexamethasone, are anti-inflammatory steroids used to manage conditions with excess inflammation, but the use of corticosteroids can have side effects. Therefore, their use for pneumonia is mostly controversial [91,92,93]. Corticosteroids reduce inflammation by reducing TNF-α, IL-6, MCP-1, IL-17, IL-8, IL-1β, and IL1R expression on neutrophils and macrophages [94,95].
5.2. Statins
Statins are competitive 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors and mainly negatively affect cholesterol synthesis and, subsequently, isoprenoid synthesis. Reduced isoprenoid synthesis furthers regulates Rac, Ras, and Rho signaling pathways which are also important in inflammation. Statins reduce expression of TNF-α, IL-6, IL-8, and IL-1β. They also affect synthesis and release of COX2, leukotrienes, and PPAR-γ. In addition, they reduce chemotaxis and oxidative bursts in neutrophils, cytokine and chemokine production by macrophages, and lymphocyte activation [96,97].
5.3. Cytokine-Targeted Therapies
Excessive production of pro-inflammatory cytokines due to bacterial or viral infection can lead to bystander cell and tissue damage. A standard strategy to negate such effects is using cytokine or receptor blockers. Tocilizumab, monoclonal antibodies against IL-6 receptor, remains the standard treatment for systemic overwhelmed cytokine production. Anakinra or canakinumab is another monoclonal antibody used to target IL-1 production. N-acetylcysteine (NAC) is another molecule used to target pro-inflammatory cytokines and chemokines like IL-6 and C-C motif chemokine ligand 5 (CCL5) [98,99].
5.4. Immunomodulatory Antibiotics
Macrolides are a group of antibiotics derived from Streptomyces species of bacteria that also have immunomodulatory effects. Erythromycin A, azithromycin, and clarithromycin are a few examples of macrolides. In the lungs, they decrease mucus’s hypersecretion by modulating MAPK signaling associated with its secretion. They also reduce IL-8 and neutrophils in bronchial lavage fluid, IL-1, IL-6, TNF-α, CD80 stimulation, and intercellular adhesion molecule 1 (ICAM) expression, thereby reducing activation of macrophages, neutrophils, mast cells, and other immune cells [100,101].
6. Therapeutic Strategies for Allergic Lung Inflammation
Several cytokines have been involved in the pathophysiology of allergic diseases, though few cytokines have an essential role in atopic inflammation. Therefore, there are a lot of potential tools at our disposal to inhibit excessive cytokine synthesis. These possible tools include humanized antibodies capable of blocking cytokines or their receptors [102], chemical medications that prevent cytokine production (tacrolimus, cyclosporine A and glucocorticoids) [103,104], soluble receptors that are capable of disposing secreted cytokines to receptor antagonists, and inhibitors of signal-transduction paths stimulated by cytokines [105].
7. Soluble Receptor/Mab Therapy
Th2 responses are the major drivers of allergic responses; therefore, any therapeutic strategies, either biological or chemical, that target Th2 cytokines are more logical options for treatment. Some examples of such therapeutic strategies are discussed below.
7.1. Anti–IL-4 Strategies
IL-4 induces IgE isotype switching in plasma cells and stimulates differentiation of ingenuous lymphocytes into Th2 cells and the consequent release of extra IL-4, IL-5, and IL-13. Two human studies assessed the effectiveness and protection of a recombinant human sIL-4R. The sIL-4R was nontoxic and well accepted, with a serum half-life of about five days. The initial study involved mature allergic patients with mild insistent asthma [106]. The nebulized sIL-4R of both 500 µg and 1500 µg or placebo was given as a dose to 19 patients. All inhaled corticosteroids were terminated one day earlier. The patients treated with the 1500 µg of sIL-4R had substantial improvement in allergic symptoms such as asthma. The 1500 µg dose of sIL-4R treatment also caused a major increase in FEV1 at four days [106]. A continuation study with 62 asthmatic patients wanting inhaled corticosteroids assessed weekly treating with sIL-4R for 12 weeks at 0.75 mg, 1.5 mg, or 3.0 mg and placebo. The patients who received a nebulized dose of 3.0 mg of sIL-4R weekly could maintain their lung functions. However, no substantial signs of progress were observed in indications or asthma exasperation. Likewise, a monoclonal antibody (mAb) against IL-4 had no verified efficacy, which ultimately resulted in further investigation into anti-IL4–definite remedies [107]. This might be because of redundancy in processes between IL-4 and some other cytokines, particularly IL-13.
7.2. IL-4Ra Receptor Antagonist
IL-4 and IL-13 bind to the IL-4Ra receptor subunit, which causes many downstream signaling effects that are significant in the biologic aspects of these cytokines. AMG-317 is a human mAb to the IL-4Ra receptor subunit developed by Amgen and is used to treat sensitive asthma [108].
7.3. Anti–IL-5 mAbs
IL-5 is produced by Th2 cells and ILC2s, which acts as an eosinophils’ activator. To treat allergic asthma, two humanized antibodies, mepolizumab and benralizumab, have been developed that block IL-5 and IL-5R function, respectively [109]. Two other humanized IL-5 mAbs, reslizumab (SCH55700) and mepolizumab, have also been studied in human subjects. In a clinical trial, asthma patients were given one dose of reslizumab (0.03 mg/kg, 0.3 mg/kg, or 1.0 mg/kg), or placebo for 3 months. Improvement in the symptoms, such as reduced serum eosinophils, was observed with high doses of reslizumab [110]. Similarly, mepolizumab treatment also significantly suppressed median serum eosinophils from baseline by 100% and airway eosinophils by 55% in asthma patients. In mild asthma patients, mepolizumab treatment does not alter the T cell function but reduces the extracellular matrix protein remodeling and airway eosinophils [111].
7.4. Anti–IL-13 mAb
IL-13 has a range of biological properties based on findings from vitro and murine studies that might be significant in the pathophysiology of asthma and other allergic lung diseases. IL-13 has been discovered in the induction of mucus fabrication, eotaxin secretion, changes in airway remodeling, and airway hyper-responsiveness. Moreover, IL-13 plays a crucial role in the synthesis of IgE. Therefore, approaches that were targeted to inhibit the effects of IL-13 might be a significant therapeutic way. The lessening of lung inflammation in anti–IL-13–treated animals was verified in rodent and primate studies. There is a minimum of three diverse humanized mAbs under production for human IL-13. These three mAbs are either in phase I or phase II human medical trials. The phase I medical trial with CAT-354 indicated that expanding single dosages of intravenously directed anti–IL-13 mAb in 34 patients having slight asthma was well endured at every dose. Moreover, no safety apprehensions were identified [112].
8. Immune Cell Treatment
In response to allergen challenge, eosinophils were stimulated, and leukocytic infiltrate with T cell activity can be seen in the lungs of allergic patients. T cells recognize antigens, which then initiate the differentiation of naive CD4+ T cells into Th2 cells and Tregs. The latter specifically suppress the extreme immunological response of Th2 cells. Asthma can occur when Tregs become dysfunctional and cannot control the Th2 cell response. In this regard, increasing the number of Tregs and improving their Th2 suppression might be a better alternative treatment for allergic patients. The most common process for exogenous Treg stimulation is antigen-specific immunotherapy (ASIT) using subcutaneous immunotherapy (SCIT) and sublingual immunotherapy (SLIT) [113]. There are ongoing clinical trials concerning the usage of SCIT in allergic lung disease, where SLIT medicines are more commonly used. Hoshino et al. presented that in 102 patients suffering from asthmatic rhinitis, treatment with house dust mite-SLIT therapy significantly decreased the eosinophilic airway tenderness and fractional exhaled nitric oxide (FeNO) as compared to pharmacotherapy [114]. SLIT and SCIT therapy upregulate CD4+ CD25+ FoxP3+ Treg expression in the asthmatic patient [115].
Contrastingly, Tregs can be manipulated ex vivo by a chimeric antigen receptor (CAR) and, thus, offer more specificity regarding various antigens. For example, an injection of CAR Tregs in a mouse OVA-induced asthma model reduced mucus hypersecretion, Th2 cell response, and air network hyper-reactivity [116]. Therefore, advanced studies should be conducted to identify airway allergy triggers and expand the treatments based on Treg functions.
9. Stem Cell Treatment
The adult bone marrow stem cells, including mesenchymal stem cells (MSCs), hematopoietic stem cells (HSCs), and endothelial progenitor cells (EPCs), hold an impressive potential for the treatment of allergic lung diseases and inflammation. MSCs have been widely functional in numerous investigational studies in asthma and other allergic lung diseases. The regenerative properties of MSCs are primarily associated with the regulation of the immune system at the site of infection. These stem cells are suitable for the regulation of the Th2 to Th1 ratio, and synthesis of IL-3, IL4, IL-13, and mucus after institution into the asthmatic place. These actions overlap with the proliferation of CD25+, CD4+, Treg FoxP3+ cells, IL-10 in bronchioalveolar release, blockage of phagocytic activity in alveolar macrophages, destruction of nitrosative stress, and reduction of mast and goblet cells [117].
Various kinds of MSC transplantation have diverse time-dependent healing consequences. The intravenous management perhaps leads to insufficient MSC recruitment to the asthmatic locus due to complications related to the ability to cross the blood-air barrier and nonspecific bio-distribution. It has also been revealed that intraperitoneal management of bone marrow MSCs is the probable approach to control the allergic responses. These stem cells migrate to the pulmonary sites shortly after the administration into the peritoneal cavity and then employ immunomodulatory actions [117]. If we suppose that the healing ability of transplant MSCs is primarily done through paracrine action, it is rational to transfer discrete cell types directly to the pulmonary locus instead of through systemic route [117]. The portion of transplant MSCs expires shortly after inoculation into exacerbated positions due to mechanical stress and provocative tissue situations. However, fewer MSCs are required in local delivery to accomplish healing efficiency.
10. Transcription Factor Inhibition
Many transcription factors are essential in regulating the gene expression of cytokines and mediators. Transcription factors are proteins that bind to promoter areas of genes and activate or prevent their transcription after binding. They are possible targets for immunomodulatory agents as transcription factors play a crucial role in producing inflammatory mediators that are essential for the pathogenesis of allergic diseases.
10.1. Syk Kinase Inhibitors
The Syk kinase is an intracellular protein tyrosine kinase that significantly activates mast cells and basophil. Consequently, inhibition of Syk kinase signifies a possible beneficial modality for allergic inflammation. In this regard, R112 is a Syk kinase inhibitor directed intranasally to patients suffering from seasonal allergic rhinitis. The administration of Syk kinase inhibitor revealed progress in symptoms over placebo with a period of action beyond 4 h. In addition, there were no critical adverse actions stated [118].
10.2. Peroxisome Proliferator-Activated Receptor (PPAR)γ Agonists
One of the key transcription factors in the manifestation of Th2 cytokines in allergic lung diseases is GATA-3. The expression of GATA-3 is augmented in the asthmatic T cells. The reduction of GATA-3 expression in the murine asthma model caused reduced lung infection and decreased production of Th2 cytokines and IgE. Currently, there are no artificial inhibitors of GATA-3, but peroxisome proliferator-activated receptor (PPAR)-γ agonists have been reported to prevent GATA-3 expression and subsequent Th2-driven provocative reactions in murine models. Numerous minor experimental trials continue to examine the actions of PPAR-γ agonists for the therapy of allergic diseases.
11. Conclusions
More detailed understanding and knowledge of the signaling pathways that regulate cell functions responsible for immune responses will pave the way for developing efficient and safe treatment of allergic inflammation in the lung. The studies described in this article help illustrate the complexities and differences of allergic and infection-associated lung inflammation and provide hope that efficient methods to target and block these signaling pathways can be developed.
Author Contributions
M.U., A.N. and S.W.; writing—original draft preparation, M.U., A.N. and S.W.; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict to interest.
References
- Passalacqua, G.; Ciprandi, G. Allergy and the lung. Clin. Exp. Immunol. 2008, 153, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Costabel, U.; Miyazaki, Y.; Pardo, A.; Koschel, D.; Bonella, F.; Spagnolo, P.; Guzman, J.; Ryerson, C.J.; Selman, M. Hypersensitivity pneumonitis. Nat. Rev. Dis. Primers 2020, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Dharmage, S.C.; Perret, J.L.; Custovic, A. Epidemiology of Asthma in Children and Adults. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Schatz, M.; Rosenwasser, L. The Allergic Asthma Phenotype. Allergy Clin. Immunol. Pract. 2014, 2, 645–648. [Google Scholar] [CrossRef]
- Holgate, S.T. Pathogenesis of Asthma. Clin. Exp. Allergy 2008, 38, 872–897. [Google Scholar] [CrossRef]
- Akinbami, L.J.; Moorman, J.E.; Bailey, C.; Zahran, H.S.; King, M.; Johnson, C.A.; Liu, X. Trends in Asthma Prevalence, Health Care Use, and Mortality in the United States, 2001–2010; US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics: Hyattsville, MD, USA, 2012; pp. 1–8.
- Possa, S.; Leick, E.; Prado, C.; Martins, M.; Tibério, I. Eosinophilic Inflammation in Allergic Asthma. Front. Pharmacol. 2013, 4, 46. [Google Scholar] [CrossRef] [Green Version]
- Bogaert, P.; Tournoy, K.G.; Naessens, T.; Grooten, J. Where asthma and hypersensitivity pneumonitis meet and differ: Noneosinophilic severe asthma. Am. J. Pathol. 2009, 174, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Bochner, B.S.; Undem, B.J.; Lichtenstein, L.M. Immunological Aspects of Allergic Asthma. Annu. Rev. Immunol. 1994, 12, 295–335. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A.; King, T.E., Jr. Hypersensitivity pneumonitis: Insights in diagnosis and pathobiology. Am. J. Respir. Crit. Care Med. 2012, 186, 314–324. [Google Scholar] [CrossRef]
- Backman, H.; Räisänen, P.; Hedman, L.; Stridsman, C.; Andersson, M.; Lindberg, A.; Lundbäck, B.; Rönmark, E. Increased prevalence of allergic asthma from 1996 to 2006 and further to 2016-results from three population surveys. Clin. Exp. Allergy 2017, 47, 1426–1435. [Google Scholar] [CrossRef]
- Stern, J.; Pier, J.; Litonjua, A.A. Asthma epidemiology and risk factors. Semin. Immunopathol. 2020, 42, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, M.L.; Gu, T.; Murray, S.; Gross, B.H.; Chughtai, A.; Sayyouh, M.; Kazerooni, E.A.; Myers, J.L.; Lagstein, A.; Konopka, K.E.; et al. Hypersensitivity Pneumonitis: Radiologic Phenotypes Are Associated With Distinct Survival Time and Pulmonary Function Trajectory. Chest 2019, 155, 699–711. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Assessing National Capacity for the Prevention and Control of Noncommunicable Diseases: Report of the 2019 Global Survey; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Beasley, R.; Crane, J.; Lai, C.K.W.; Pearce, N. Prevalence and etiology of asthma. J. Allergy Clin. Immunol. 2000, 105, S466–S472. [Google Scholar] [CrossRef]
- Pakkasela, J.; Ilmarinen, P.; Honkamäki, J.; Tuomisto, L.E.; Andersén, H.; Piirilä, P.; Hisinger-Mölkänen, H.; Sovijärvi, A.; Backman, H.; Lundbäck, B.; et al. Age-specific incidence of allergic and non-allergic asthma. BMC Pulm. Med. 2020, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Perez, E.R.; Kong, A.M.; Raimundo, K.; Koelsch, T.L.; Kulkarni, R.; Cole, A.L. Epidemiology of Hypersensitivity Pneumonitis among an Insured Population in the United States: A Claims-based Cohort Analysis. Ann. Am. Thorac. Soc. 2018, 15, 460–469. [Google Scholar] [CrossRef]
- Bang, K.M.; Weissman, D.N.; Pinheiro, G.A.; Antao, V.C.; Wood, J.M.; Syamlal, G. Twenty-three years of hypersensitivity pneumonitis mortality surveillance in the United States. Am. J. Ind. Med. 2006, 49, 997–1004. [Google Scholar] [CrossRef]
- Murdoch, J.R.; Lloyd, C.M. Chronic inflammation and asthma. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2010, 690, 24–39. [Google Scholar] [CrossRef]
- Cano-Jiménez, E.; Acuña, A.; Botana, M.I.; Hermida, T.; González, M.G.; Leiro, V.; Martín, I.; Paredes, S.; Sanjuán, P. Revisión de la enfermedad del pulmón de granjero. Arch. De Bronconeumol. 2016, 52, 321–328. [Google Scholar] [CrossRef]
- Wollin, L.; Distler, J.H.W.; Redente, E.F.; Riches, D.W.H.; Stowasser, S.; Schlenker-Herceg, R.; Maher, T.M.; Kolb, M. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur. Respir. J. 2019, 54. [Google Scholar] [CrossRef]
- Just, J.; Deschildre, A.; Lejeune, S.; Amat, F. New perspectives of childhood asthma treatment with biologics. Pediatr. Allergy Immunol. 2019, 30, 159–171. [Google Scholar] [CrossRef]
- Agache, I.; Rocha, C.; Beltran, J.; Song, Y.; Posso, M.; Solà, I.; Alonso-Coello, P.; Akdis, C.; Akdis, M.; Canonica, G.W.; et al. Efficacy and safety of treatment with biologicals (benralizumab, dupilumab and omalizumab) for severe allergic asthma: A systematic review for the EAACI Guidelines-recommendations on the use of biologicals in severe asthma. Allergy 2020, 75, 1043–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.; Price, D. Treatment Adherence in Adolescents with Asthma. J. Asthma Allergy 2020, 13, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Efficacy of inhaled corticosteroids in asthma. J. Allergy Clin. Immunol. 1998, 102, 531–538. [Google Scholar] [CrossRef]
- Vasakova, M.; Selman, M.; Morell, F.; Sterclova, M.; Molina-Molina, M.; Raghu, G. Hypersensitivity Pneumonitis: Current Concepts of Pathogenesis and Potential Targets for Treatment. Am. J. Respir. Crit. Care Med. 2019, 200, 301–308. [Google Scholar] [CrossRef]
- Lacasse, Y.; Selman, M.; Costabel, U.; Dalphin, J.C.; Ando, M.; Morell, F.; Erkinjuntti-Pekkanen, R.; Muller, N.; Colby, T.V.; Schuyler, M.; et al. Clinical diagnosis of hypersensitivity pneumonitis. Am. J. Respir. Crit. Care Med. 2003, 168, 952–958. [Google Scholar] [CrossRef]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Shibata, S.; Furusawa, H.; Inase, N. Pirfenidone in chronic hypersensitivity pneumonitis: A real-life experience. Sarcoidosis Vasc. Diffus. Lung Dis. 2018, 35, 139–142. [Google Scholar] [CrossRef]
- Bartlett, J.G. Anaerobic bacterial infection of the lung. Anaerobe 2012, 18, 235–239. [Google Scholar] [CrossRef]
- Nogueira, R.; Melo, N.; Novais, E.B.H.; Martins, N.; Delgado, L.; Morais, A.; P, C.M. Hypersensitivity pneumonitis: Antigen diversity and disease implications. Pulmonology 2019, 25, 97–108. [Google Scholar] [CrossRef]
- Christensen, L.T.; Schmidt, C.D.; Robbins, L. Pigeon breeders’ disease—A prevalence study and review. Clin. Allergy 1975, 5, 417–430. [Google Scholar] [CrossRef]
- Eddens, T.; Kolls, J.K. Host defenses against bacterial lower respiratory tract infection. Curr. Opin. Immunol. 2012, 24, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartlett, J.G. Diagnostic tests for agents of community-acquired pneumonia. Clin. Infect. Dis. 2011, 52 (Suppl. S4), S296–S304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruganandah, V.; Kupz, A. Immune responses to bacterial lung infections and their implications for vaccination. Int. Immunol. 2021, 34, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Balamayooran, T.; Balamayooran, G.; Jeyaseelan, S. Review: Toll-like receptors and NOD-like receptors in pulmonary antibacterial immunity. Innate Immun. 2010, 16, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opitz, B.; van Laak, V.; Eitel, J.; Suttorp, N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am. J. Respir. Crit. Care Med. 2010, 181, 1294–1309. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Mindru, C.; Botha, J.F.; Grant, W.J.; Mercer, D.F.; Olivera, M.A.; McCartan, M.A.; McCashland, T.M.; Langnas, A.N.; Florescu, D.F. Risk of cytomegalovirus disease in high-risk liver transplant recipients on valganciclovir prophylaxis: A systematic review and meta-analysis. Liver Transpl. 2012, 18, 1440–1447. [Google Scholar] [CrossRef]
- Pritt, B.S.; Aubry, M.C. Histopathology of viral infections of the lung. Semin. Diagn. Pathol. 2017, 34, 510–517. [Google Scholar] [CrossRef]
- Jennings, L.C.; Anderson, T.P.; Beynon, K.A.; Chua, A.; Laing, R.T.; Werno, A.M.; Young, S.A.; Chambers, S.T.; Murdoch, D.R. Incidence and characteristics of viral community-acquired pneumonia in adults. Thorax 2008, 63, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [Green Version]
- Coro, E.S.; Chang, W.L.; Baumgarth, N. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J. Immunol. 2006, 176, 4343–4351. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Role of type I interferons in the activation of autoreactive B cells. Immunol. Cell Biol. 2012, 90, 498–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crouse, J.; Kalinke, U.; Oxenius, A. Regulation of antiviral T cell responses by type I interferons. Nat. Rev. Immunol. 2015, 15, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Xu, H.C.; Lang, P.A.; Oxenius, A. NK cells regulating T cell responses: Mechanisms and outcome. Trends Immunol. 2015, 36, 49–58. [Google Scholar] [CrossRef]
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latgé, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar] [CrossRef] [Green Version]
- Kronstad, J.W.; Attarian, R.; Cadieux, B.; Choi, J.; D’Souza, C.A.; Griffiths, E.J.; Geddes, J.M.; Hu, G.; Jung, W.H.; Kretschmer, M.; et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 2011, 9, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Lu, G.; Meng, G. Pathogenic Fungal Infection in the Lung. Front. Immunol. 2019, 10, 1524. [Google Scholar] [CrossRef] [Green Version]
- Sorrell, T.C.; Chen, S.C. Fungal-derived immune modulating molecules. Adv. Exp. Med. Biol. 2009, 666, 108–120. [Google Scholar] [CrossRef]
- Hatinguais, R.; Willment, J.A.; Brown, G.D. PAMPs of the Fungal Cell Wall and Mammalian PRRs. Curr. Top. Microbiol. Immunol. 2020, 425, 187–223. [Google Scholar] [CrossRef]
- Osterholzer, J.J.; Milam, J.E.; Chen, G.H.; Toews, G.B.; Huffnagle, G.B.; Olszewski, M.A. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans. Infect. Immun. 2009, 77, 3749–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311. [Google Scholar] [CrossRef] [PubMed]
- McDermott, A.J.; Klein, B.S. Helper T-cell responses and pulmonary fungal infections. Immunology 2018, 155, 155–163. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Seeger, W.; Savai, R. Classical IL-6 signaling: A promising therapeutic target for pulmonary arterial hypertension. J. Clin. Investig. 2018, 128, 1720–1723. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef]
- Pellegrini, E.; Desfosses, A.; Wallmann, A.; Schulze, W.M.; Rehbein, K.; Mas, P.; Signor, L.; Gaudon, S.; Zenkeviciute, G.; Hons, M.; et al. RIP2 filament formation is required for NOD2 dependent NF-kappaB signalling. Nat. Commun. 2018, 9, 4043. [Google Scholar] [CrossRef] [Green Version]
- Saxena, M.; Yeretssian, G. NOD-Like Receptors: Master Regulators of Inflammation and Cancer. Front. Immunol. 2014, 5, 327. [Google Scholar] [CrossRef] [Green Version]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Colgan, J.D.; Hankel, I.L. Signaling pathways critical for allergic airway inflammation. Curr. Opin. Allergy Clin. Immunol. 2010, 10, 42–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills-Karp, M. Immunologic basis of antigen-induced airway hyperresponsiveness. Annu Rev. Immunol. 1999, 17, 255–281. [Google Scholar] [CrossRef] [PubMed]
- Calven, J.; Ax, E.; Radinger, M. The Airway Epithelium-A Central Player in Asthma Pathogenesis. Int. J. Mol. Sci. 2020, 21, 8907. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, I.; Steer, C.A.; Takei, F. Lung ILC2s link innate and adaptive responses in allergic inflammation. Trends Immunol. 2015, 36, 189–195. [Google Scholar] [CrossRef]
- Toki, S.; Goleniewska, K.; Zhang, J.; Zhou, W.; Newcomb, D.C.; Zhou, B.; Kita, H.; Boyd, K.L.; Peebles, R.S., Jr. TSLP and IL-33 reciprocally promote each other’s lung protein expression and ILC2 receptor expression to enhance innate type-2 airway inflammation. Allergy 2020, 75, 1606–1617. [Google Scholar] [CrossRef]
- Barnes, H.; Troy, L.; Lee, C.T.; Sperling, A.; Strek, M.; Glaspole, I. Hypersensitivity pneumonitis: Current concepts in pathogenesis, diagnosis, and treatment. Allergy 2021. [Google Scholar] [CrossRef]
- Girard, M.; Israel-Assayag, E.; Cormier, Y. Impaired function of regulatory T-cells in hypersensitivity pneumonitis. Eur. Respir. J. 2011, 37, 632–639. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G723–G728. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Kim, H.Y.; Chang, Y.J.; DeKruyff, R.H.; Umetsu, D.T. Innate lymphoid cells and asthma. J. Allergy Clin. Immunol. 2014, 133, 943–950. [Google Scholar] [CrossRef]
- Halim, T.Y.; Krauss, R.H.; Sun, A.C.; Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Bartemes, K.R.; Iijima, K.; Kobayashi, T.; Kephart, G.M.; McKenzie, A.N.; Kita, H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J. Immunol. 2012, 188, 1503–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.N.; Guo, Y.B.; Li, X.; Li, C.L.; Tan, W.P.; Fan, X.L.; Qin, Z.L.; Chen, D.; Wen, W.P.; Zheng, S.G.; et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy 2018, 73, 1860–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, T.; Pesic, J.; Pardali, K.; Gaestel, M.; Arthur, J.S.C. p38 MAPK signalling regulates cytokine production in IL-33 stimulated Type 2 Innate Lymphoid cells. Sci. Rep. 2020, 10, 3479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, B.; Martinez, F.D.; Halonen, M.; Barbee, R.A.; Cline, M.G. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N. Engl. J. Med. 1989, 320, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Manabe, T.; Niki, Y.; Matsushima, T. Summer-type hypersensitivity pneumonitis in a patient with a positive skin test (15 minutes) for Trichosporon mucoides and a high serum IgE level. Nihon Kyobu Shikkan Gakkai Zasshi 1996, 34, 1168–1173. [Google Scholar] [PubMed]
- Siraganian, R.P. Mast cell signal transduction from the high-affinity IgE receptor. Curr. Opin. Immunol. 2003, 15, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Nakae, S.; Suto, H.; Iikura, M.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast cells enhance T cell activation: Importance of mast cell costimulatory molecules and secreted TNF. J. Immunol. 2006, 176, 2238–2248. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.C. Immunoglobulin E receptor signaling and asthma. J. Biol. Chem. 2011, 286, 32891–32897. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Kurashima, Y. Two Sides of the Coin: Mast Cells as a Key Regulator of Allergy and Acute/Chronic Inflammation. Cells 2021, 10, 1615. [Google Scholar] [CrossRef]
- Rivera, J.; Fierro, N.A.; Olivera, A.; Suzuki, R. New insights on mast cell activation via the high affinity receptor for IgE. Adv. Immunol. 2008, 98, 85–120. [Google Scholar] [CrossRef] [Green Version]
- Huber, M.; Helgason, C.D.; Damen, J.E.; Liu, L.; Humphries, R.K.; Krystal, G. The src homology 2-containing inositol phosphatase (SHIP) is the gatekeeper of mast cell degranulation. Proc. Natl. Acad. Sci. USA 1998, 95, 11330–11335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, J.T. Basophils beyond effector cells of allergic inflammation. Adv. Immunol. 2009, 101, 123–161. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Sharma, J.; Raju, R.; Palapetta, S.M.; Prasad, T.S.; Huang, T.C.; Yoda, A.; Tyner, J.W.; van Bodegom, D.; Weinstock, D.M.; et al. TSLP signaling pathway map: A platform for analysis of TSLP-mediated signaling. Database 2014, 2014, bau007. [Google Scholar] [CrossRef]
- Markovic, I.; Savvides, S.N. Modulation of Signaling Mediated by TSLP and IL-7 in Inflammation, Autoimmune Diseases, and Cancer. Front. Immunol. 2020, 11, 1557. [Google Scholar] [CrossRef]
- Nakajima, H.; Iwamoto, I.; Tomoe, S.; Matsumura, R.; Tomioka, H.; Takatsu, K.; Yoshida, S. CD4+ T-lymphocytes and interleukin-5 mediate antigen-induced eosinophil infiltration into the mouse trachea. Am. Rev. Respir. Dis. 1992, 146, 374–377. [Google Scholar] [CrossRef]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Deindl, S.; Kadlecek, T.A.; Brdicka, T.; Cao, X.; Weiss, A.; Kuriyan, J. Structural basis for the inhibition of tyrosine kinase activity of ZAP-70. Cell 2007, 129, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Müller-Redetzky, H.; Lienau, J.; Suttorp, N.; Witzenrath, M. Therapeutic strategies in pneumonia: Going beyond antibiotics. Eur. Respir. Rev. 2015, 24, 516–524. [Google Scholar] [CrossRef]
- Blum, C.A.; Nigro, N.; Briel, M.; Schuetz, P.; Ullmer, E.; Suter-Widmer, I.; Winzeler, B.; Bingisser, R.; Elsaesser, H.; Drozdov, D.; et al. Adjunct prednisone therapy for patients with community-acquired pneumonia: A multicentre, double-blind, randomised, placebo-controlled trial. Lancet 2015, 385, 1511–1518. [Google Scholar] [CrossRef]
- Pliakos, E.E.; Andreatos, N.; Tansarli, G.S.; Ziakas, P.D.; Mylonakis, E. The Cost-Effectiveness of Corticosteroids for the Treatment of Community-Acquired Pneumonia. Chest 2019, 155, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Remmelts, H.H.; Meijvis, S.C.; Biesma, D.H.; van Velzen-Blad, H.; Voorn, G.P.; Grutters, J.C.; Bos, W.J.; Rijkers, G.T. Dexamethasone downregulates the systemic cytokine response in patients with community-acquired pneumonia. Clin. Vaccine Immunol. 2012, 19, 1532–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihailidou, I.; Pelekanou, A.; Pistiki, A.; Spyridaki, A.; Tzepi, I.M.; Damoraki, G.; Giamarellos-Bourboulis, E.J. Dexamethasone Down-Regulates Expression of Triggering Receptor Expressed on Myeloid Cells-1: Evidence for a TNFα-Related Effect. Front. Public Health 2013, 1, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, J.D.; Short, P.M.; Mandal, P.; Akram, A.R.; Hill, A.T. Statins in community acquired pneumonia: Evidence from experimental and clinical studies. Respir. Med. 2010, 104, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parihar, S.P.; Guler, R.; Brombacher, F. Statins: A viable candidate for host-directed therapy against infectious diseases. Nat. Rev. Immunol. 2019, 19, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Lukan, N. “Cytokine storm”, not only in COVID-19 patients. Mini-review. Immunol. Lett. 2020, 228, 38–44. [Google Scholar] [CrossRef]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.W.; Cinatl, J. N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem. Pharm. 2010, 79, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Kanoh, S.; Rubin, B.K. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin. Microbiol. Rev. 2010, 23, 590–615. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowska, B.; Maślińska, M. Macrolide therapy in chronic inflammatory diseases. Mediat. Inflamm. 2012, 2012, 636157. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Dong, C. Therapeutic antibodies that target inflammatory cytokines in autoimmune diseases. Int. Immunol. 2016, 28, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Tauber, P.A.; Pickl, W.F. Pharmacological targeting of allergen-specific T lymphocytes. Immunol. Lett. 2017, 189, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Therapeutic strategies for allergic diseases. Nature 1999, 402, B31–B38. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Botran, R.; Crespo, F.A.; Sun, X. Soluble cytokine receptors in biological therapy. Expert Opin. Biol. 2002, 2, 585–605. [Google Scholar] [CrossRef] [PubMed]
- Borish, L.C.; Nelson, H.S.; Lanz, M.J.; Claussen, L.; Whitmore, J.B.; Agosti, J.M.; Garrison, L. Interleukin-4 receptor in moderate atopic asthma. A phase I/II randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 1999, 160, 1816–1823. [Google Scholar] [CrossRef]
- Borish, L.C.; Nelson, H.S.; Corren, J.; Bensch, G.; Busse, W.W.; Whitmore, J.B.; Agosti, J.M.; Group, I.-R.A.S. Efficacy of soluble IL-4 receptor for the treatment of adults with asthma. J. Allergy Clin. Immunol. 2001, 107, 963–970. [Google Scholar] [CrossRef]
- Corren, J.; Busse, W.; Meltzer, E.O.; Mansfield, L.; Bensch, G.; Fahrenholz, J.; Wenzel, S.E.; Chon, Y.; Dunn, M.; Weng, H.H.; et al. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am. J. Respir. Crit. Care Med. 2010, 181, 788–796. [Google Scholar] [CrossRef]
- Nagase, H.; Ueki, S.; Fujieda, S. The roles of IL-5 and anti-IL-5 treatment in eosinophilic diseases: Asthma, eosinophilic granulomatosis with polyangiitis, and eosinophilic chronic rhinosinusitis. Allergol. Int. 2020, 69, 178–186. [Google Scholar] [CrossRef]
- Kips, J.C.; O’Connor, B.J.; Langley, S.J.; Woodcock, A.; Kerstjens, H.A.; Postma, D.S.; Danzig, M.; Cuss, F.; Pauwels, R.A. Effect of SCH55700, a humanized anti-human interleuki.in-5 antibody, in severe persistent asthma: A pilot study. Am. J. Respir. Crit. Care Med. 2003, 167, 1655–1659. [Google Scholar] [CrossRef]
- Flood-Page, P.T.; Menzies-Gow, A.N.; Kay, A.B.; Robinson, D.S. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am. J. Respir. Crit. Care Med. 2003, 167, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Kane, B.; Molfino, N.A.; Faggioni, R.; Roskos, L.; Woodcock, A. A phase 1 study evaluating the pharmacokinetics, safety and tolerability of repeat dosing with a human IL-13 antibody (CAT-354) in subjects with asthma. BMC Pulm. Med. 2010, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Alvaro-Lozano, M.; Akdis, C.A.; Akdis, M.; Alviani, C.; Angier, E.; Arasi, S.; Arzt-Gradwohl, L.; Barber, D.; Bazire, R.; Cavkaytar, O.; et al. EAACI Allergen Immunotherapy User’s Guide. Pediatr. Allergy Immunol. 2020, 31 (Suppl. S25), 1–101. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, M.; Akitsu, K.; Kubota, K. Effect of Sublingual Immunotherapy on Airway Inflammation and Airway Wall Thickness in Allergic Asthma. J. Allergy Clin. Immunol. Pr. 2019, 7, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Xian, M.; Feng, M.; Dong, Y.; Wei, N.; Su, Q.; Li, J. Changes in CD4+CD25+FoxP3+ Regulatory T Cells and Serum Cytokines in Sublingual and Subcutaneous Immunotherapy in Allergic Rhinitis with or without Asthma. Int. Arch. Allergy Immunol. 2020, 181, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Skuljec, J.; Chmielewski, M.; Happle, C.; Habener, A.; Busse, M.; Abken, H.; Hansen, G. Chimeric Antigen Receptor-Redirected Regulatory T Cells Suppress Experimental Allergic Airway Inflammation, a Model of Asthma. Front. Immunol. 2017, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Mirershadi, F.; Ahmadi, M.; Rezabakhsh, A.; Rajabi, H.; Rahbarghazi, R.; Keyhanmanesh, R. Unraveling the therapeutic effects of mesenchymal stem cells in asthma. Stem Cell Res. 2020, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, E.O.; Berkowitz, R.B.; Grossbard, E.B. An intranasal Syk-kinase inhibitor (R112) improves the symptoms of seasonal allergic rhinitis in a park environment. J. Allergy Clin. Immunol. 2005, 115, 791–796. [Google Scholar] [CrossRef] [PubMed]
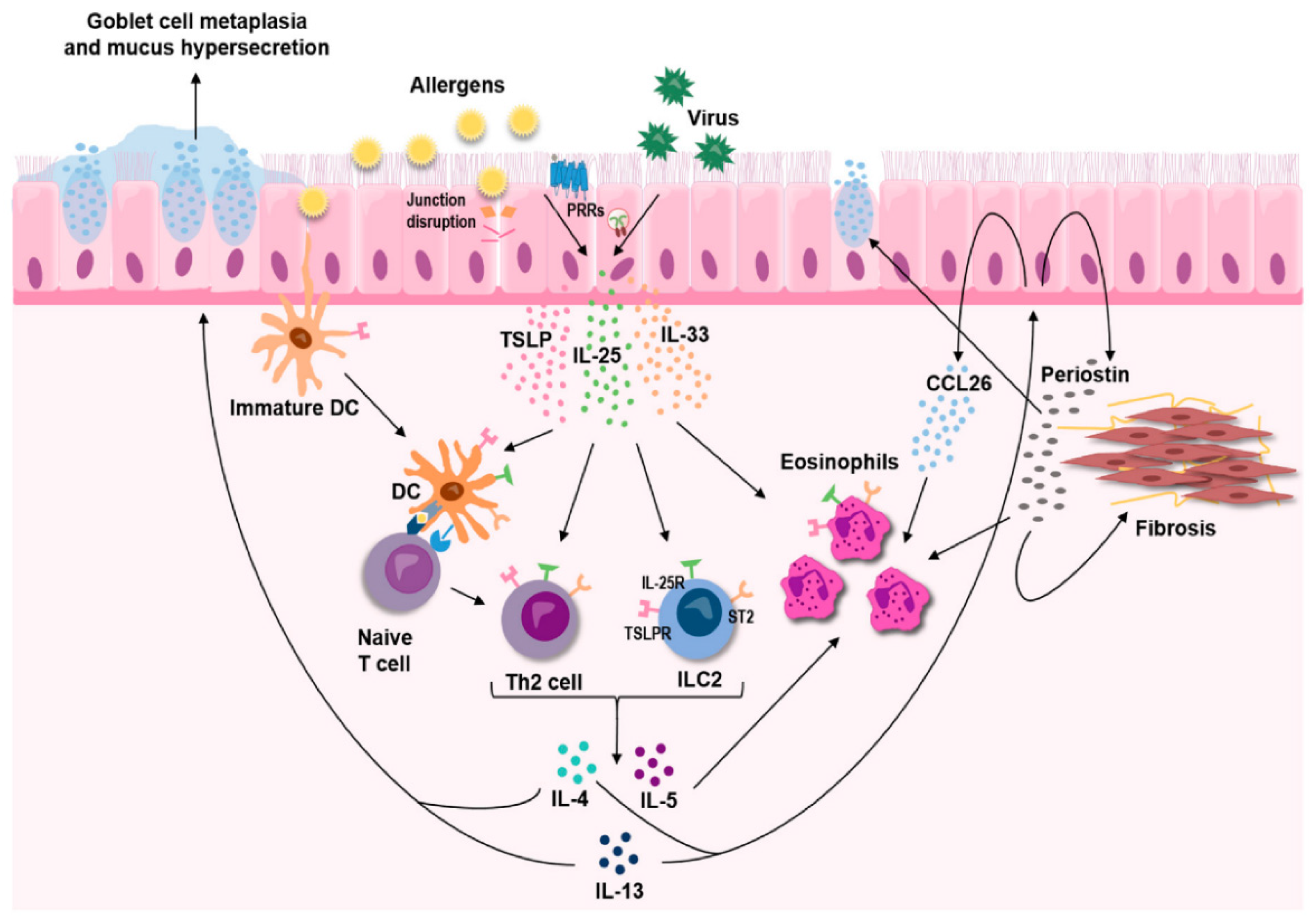
Figure 1.
In response to allergens, thymic stromal lymphopoietin (TSLP), interleukin-33 (IL-33), and interleukin-25 (IL-25) cytokines are released that mediate type 2 inflammation in the airways through effects on both innate and adaptive immune cells. Figure adapted from Calven et al. [65].
Figure 1.
In response to allergens, thymic stromal lymphopoietin (TSLP), interleukin-33 (IL-33), and interleukin-25 (IL-25) cytokines are released that mediate type 2 inflammation in the airways through effects on both innate and adaptive immune cells. Figure adapted from Calven et al. [65].

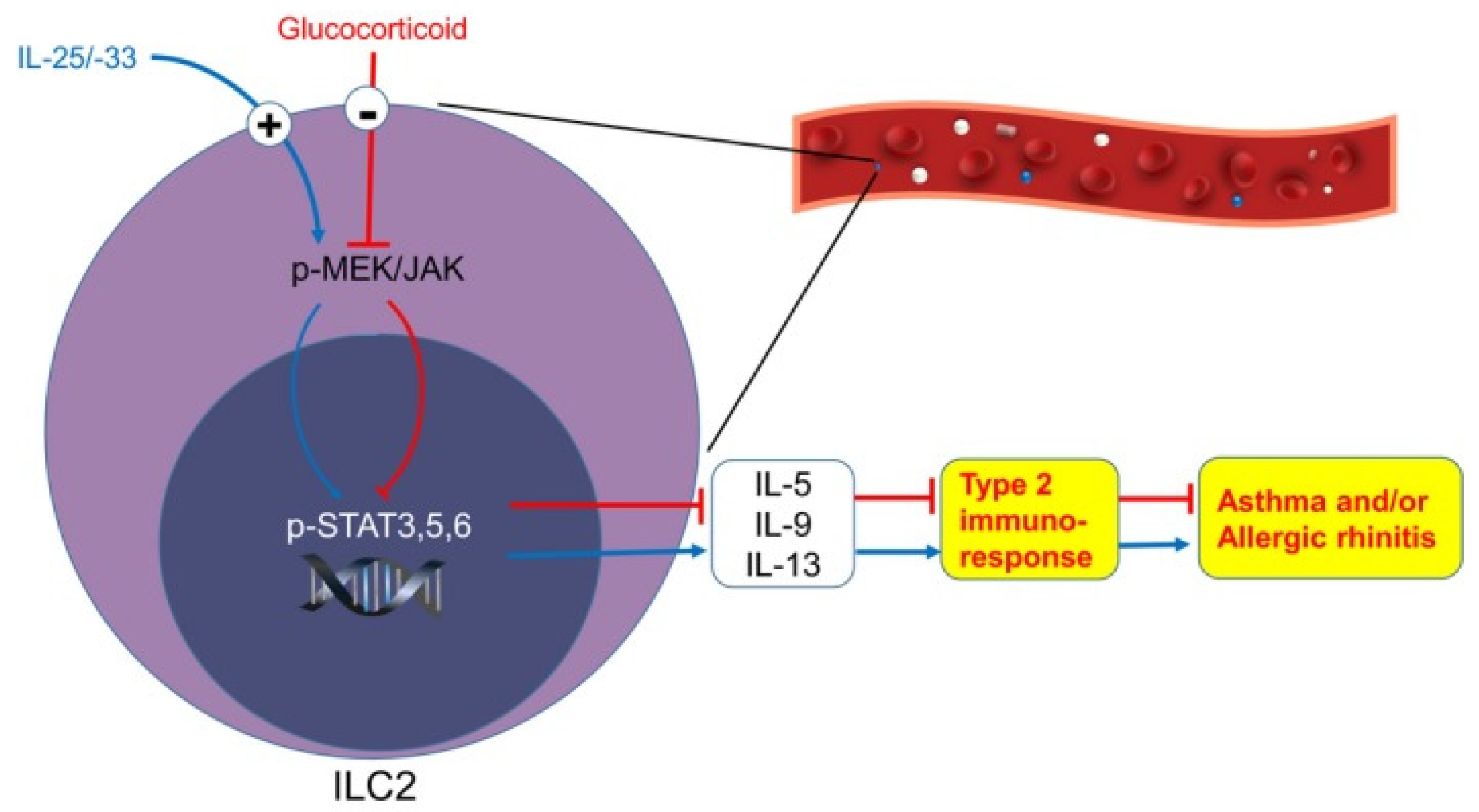
Figure 2.
High levels of IL-5 and IL-13 produced by ILC2s via STAT3, STAT5, and STAT6 signaling pathways. Image of inflammatory signaling pathway associated with ILC2 activation adapted from Yu et al. [74].
Figure 2.
High levels of IL-5 and IL-13 produced by ILC2s via STAT3, STAT5, and STAT6 signaling pathways. Image of inflammatory signaling pathway associated with ILC2 activation adapted from Yu et al. [74].

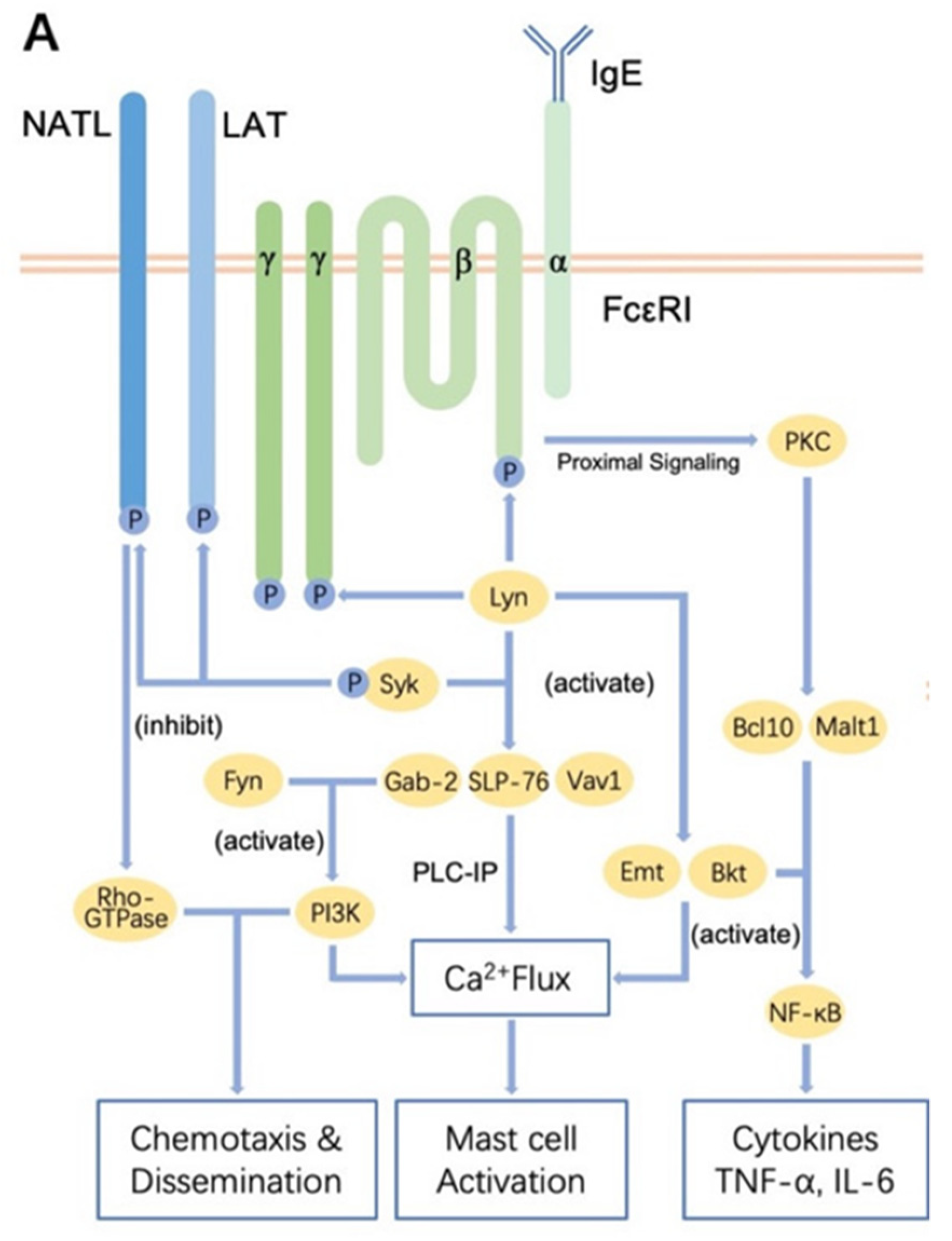
Figure 3.
Regulation of receptor-mediated mast cell activation. IgE-FcεRI is the most classical mechanism of mast cell activation, which mainly relies on the Lyn-Syk and Fyn pathways to activate mast cells. Image of mast cell activation adapted and modified from Zhang, et al. [81].
Figure 3.
Regulation of receptor-mediated mast cell activation. IgE-FcεRI is the most classical mechanism of mast cell activation, which mainly relies on the Lyn-Syk and Fyn pathways to activate mast cells. Image of mast cell activation adapted and modified from Zhang, et al. [81].


{kind=link}
{kind=link}
{kind=link}
Table 1.
Infectious agents-derived PAMPs and their PRRs.
| Infectious Agent | PAMPs | PRR |
|---|---|---|
| Bacteria | LPS | TLR4 |
| Lipoproteins, peptidoglycan | TLR2,6 | |
| Flagellin | TLR5 | |
| Bacterial DNA (CpG) | TLR9 | |
| Muramyl dipeptide (MDP) | NOD2 | |
| Diaminopimelic acid | NOD1 | |
| Viruses | Haemagglutinin (HA) | TLR2 |
| DsRNA | TLR3 | |
| Short dsRNA | RIG-1 | |
| Long dsRNA | MDA-5 | |
| ssRNA | TLR7/8 | |
| dsDNA | TLR9, NLRP3 | |
| Fungi | β-Glucan | TLR2, Dectin-1 |
| phospholipomannon | TLR2 | |
| Mannose | Mannose receptors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Upadhyay, M.; Nehme, A.; Wasnik, S. Title-Inflammatory Signaling Pathways in Allergic and Infection-Associated Lung Diseases. Allergies 2022, 2, 57-74. https://doi.org/10.3390/allergies2020006
AMA Style
Upadhyay M, Nehme A, Wasnik S. Title-Inflammatory Signaling Pathways in Allergic and Infection-Associated Lung Diseases. Allergies. 2022; 2(2):57-74. https://doi.org/10.3390/allergies2020006
Chicago/Turabian StyleUpadhyay, Mala, Antoine Nehme, and Samiksha Wasnik. 2022. "Title-Inflammatory Signaling Pathways in Allergic and Infection-Associated Lung Diseases" Allergies 2, no. 2: 57-74. https://doi.org/10.3390/allergies2020006