
Density Functional Theory Study of Oxygen Evolution Reaction Mechanism on Rare Earth Sc-Doped Graphene
 ,
,  and
and
Abstract
:1. Introduction
2. Computational Details
2.1. Methods
2.2. Models
2.3. Computational Contents
- (1)
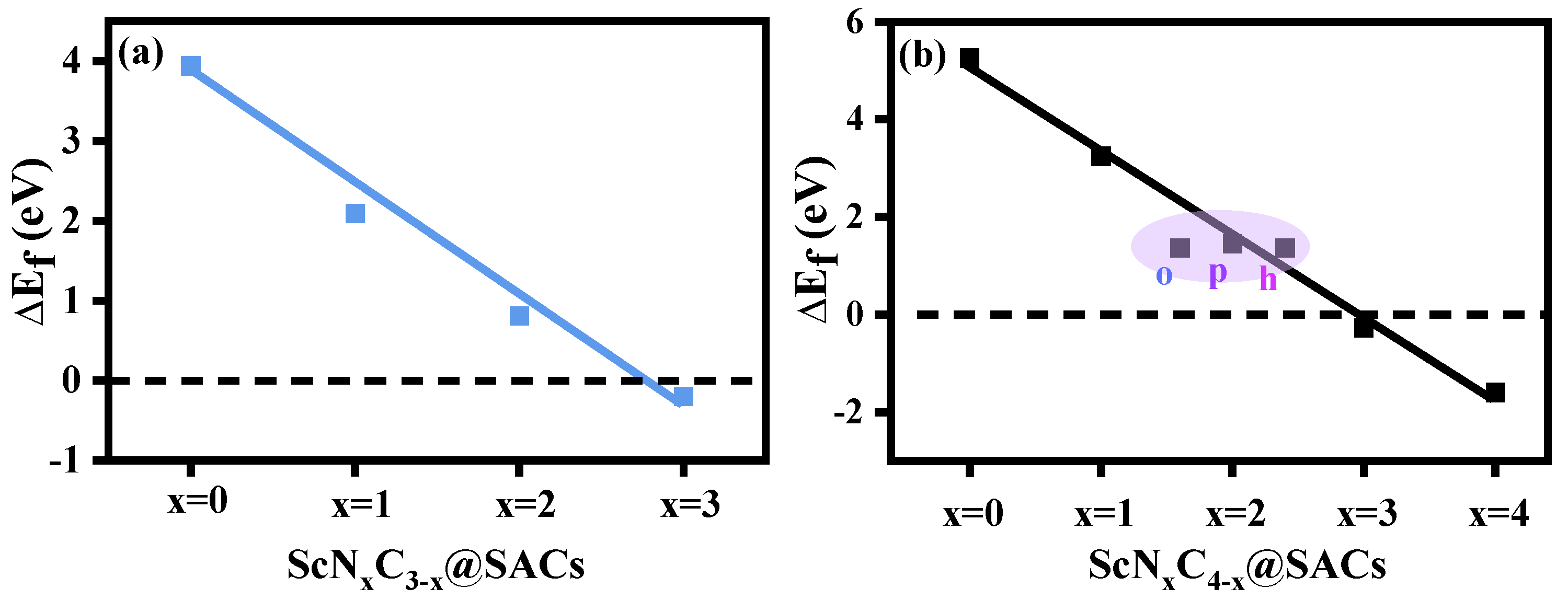
- Formation energy ()
- (2)
- Adsorption energy ()
- (3)
- Gibbs free energy variation ()
- (4)
- Reaction steps of the OER in an alkaline environmentOH + *→OH* + eOH* + OH→O* + H2O (l) + eO* + OH→OOH* + eOOH* + OH→O2 (g) + * + H2O + e
3. Results and Discussion
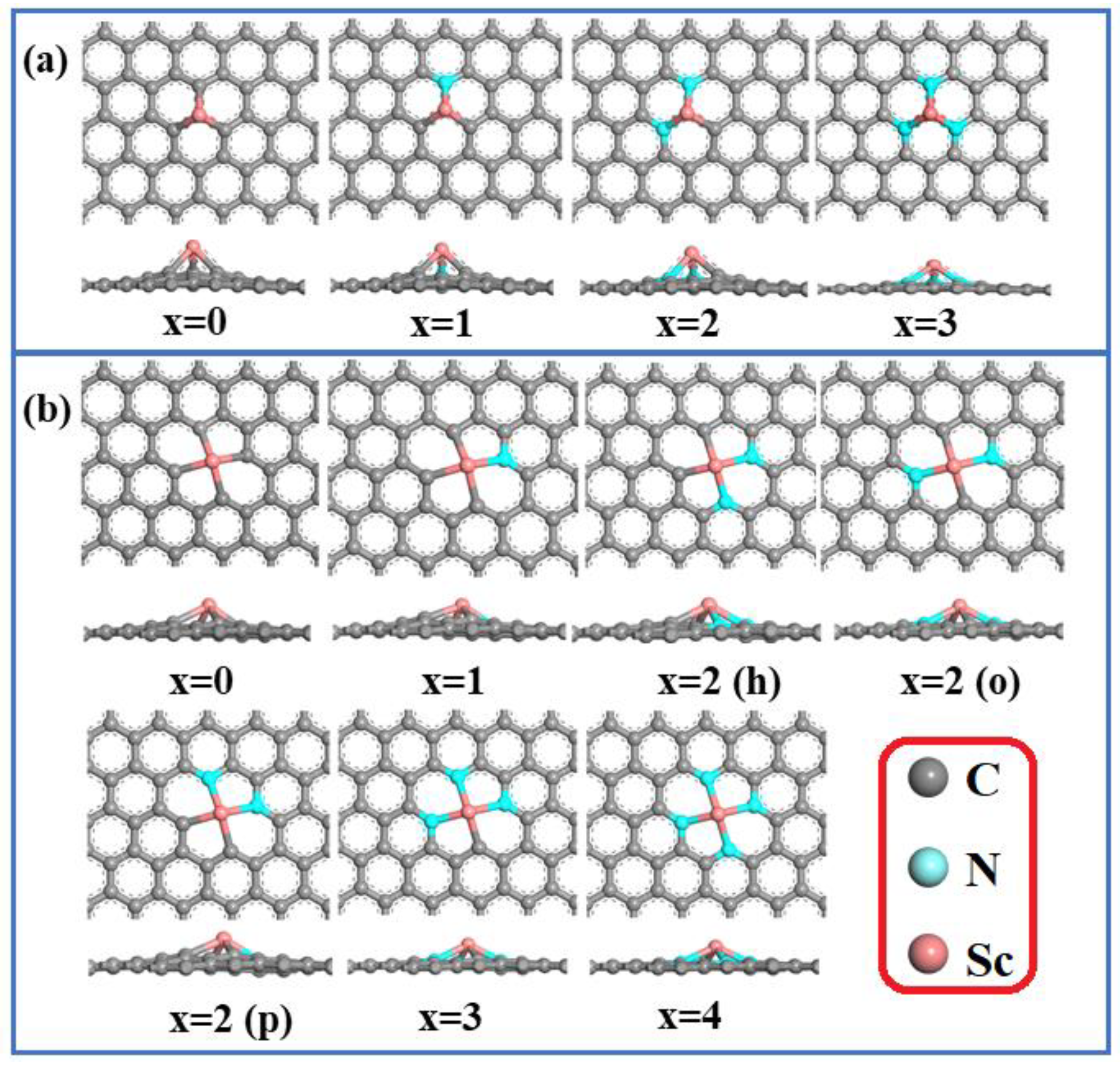
3.1. Stability of Sc and N Co-Doped Graphene
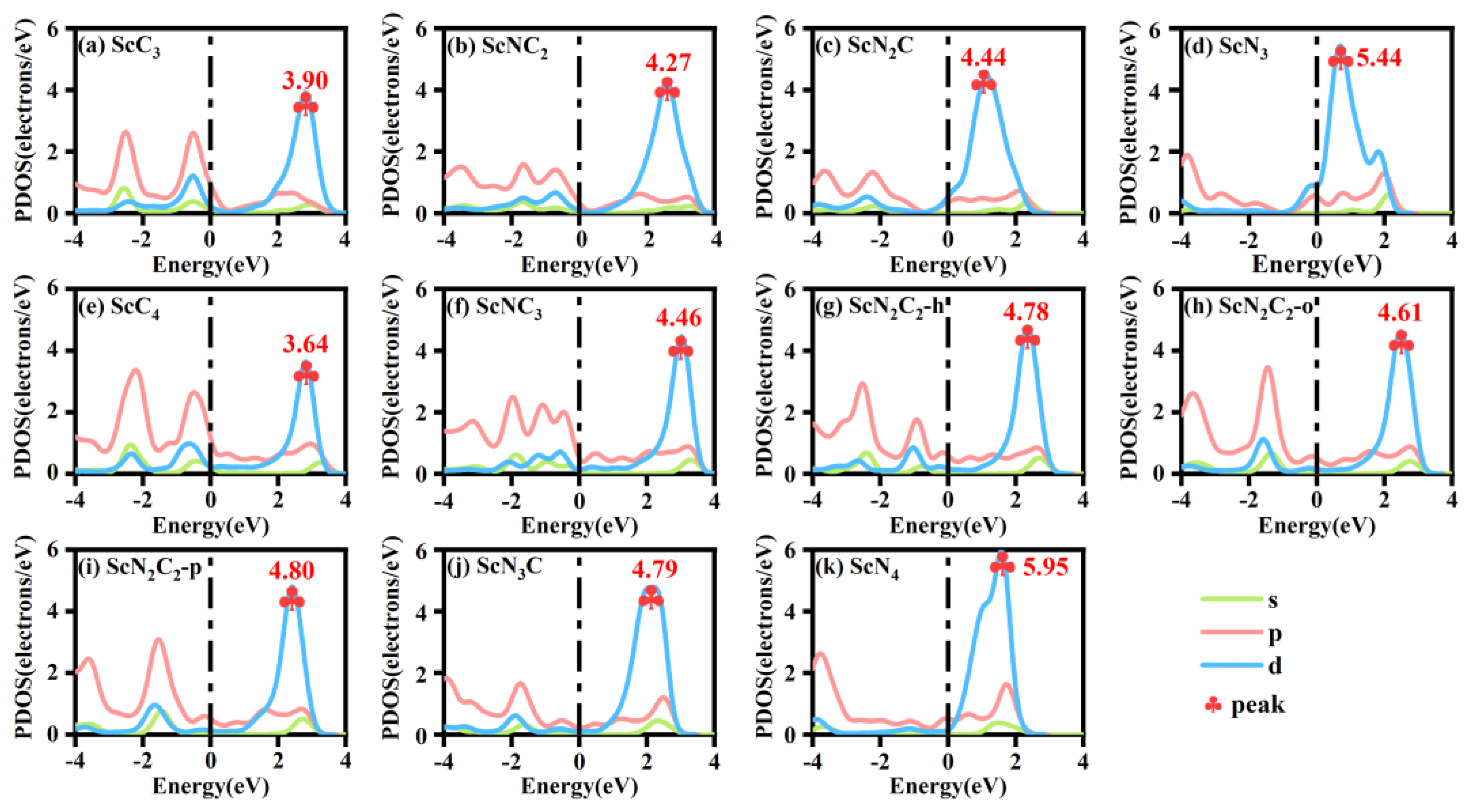
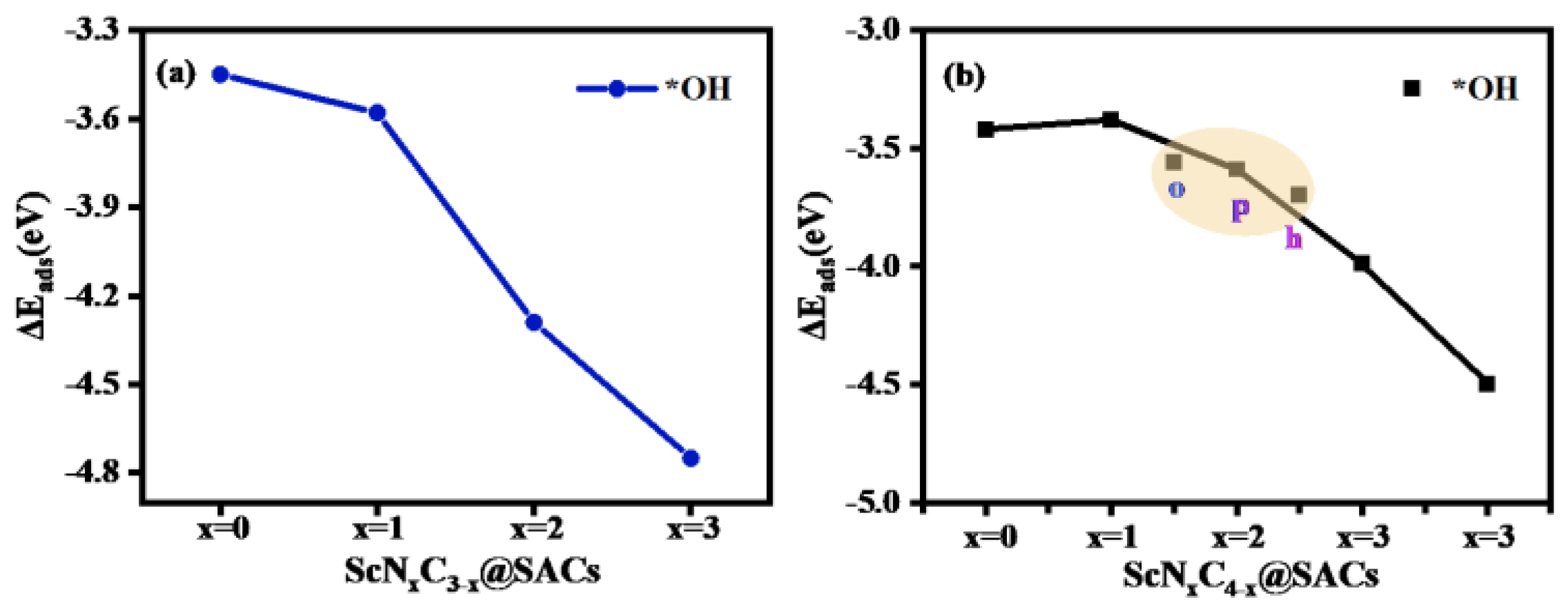
3.2. Adsorption Properties of the Intermediates
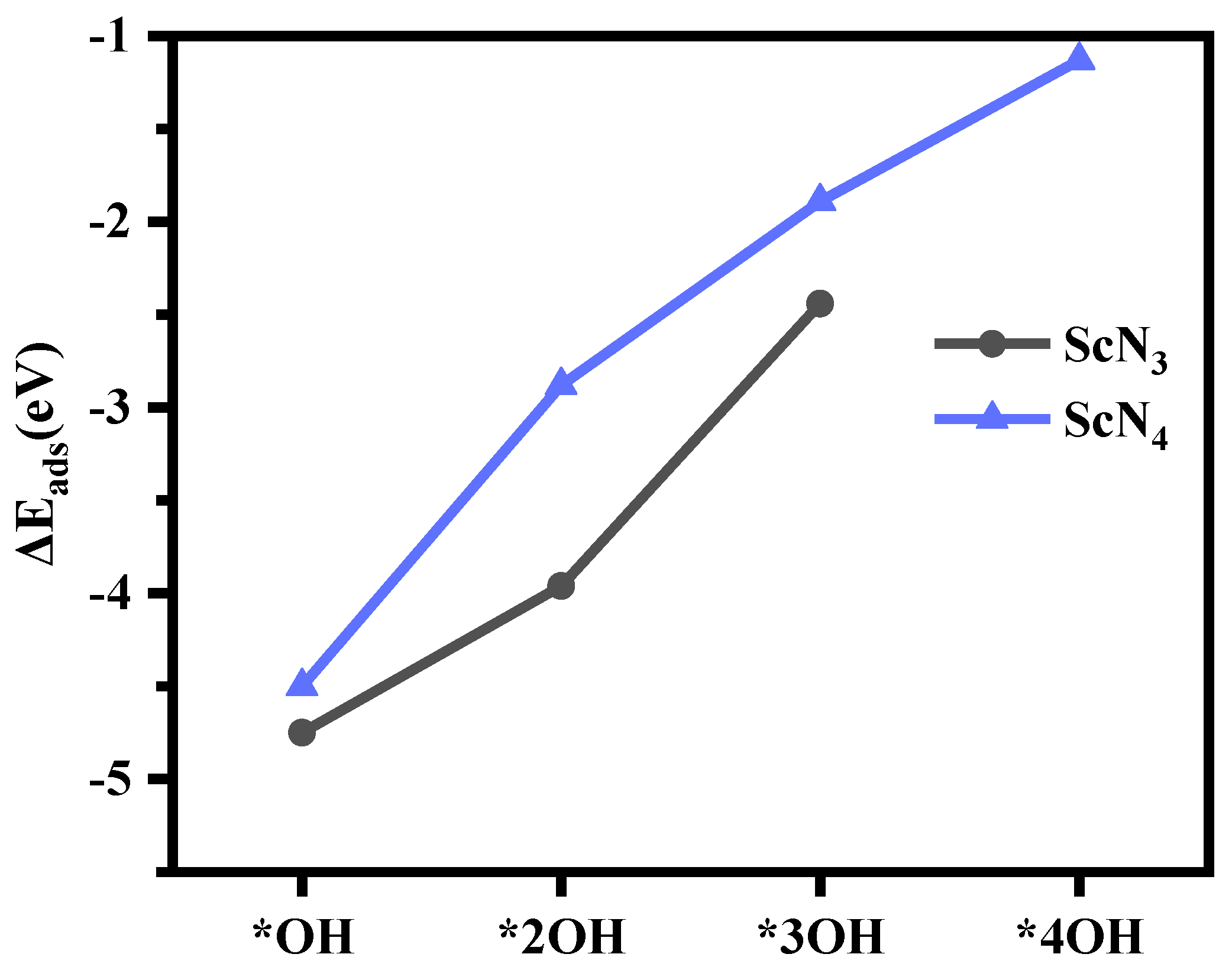
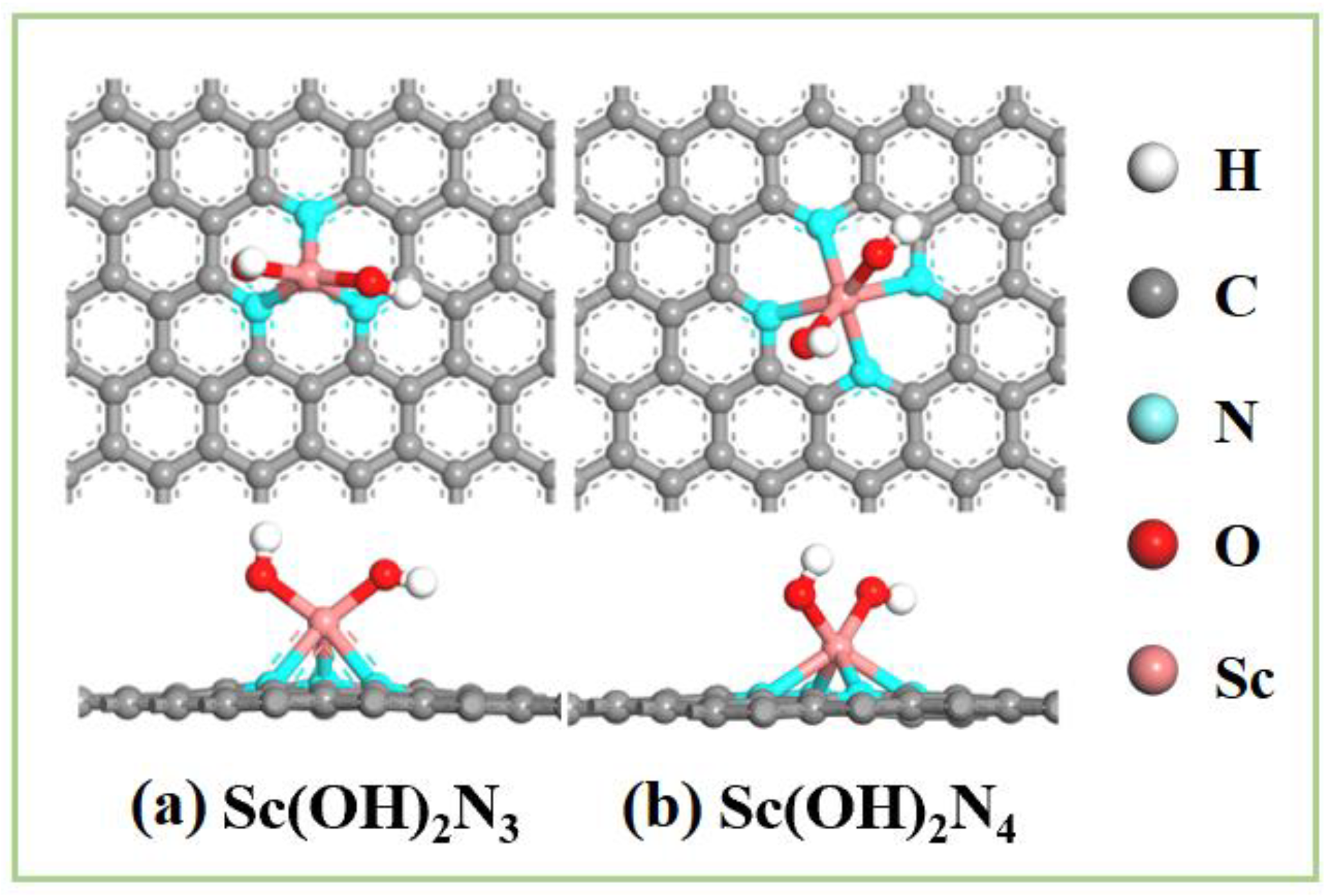
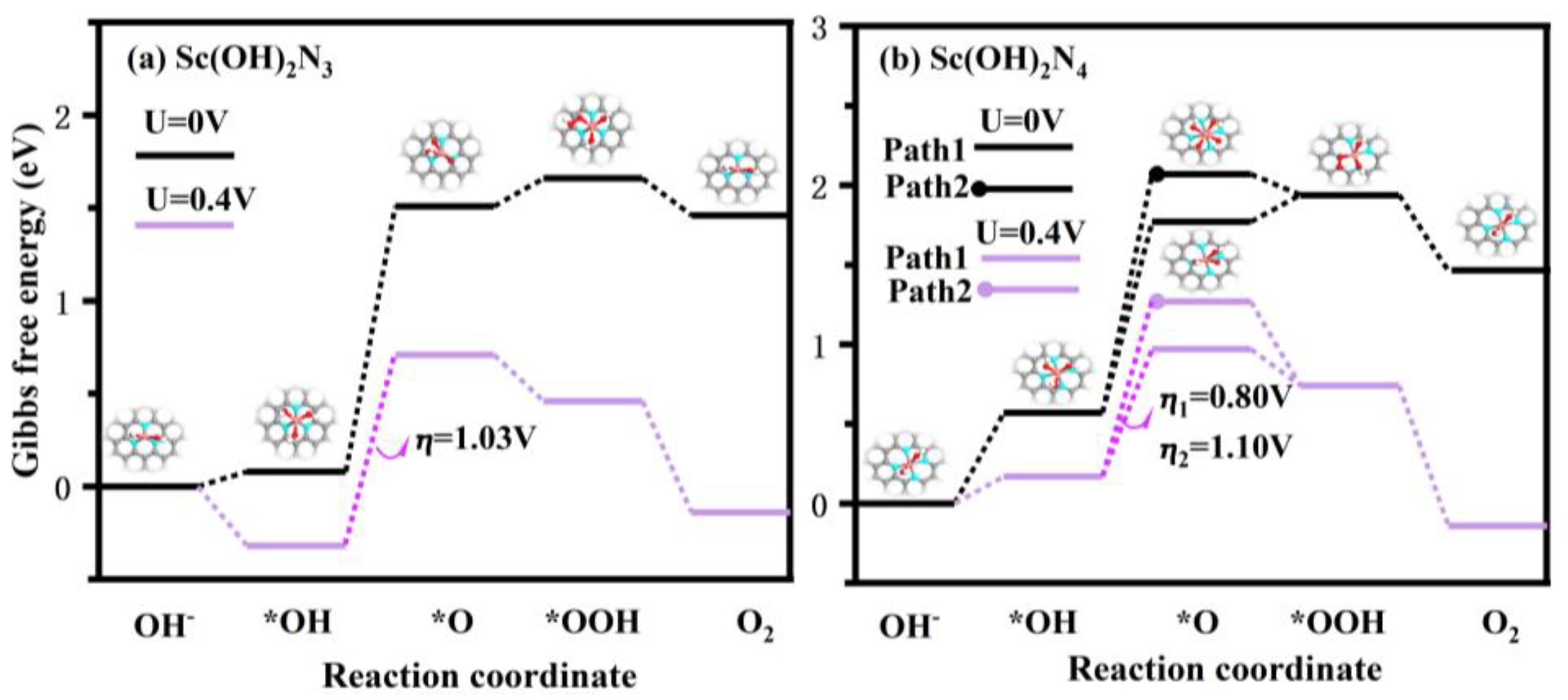
3.3. The Catalytic Activity of Sc(OH)2N3 and Sc(OH)2N4
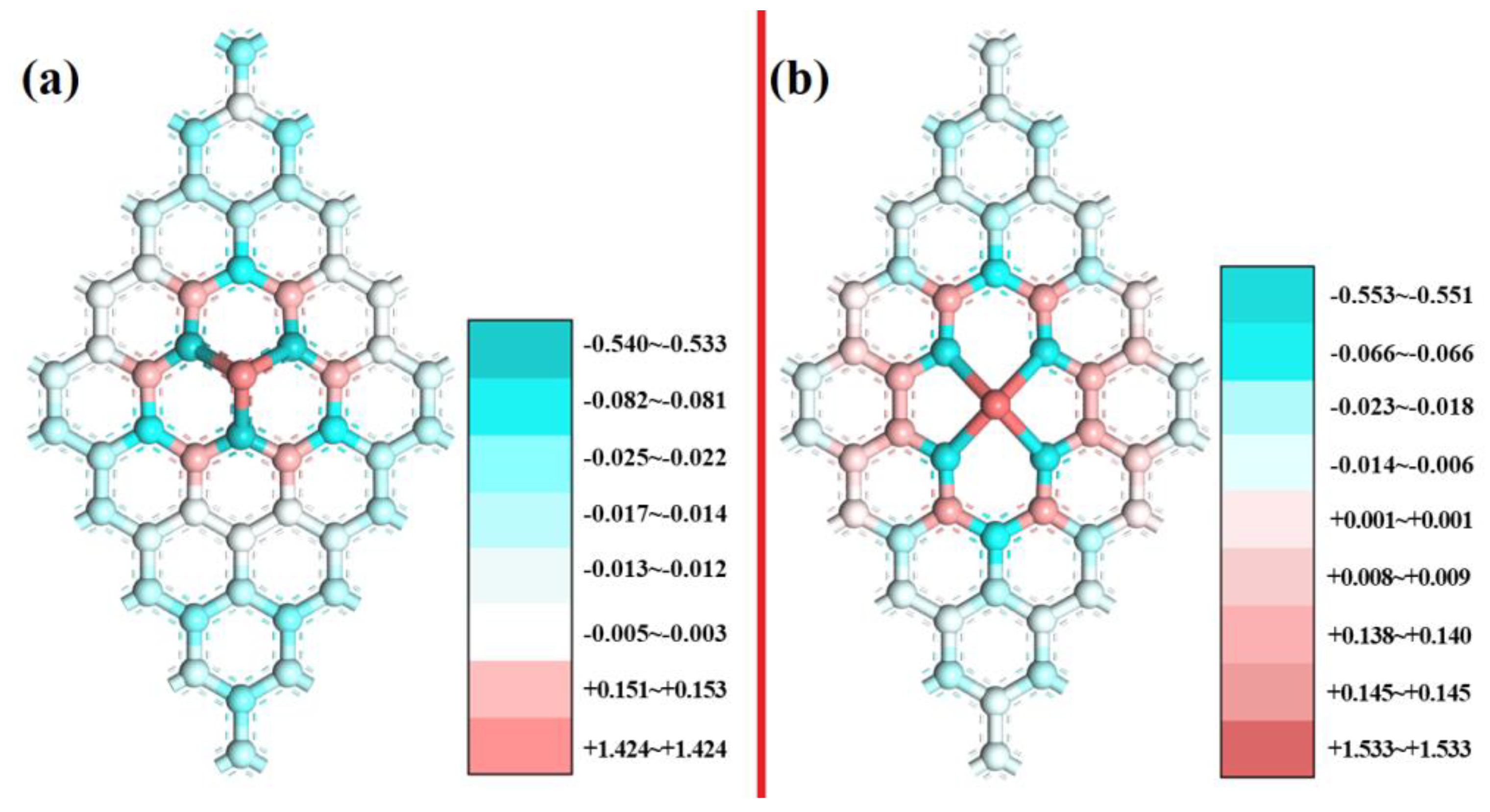
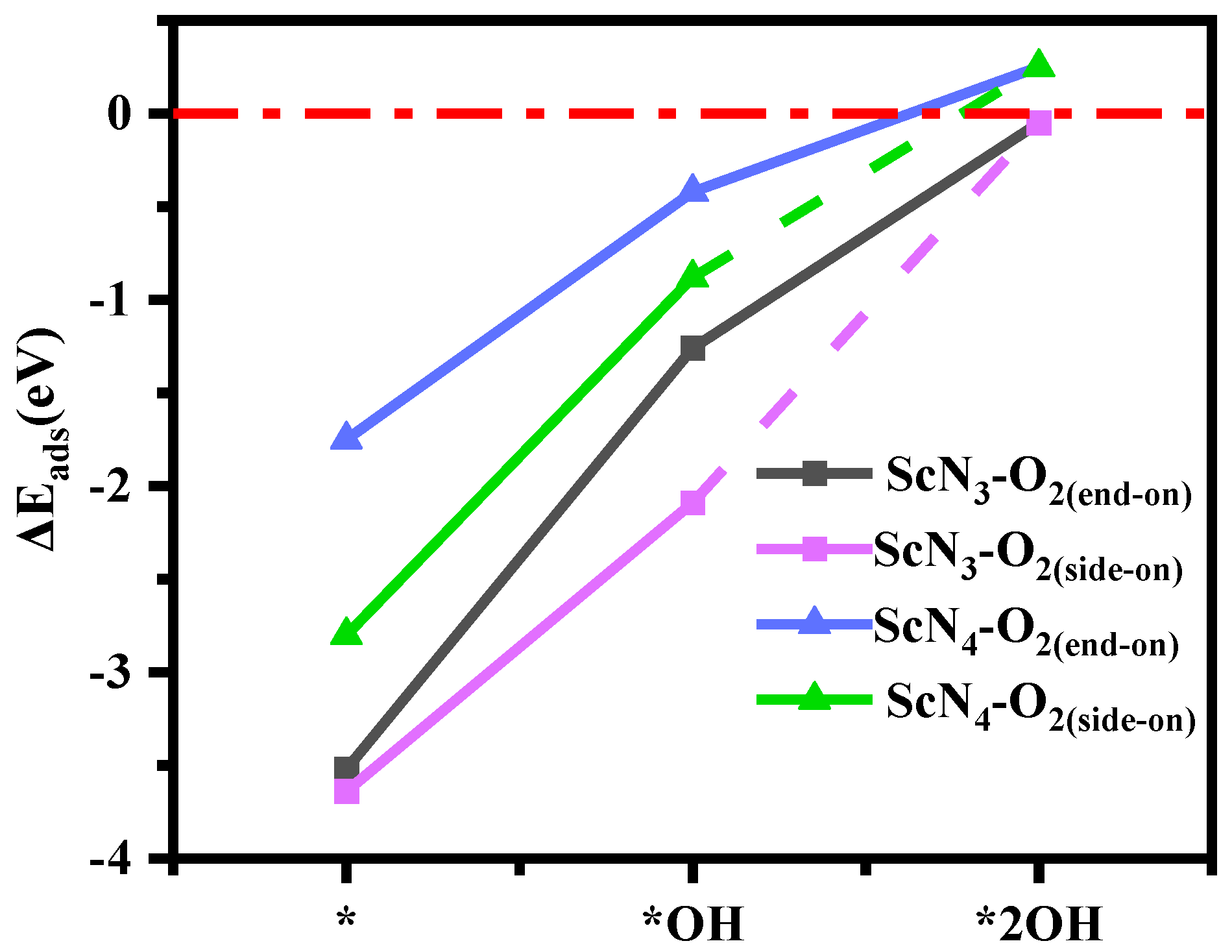
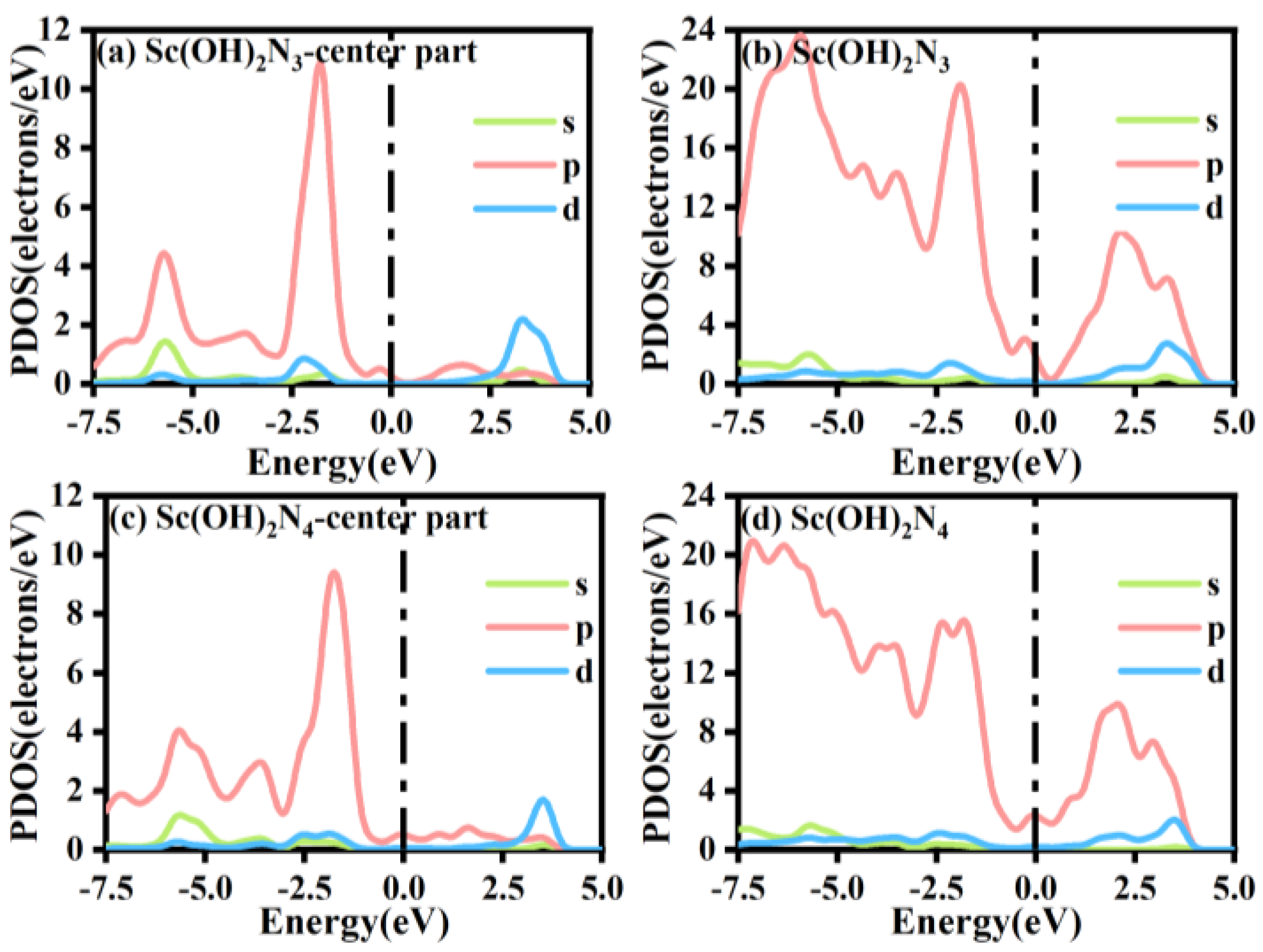
3.4. The Origin of Catalytic Activity of Sc(OH)2N3 and Sc(OH)2N4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Güney, T. Renewable energy consumption and sustainable development in high-income countries. Int. J. Sustain. Dev. World Ecol. 2021, 28, 376–385. [Google Scholar] [CrossRef]
- Güney, T. Renewable energy, non-renewable energy and sustainable development. Int. J. Sustain. Dev. World Ecol. 2019, 26, 389–397. [Google Scholar] [CrossRef]
- Hao, J.; Yuan, L.; Zhu, Y.; Jaroniec, M.; Qiao, S. Triple-Function Electrolyte Regulation toward Advanced Aqueous Zn-Ion Batteries. Adv. Mater. 2022, 34, 2206963. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.Z.; Rehman, A.-U.; Siddique, S. Prospects and challenges of graphene based fuel cells. J. Energy Chem. 2019, 39, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xia, Z.; Dai, L. Carbon-based electrocatalysts for advanced energy conversion and storage. Sci. Adv. 2015, 1, e1500564. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wang, X.; Li, Z.; Yang, B.; Ling, M.; Gao, X.; Lu, J.; Shi, Q.; Lei, L.; Wu, G.; et al. Designing 3d dual transition metal electrocatalysts for oxygen evolution reaction in alkaline electrolyte: Beyond oxides. Nano Energy 2020, 77, 105162. [Google Scholar] [CrossRef]
- Dang, N.K.; Tiwari, J.N.; Sultan, S.; Meena, A.; Kim, K.S. Multi-site catalyst derived from Cr atoms-substituted CoFe nanoparticles for high-performance oxygen evolution activity. Chem. Eng. J. 2021, 404, 126513. [Google Scholar] [CrossRef]
- Zheng, X.; Yang, J.; Xu, Z.; Wang, Q.; Wu, J.; Zhang, E.; Dou, S.; Sun, W.; Wang, D.; Li, Y. Ru–Co Pair Sites Catalyst Boosts the Energetics for the Oxygen Evolution Reaction. Angew. Chem. Int. Ed. 2022, 61, e202205946. [Google Scholar] [CrossRef]
- An, L.; Wei, C.; Lu, M.; Liu, H.; Chen, Y.; Scherer, G.G.; Fisher, A.C.; Xi, P.; Xu, Z.J.; Yan, C.H. Recent Development of Oxygen Evolution Electrocatalysts in Acidic Environment. Adv. Mater. 2021, 33, e2006328. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, Y.; Wang, Y.; Zhang, H.-N.; Zhu, L.-H.; Wang, X.-C. Electronic properties of double-atom catalysts for electrocatalytic oxygen evolution reaction in alkaline solution: A DFT study. Nanoscale 2022, 14, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Long, X.; Xiao, J.; Zhang, Z.; Liu, G.; Tong, H.; Liu, Z.; Li, N.; Qian, D.; Li, J.; et al. Rationally constructing CoO and CoSe2 hybrid with CNTs-graphene for impressively enhanced oxygen evolution and DFT calculations. Chem. Eng. J. 2021, 422, 129982. [Google Scholar] [CrossRef]
- Charles, V.; Anumah, A.O.; Adegoke, K.A.; Adesina, M.O.; Ebuka, I.P.; Gaya, N.A.; Ogwuche, S.; Yakubu, M.O. Progress and challenges pertaining to the earthly-abundant electrocatalytic materials for oxygen evolution reaction. Sustain. Mater. Technol. 2021, 28, e00252. [Google Scholar] [CrossRef]
- Ji, Y.; Dong, H.; Liu, C.; Li, Y. The progress of metal-free catalysts for the oxygen reduction reaction based on theoretical simulations. J. Mater. Chem. A 2018, 6, 13489–13508. [Google Scholar] [CrossRef]
- Suen, N.-T.; Hung, S.-F.; Quan, Q.; Zhang, N.; Xu, Y.-J.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, X.F.; Zang, S.; Lou, X.W. Non-Noble-Metal-Based Electrocatalysts toward the Oxygen Evolution Reaction. Adv. Funct. Mater. 2020, 30, 1910274. [Google Scholar] [CrossRef]
- Sun, W.; Zhou, Z.; Zaman, W.Q.; Cao, L.-M.; Yang, J. Rational Manipulation of IrO2 Lattice Strain on α-MnO2 Nanorods as a Highly Efficient Water-Splitting Catalyst. ACS Appl. Mater. Interfaces 2017, 9, 41855–41862. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef]
- Chen, Y.; Rui, K.; Zhu, J.; Dou, S.X.; Sun, W. Recent Progress on Nickel-Based Oxide/(Oxy)Hydroxide Electrocatalysts for the Oxygen Evolution Reaction. Chem. Eur. J. 2019, 25, 703–713. [Google Scholar] [CrossRef]
- Ďurovič, M.; Hnát, J.; Bouzek, K. Electrocatalysts for the hydrogen evolution reaction in alkaline and neutral media. A comparative review. J. Power Sources 2021, 493, 229708. [Google Scholar] [CrossRef]
- Wang, C.; Tong, H.; Lu, J.; Liu, B.; Zheng, F.; Tao, W.; Zhang, W.; Chen, Q. Boosting oxygen evolution reaction on graphene through engineering electronic structure. Carbon 2020, 170, 414–420. [Google Scholar] [CrossRef]
- Sadeghi, S.; Amani, M. Co-doped triel–pnicogen graphene as metal-free catalyst for CO oxidation: Role of multi-center covalency. J. Mol. Model. 2019, 25, 77. [Google Scholar] [CrossRef]
- Zhou, X.; Kang, L. A DFT study of graphene-FeNx (x = 4, 3, 2, 1) catalysts for acetylene hydrochlorination. Colloids Surfaces A Physicochem. Eng. Asp. 2021, 618, 126495. [Google Scholar] [CrossRef]
- Ede, S.R.; Luo, Z. Tuning the intrinsic catalytic activities of oxygen-evolution catalysts by doping: A comprehensive review. J. Mater. Chem. A 2021, 9, 20131–20163. [Google Scholar] [CrossRef]
- Li, F.; Shu, H.; Liu, X.; Shi, Z.; Liang, P.; Chen, X. Electrocatalytic Activity and Design Principles of Heteroatom-Doped Graphene Catalysts for Oxygen-Reduction Reaction. J. Phys. Chem. C 2017, 121, 14434–14442. [Google Scholar] [CrossRef]
- Bai, J.; Zhu, Q.; Lv, Z.; Dong, H.; Yu, J.; Dong, L. Nitrogen-doped graphene as catalysts and catalyst supports for oxygen reduction in both acidic and alkaline solutions. Int. J. Hydrogen Energy 2013, 38, 1413–1418. [Google Scholar] [CrossRef]
- Wang, T.; Sang, X.; Zheng, W.; Yang, B.; Yao, S.; Lei, C.; Li, Z.; He, Q.; Lu, J.; Lei, L.; et al. Gas Diffusion Strategy for Inserting Atomic Iron Sites into Graphitized Carbon Supports for Unusually High-Efficient CO 2 Electroreduction and High-Performance Zn–CO2 Batteries. Adv. Mater. 2020, 32, 2002430. [Google Scholar] [CrossRef]
- Lee, W.H.; Ko, Y.-J.; Kim, J.-Y.; Min, B.K.; Hwang, Y.J.; Oh, H.-S. Single-atom catalysts for the oxygen evolution reaction: Recent developments and future perspectives. Chem. Commun. 2020, 56, 12687–12697. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Lee, Y.J.; Jan, A.; Choi, S.M.; Park, M.Y.; Choi, S.; Hwang, J.Y.; Hong, S.; Park, S.G.; Chang, H.J.; et al. Highly active and thermally stable single-atom catalysts for high-temperature electrochemical devices. Energy Environ. Sci. 2020, 13, 4903–4920. [Google Scholar] [CrossRef]
- Kim, J.; Roh, C.-W.; Sahoo, S.K.; Yang, S.; Bae, J.; Han, J.W.; Lee, H. Highly Durable Platinum Single-Atom Alloy Catalyst for Electrochemical Reactions. Adv. Energy Mater. 2018, 8, 1701476. [Google Scholar] [CrossRef]
- Zou, L.; Wei, Y.-S.; Hou, C.-C.; Li, C.; Xu, Q. Single-Atom Catalysts Derived from Metal–Organic Frameworks for Electrochemical Applications. Small 2021, 17, 2004809. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, J.; Chen, J.; Chen, Y.; Zhang, C.; Luo, Y.; Wang, G.; Wang, R. Bi-functional electrocatalysis through synergetic coupling strategy of atomically dispersed Fe and Co active sites anchored on 3D nitrogen-doped carbon sheets for Zn-air battery. J. Catal. 2021, 397, 223–232. [Google Scholar] [CrossRef]
- Kattel, S.; Wang, G. Reaction Pathway for Oxygen Reduction on FeN4 Embedded Graphene. J. Phys. Chem. Lett. 2014, 5, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Kattel, S.; Wang, G. A density functional theory study of oxygen reduction reaction on Me–N4 (Me = Fe, Co, or Ni) clusters between graphitic pores. J. Mater. Chem. A 2013, 1, 10790–10797. [Google Scholar] [CrossRef]
- Ding, S.; Lyu, Z.; Sarnello, E.; Xu, M.; Fang, L.; Tian, H.; Karcher, S.E.; Li, T.; Pan, X.; McCloy, J.; et al. A MnOx enhanced atomically dispersed iron–nitrogen–carbon catalyst for the oxygen reduction reaction. J. Mater. Chem. A 2022, 10, 5981–5989. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Lu, Z.; Wang, W. Bifunctional CoNx embedded graphene electrocatalysts for OER and ORR: A theoretical evaluation. Carbon 2018, 130, 112–119. [Google Scholar] [CrossRef]
- Wang, R.; Lu, G.; Qiao, W.; Sun, Z.; Zhuang, H.; Yu, J. Catalytic effect of praseodymium oxide additive on the microstructure and electrical property of graphite anode. Carbon 2015, 95, 940–948. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Shin, D.; Jeong, H.; Jang, M.G.; Lee, H.; Han, J.W. Design of an Ultrastable and Highly Active Ceria Catalyst for CO Oxidation by Rare-Earth- and Transition-Metal Co-Doping. ACS Catal. 2020, 10, 14877–14886. [Google Scholar] [CrossRef]
- Li, J.; Chu, B.; Xie, Z.; Deng, Y.; Zhou, Y.; Dong, L.; Li, B.; Chen, Z. Mechanism and DFT Study of Degradation of Organic Pollutants on Rare Earth Ions Doped TiO2 Photocatalysts Prepared by Sol-Hydrothermal Synthesis. Catal. Lett. 2022, 152, 489–502. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, C.; Zhang, J.; Xu, J.; Xu, L.; Xu, X.; Li, J.; Cui, C. Rare-Earth-Catalyzed Selective 1,4-Hydrosilylation of Branched 1,3-Enynes Giving Tetrasubstituted Silylallenes. J. Am. Chem. Soc. 2021, 143, 12913–12918. [Google Scholar] [CrossRef]
- Abbott, D.F.; Pittkowski, R.K.; Macounova, K.; Nebel, R.; Marelli, E.; Fabbri, E.; Castelli, I.E.; Krtil, P.; Schmidt, T.J. Design and Synthesis of Ir/Ru Pyrochlore Catalysts for the Oxygen Evolution Reaction Based on Their Bulk Thermodynamic Properties. ACS Appl. Mater. Interfaces 2019, 11, 37748–37760. [Google Scholar] [CrossRef]
- Zhu, M.; Zhao, C.; Liu, X.; Wang, X.; Zhou, F.; Wang, J.; Hu, Y.; Zhao, Y.; Yao, T.; Yang, L.-M.; et al. Single Atomic Cerium Sites with a High Coordination Number for Efficient Oxygen Reduction in Proton-Exchange Membrane Fuel Cells. ACS Catal. 2021, 11, 3923–3929. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, J.; Jin, C.; Wang, N.; Wang, F. Hexagonal La2O3 Nanocrystals Chemically Coupled with Nitrogen-Doped Porous Carbon as Efficient Catalysts for the Oxygen Reduction Reaction. Chem. A Eur. J. 2020, 26, 12606–12614. [Google Scholar] [CrossRef] [PubMed]
- Murr, L.E. Summarizing Atom and Ion Structure: The Periodic Table of the Elements. In Handbook of Materials Structures, Properties, Processing and Performance; Springer International Publishing: Cham, Switzerland, 2015; pp. 83–95. [Google Scholar] [CrossRef]
- Gismondi, P.; Kuzmin, A.; Unsworth, C.; Rangan, S.; Khalid, S.; Saha, D. Understanding the Adsorption of Rare-Earth Elements in Oligo-Grafted Mesoporous Carbon. Langmuir 2022, 38, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, Y.; Lee, J.-M.; Fu, G. Recent advances in rare-earth-based materials for electrocatalysis. Chem Catal. 2022, 2, 967–1008. [Google Scholar] [CrossRef]
- Liu, J.; Kong, X.; Zheng, L.; Guo, X.; Liu, X.; Shui, J. Rare Earth Single-Atom Catalysts for Nitrogen and Carbon Dioxide Reduction. ACS Nano 2020, 14, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.-Z. Graphdiyne-supported single-atom Sc and Ti catalysts for high-efficient CO oxidation. Carbon 2016, 108, 343–350. [Google Scholar] [CrossRef]
- Wang, Q.-Y.; Tong, Y.-C.; Yan, P.-J.; Xu, X.-J.; Li, Z. Attachment of CO to a (6, 6) CNT with a Sc adsorbate atom. Struct. Chem. 2019, 30, 399–408. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Delley, B. The conductor-like screening model for polymers and surfaces. Mol. Simul. 2006, 32, 117–123. [Google Scholar] [CrossRef]
- Yang, K.; Zaffran, J.; Yang, B. Fast prediction of oxygen reduction reaction activity on carbon nanotubes with a localized geometric descriptor. Phys. Chem. Chem. Phys. 2020, 22, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhu, G.; Yang, D.; Jia, D.; Jin, F.; Wang, W. Systematic exploration of N, C configurational effects on the ORR performance of Fe–N doped graphene catalysts based on DFT calculations. RSC Adv. 2019, 9, 22656–22667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Luo, M.; Chen, M.; Qi, X.; Liu, J.; Liu, C.; Peera, S.G.; Liang, T. Exploring the oxygen electrode bi-functional activity of Ni–N–C-doped graphene systems with N, C co-ordination and OH ligand effects. J. Mater. Chem. A 2020, 8, 20453–20462. [Google Scholar] [CrossRef]
- Revanappa, S.K.; Soni, I.; Siddalinganahalli, M.; Jayaprakash, G.K.; Flores-Moreno, R.; Nanjegowda, C.B. A Fukui Analysis of an Arginine-Modified Carbon Surface for the Electrochemical Sensing of Dopamine. Materials 2022, 15, 6337. [Google Scholar] [CrossRef]
- Jayaprakash, G.K.; Swamy, B.E.K.; Flores-Moreno, R.; Pineda-Urbina, K. Theoretical and Cyclic Voltammetric Analysis of Asparagine and Glutamine Electrocatalytic Activities for Dopamine Sensing Applications. Catalysts 2023, 13, 100. [Google Scholar] [CrossRef]
- Qin, R.; Zhou, L.; Liu, P.; Gong, Y.; Liu, K.; Xu, C.; Zhao, Y.; Gu, L.; Fu, G.; Zheng, N. Alkali ions secure hydrides for catalytic hydrogenation. Nat. Catal. 2020, 3, 703–709. [Google Scholar] [CrossRef]
- Liang, Z.; Luo, M.; Chen, M.; Liu, C.; Peera, S.G.; Qi, X.; Liu, J.; Kumar, U.P.; Liang, T.L.T. Evaluating the catalytic activity of transition metal dimers for the oxygen reduction reaction. J. Colloid Interface Sci. 2020, 568, 54–62. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sc(OH)2N3 | −2.44 | −2.98 | −1.21 | −0.05 | −0.70 |
| Sc(OH)2N4 | −1.89 | −2.67 | −0.94 | 0.25 | −0.51 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Liao, M.; Hu, Y.; Lee, T.-G.; Koutavarapu, R.; Peera, S.G.; Liu, C. Density Functional Theory Study of Oxygen Evolution Reaction Mechanism on Rare Earth Sc-Doped Graphene. Batteries 2023, 9, 175. https://doi.org/10.3390/batteries9030175
Liu Y, Liao M, Hu Y, Lee T-G, Koutavarapu R, Peera SG, Liu C. Density Functional Theory Study of Oxygen Evolution Reaction Mechanism on Rare Earth Sc-Doped Graphene. Batteries. 2023; 9(3):175. https://doi.org/10.3390/batteries9030175
Chicago/Turabian StyleLiu, Yiwen, Mengqi Liao, Yuting Hu, Tae-Gwan Lee, Ravindranadh Koutavarapu, Shaik Gouse Peera, and Chao Liu. 2023. "Density Functional Theory Study of Oxygen Evolution Reaction Mechanism on Rare Earth Sc-Doped Graphene" Batteries 9, no. 3: 175. https://doi.org/10.3390/batteries9030175