
Zero-Field Splitting in Hexacoordinate Co(II) Complexes



Abstract
:1. Introduction
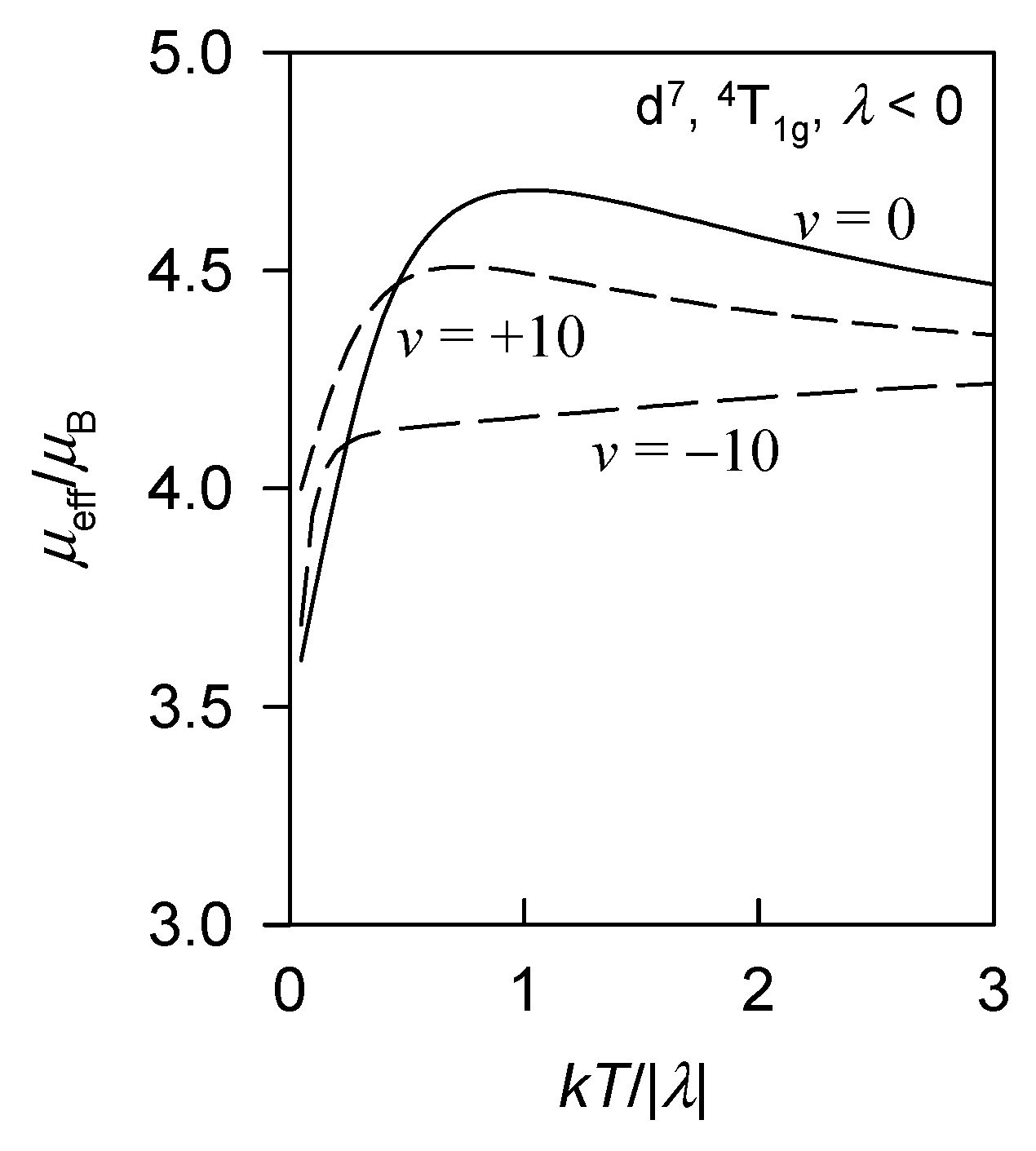
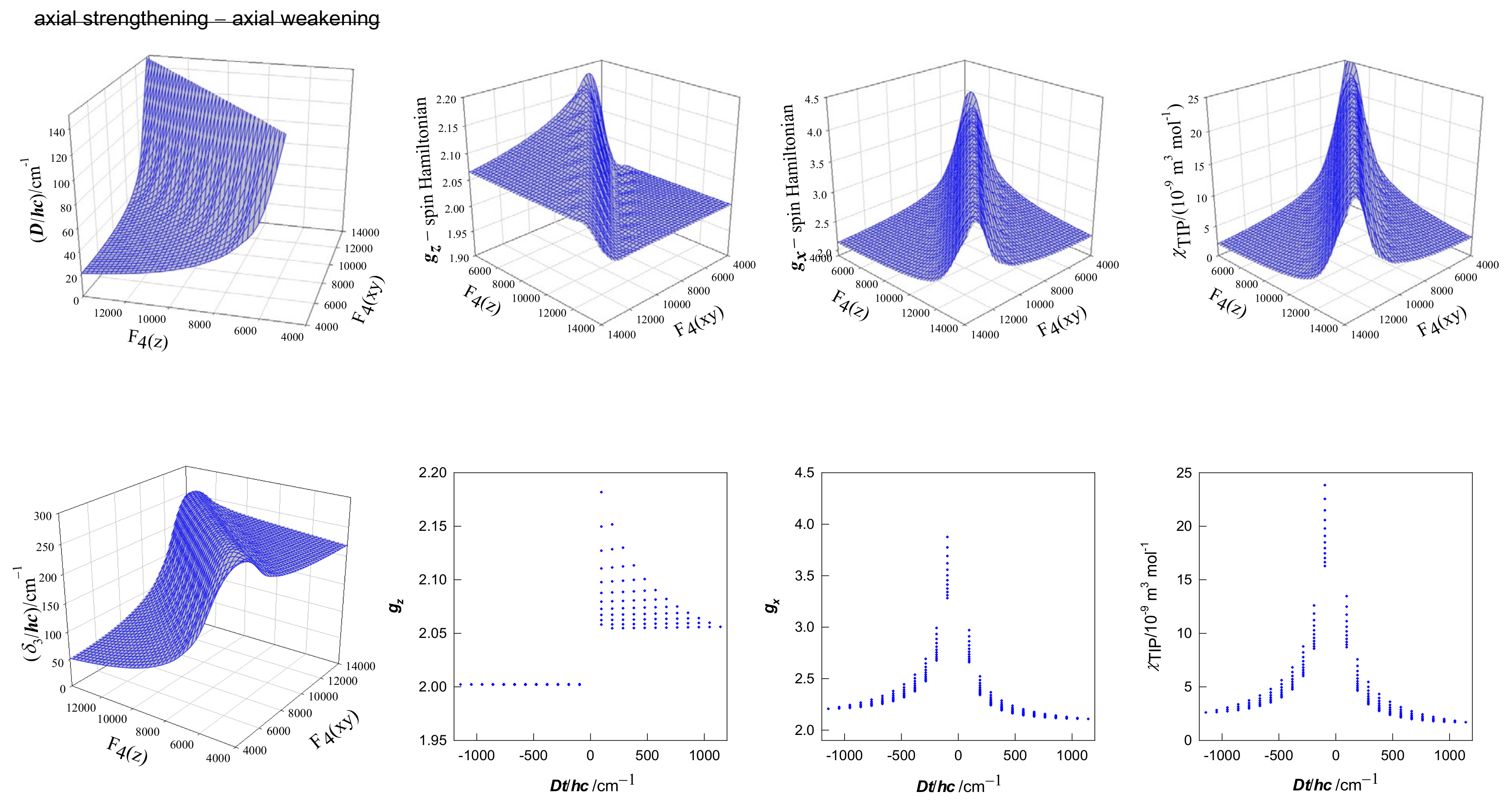
2. Theoretical Analysis
3. Methods and Modelling
3.1. Spin Hamiltonian
3.2. Griffith–Figgis Model
3.3. Ab Initio Calculations
3.4. Generalized Crystal-Field Theory
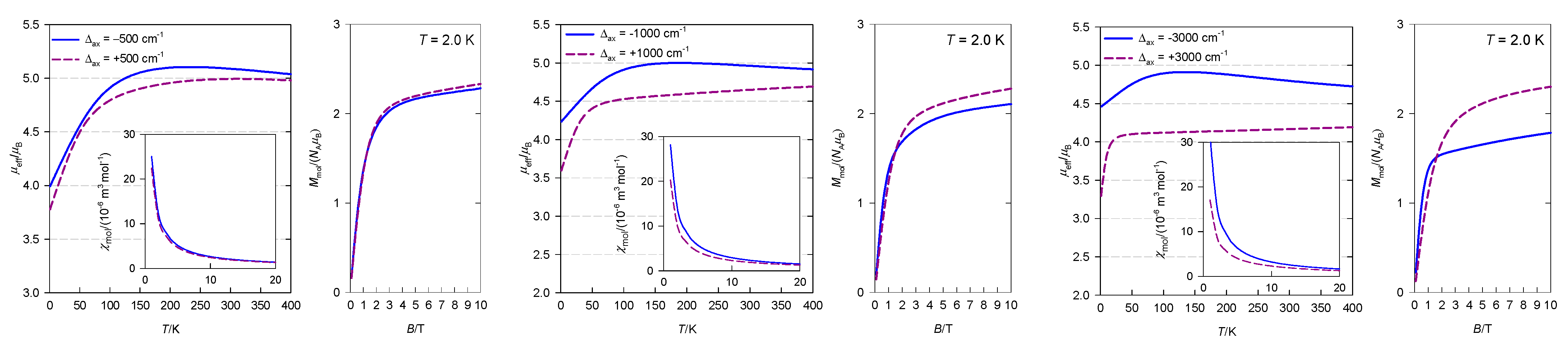
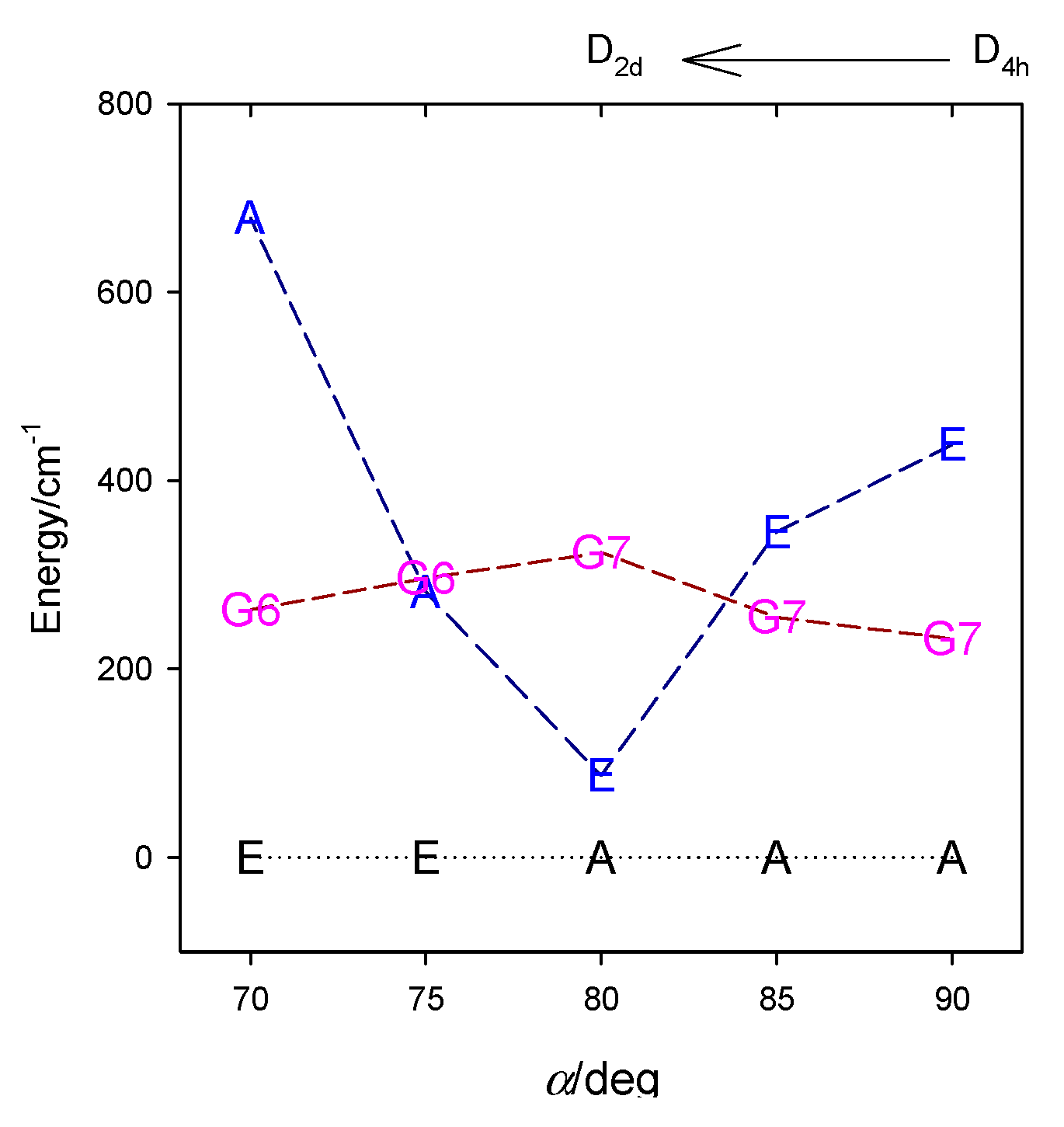
4. Results and Discussion
4.1. Geometry of Complexes
4.2. Elongated Tetragonal Bipyramid
4.3. Nearly Octahedral Systems
4.4. Compressed Tetragonal Bipyramid
4.5. Miscellaneous Geometry
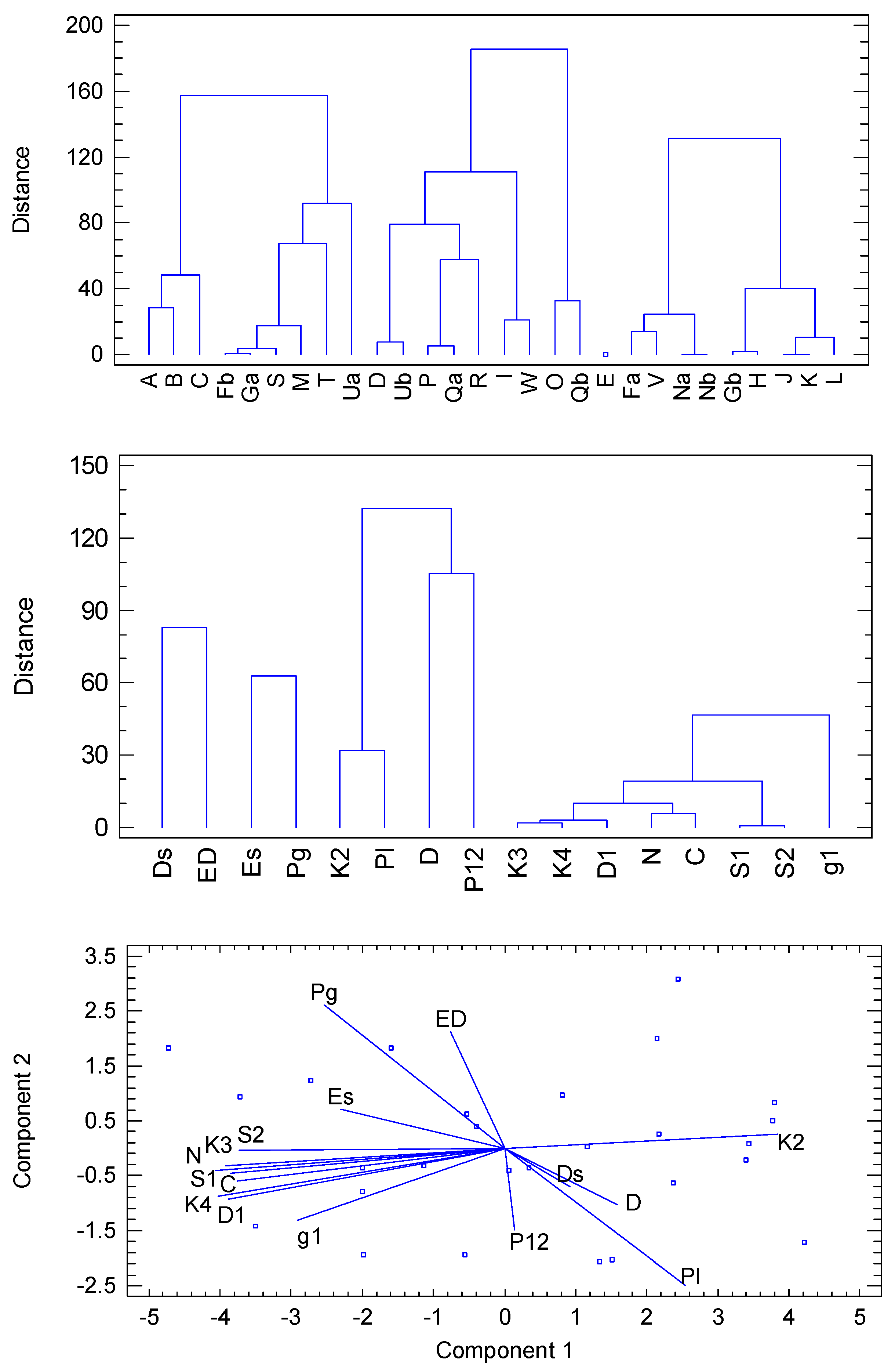
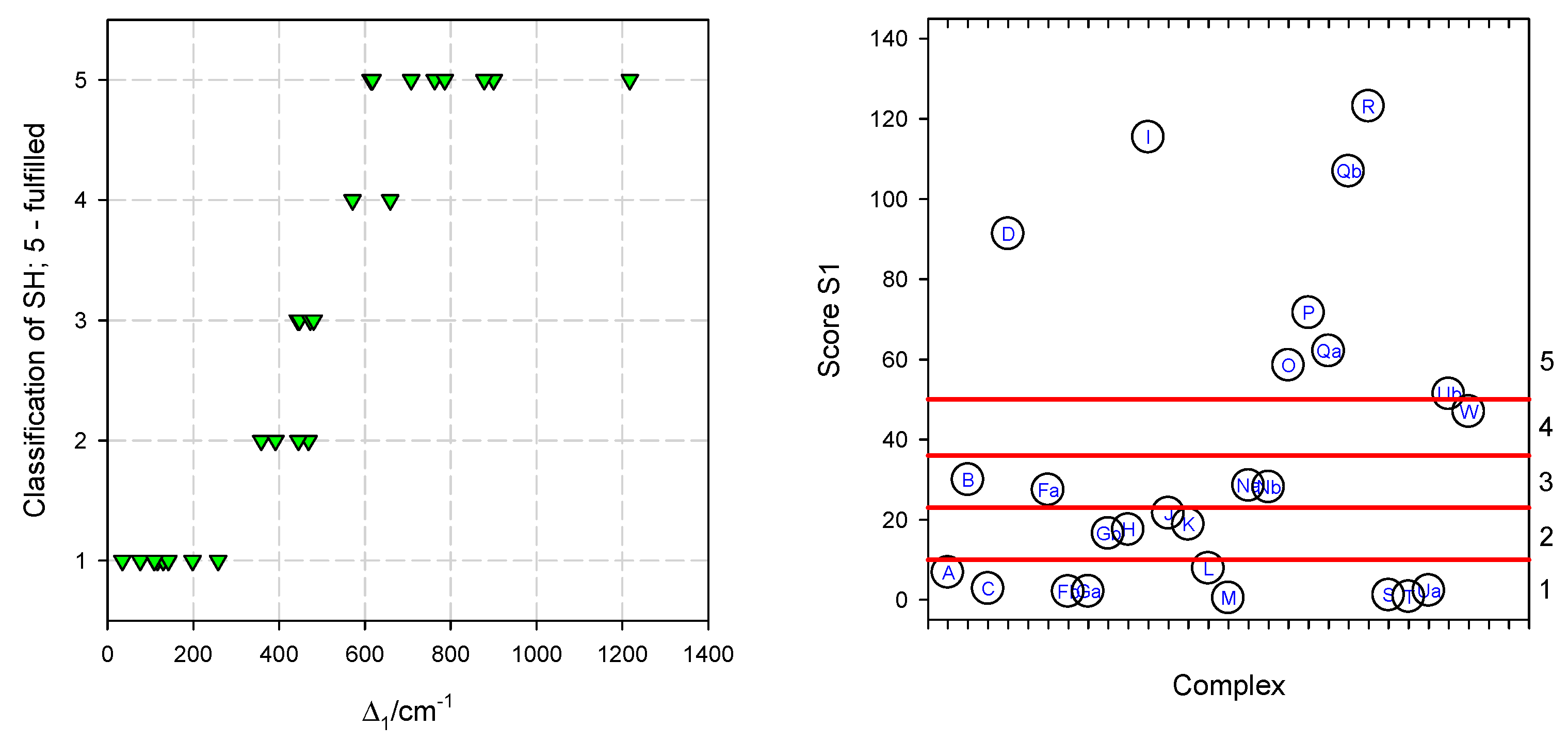
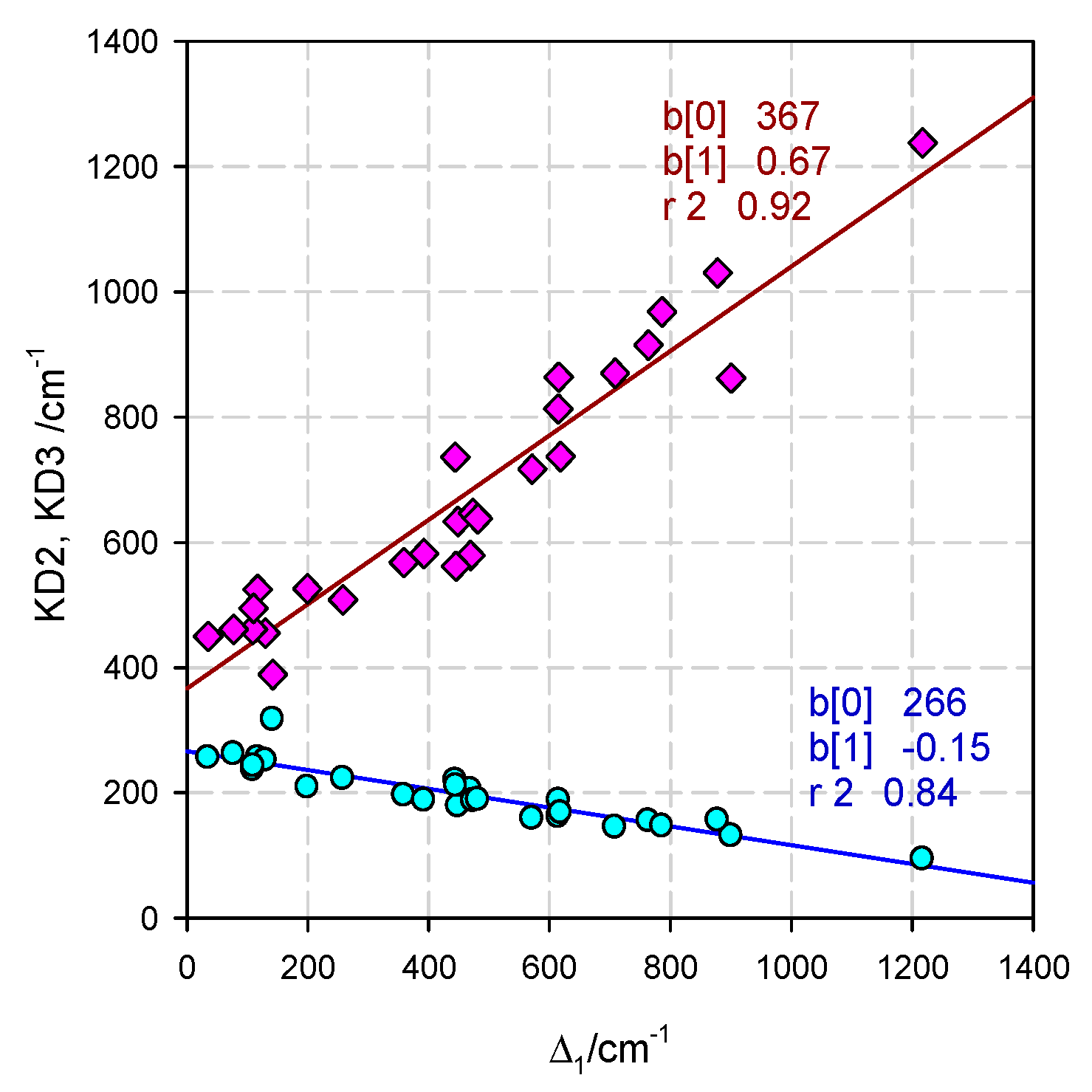
5. Statistical Analysis
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| abpt | 4-amino-3,5-bis(2-pyridyl)-1,2,4-triazol |
| ac | acetato(1-) ligand |
| ampyd | 2-aminopyrimidine |
| bz | benzoato(1-) ligand |
| bzpy | 4-benzylpyridine |
| bzpyCl | 4-(4-Chlorobenzyl)pyridine |
| dca | dicyanamide(1-) |
| dmphen | 2,9-dimethyl-1,10-phenanthroline |
| dnbz | 3,5-dinitrobenzoato(1-) |
| dppmO,O | bis-(diphenylphosphanoxido)methane |
| etpy | 4-ethylpyridine |
| fm | formiate(1-) ion |
| hfac | hexafluoroacetylacetonato(1-) |
| im, iz | 1H-imidazole |
| L1H2 | 2-{[(2-hydroxy-3-methoxyphenyl)-methylene]amino}-2-(hydroxymethyl)-1,3-propanediol |
| L2 | 2-[(2,2-diphenylethylimino)methyl]pyridine-1-oxide |
| mdnbz | 3,5-dinitrobenzoato(1-) |
| MeIm | N-methylimidazole |
| OHnic | 6-hydroxynicotinate |
| pydca | pyridine-2,6-dicarboxylato(1-) |
| pydm, dmpy | 2,6-pyridinedimethanol |
| pypz | 2,6-bis(pyrazol-1-yl)pyridine |
| tcm | tricyanomethanide(1-) |
| w | aqua ligand |
References
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006; ISBN 9780198567530. [Google Scholar]
- Winpenny, R. (Ed.) Single Molecule Magnets and Related Phenomena; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 9783642069833. [Google Scholar]
- Boča, R. Zero-field Splitting in Metal Complexes. Coord. Chem. Rev. 2004, 248, 757–815. [Google Scholar] [CrossRef]
- Bone, A.N.; Widener, C.N.; Moseley, D.H.; Liu, Z.; Lu, Z.; Cheng, Y.; Daemen, L.L.; Ozerov, M.; Telser, J.; Thirunavukkuarasu, K.; et al. Applying Unconventional Spectroscopies to the Single-Molecule Magnets, Co(PPh3)2X2 (X=Cl, Br, I): Unveiling Magnetic Transitions and Spin-Phonon Coupling. Chem. Eur. J. 2021, 27, 11110–11125. [Google Scholar] [CrossRef] [PubMed]
- Orton, J.W. Electron Paramagnetic Resonance: An Introduction to Transition Group Ions in Crystals; Iliffe Books: London, UK, 1968; ISBN 9780592050416. [Google Scholar]
- Abragam, A.; Bleaney, B. Electron Paramagnetic Resonance of Transition Ions; Clarendon Press: Oxford, UK, 1970; ISBN 9780199651528. [Google Scholar]
- Poole, C.P., Jr.; Farach, H.A. The Theory of Magnetic Resonance; Wiley: New York, NY, USA, 1972; ISBN 9780471815303. [Google Scholar]
- Wetz, J.E.; Bolton, J.R. Electron Spin Resonance: Elementary Theory and Practical Applications; McGraw Hill: New York, NY, USA, 1972; ISBN 9780471572343. [Google Scholar]
- Mabbs, F.E.; Machin, D.J. Magnetism and Transition Metal Complexes; Chapman and Hall: London, UK, 1973; ISBN 9780412112300. [Google Scholar]
- Carlin, R.L.; van Duyneveldt, A.J. Magnetic Properties of Transition Metal Compounds; Springer: New York, NY, USA, 1977; ISBN 9783540085843. [Google Scholar]
- Pilbrow, J.R. Transition Ion Electron Paramagnetic Resonance; Claredon: Oxford, UK, 1990; ISBN 0198552149. [Google Scholar]
- Mabbs, F.E.; Collison, D. Electron Paramagnetic Resonance of d Transition Metal Compounds; Elsevier: Amsterdam, The Netherlands, 1992; ISBN 9781483291499. [Google Scholar]
- Kahn, O. Molecular Magnetism; Wiley-VCH: New York, NY, USA, 1993; ISBN 9780471188384. [Google Scholar]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. ORCA-An Ab Initio, Density Functional and Semi-Empirical Program Package, version 5.0.2; HKU: Hon Kong, China, 2022.
- Aquilante, F.; Autschbach, J.; Carlson, R.K.; Chibotaru, L.F.; Delcey, M.G.; De Vico, L.; Fdez Galván, I.; Ferré, N.; Frutos, L.M.; Gagliardi, L.; et al. Molcas 8: New capabilities for multiconfigurational quantum chemical calculations across the periodic table. J. Comput. Chem. 2016, 37, 506–541. [Google Scholar] [CrossRef] [Green Version]
- Lang, L.; Atanasov, M.; Neese, F. Improvement of Ab Initio Ligand Field Theory by Means of Multistate Perturbation Theory. J. Phys. Chem. A. 2020, 124, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Boča, R. Magnetic Parameters and MagneticFunctions in Mononuclear Complexes beyond the Spin-Hamiltonian Formalism; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 9783540260790. [Google Scholar]
- Boča, R.; Rajnák, C.; Titiš, J. Quantified Quasi-symmetry in Metal Complexes. Inorg. Chem. 2022, 61, 17848–17854. [Google Scholar] [CrossRef]
- Abragam, A.; Pryce, M.H.L. Theory of the Nuclear Hyperfine Structure of Paramagnetic Resonance Spectra in Crystals. Proc. R. Soc. A. 1951, 205, 135–153. [Google Scholar] [CrossRef]
- Boča, R. A Handbook of Magnetochemical Formulae; Elsevier: Amsterdam, The Netherlands, 2012; ISBN 9780124160149. [Google Scholar]
- Sugano, S.; Tanabe, Y.; Kanimura, H. Multiplets of Transition Metal Ions in Crystals; Academic Press: New York, NY, USA, 1970; ISBN 9780124316751. [Google Scholar]
- Salthouse, J.A.; Ware, M.J. Point Group Character Tables and Related Data; University Press Cambridge: Cambridge, UK, 1972; ISBN 9780521081399. [Google Scholar]
- Bradley, C.J.; Cracknell, A.P. The Mathematical Theory of Symmetry in Solids; Claredon: Oxford, UK, 1972; ISBN 9780199582587. [Google Scholar]
- Figgis, B.N. Introduction to Ligand Fields; Wiley: New York, NY, USA, 1966; ISBN 9780470258804. [Google Scholar]
- Lloret, F.; Julve, M.; Cano, J.; Ruiz-Garcia, R.; Pardo, E. Magnetic Properties of Six-coordinated High-spin Cobalt(II) Complexes: Theoretical Background and its Application. Inorg. Chim. Acta 2008, 361, 3432–3445. [Google Scholar] [CrossRef]
- Singh, S.K.; Eng, J.; Atanasov, M.; Neese, F. Covalency and chemical bonding in transition metal complexes: An ab initio based ligand field perspective. Coord. Chem. Rev. 2017, 344, 2–25. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.-P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.-P. N-electron valence state perturbation theory: A fast implementation of the strongly contracted variant. Chem. Phys. Lett. 2001, 350, 297–305. [Google Scholar] [CrossRef]
- Dyall, K.G. The choice of a zeroth-order Hamiltonian for second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1995, 102, 4909. [Google Scholar] [CrossRef]
- Atanasov, M.; Aravena, D.; Suturina, E.; Bill, E.; Maganas, D.; Neese, F. First principles approach to the electronic structure, magnetic anisotropy and spin relaxation in mononuclear 3d-transition metal single molecule magnets. Coord. Chem. Rev. 2015, 289, 177–214. [Google Scholar] [CrossRef]
- Hess, B.A.; Marian, C.M.; Wahlgren, U.; Gropen, O. A mean-field spin-orbit method applicable to correlated wavefunctions. Chem. Phys. Lett. 1996, 251, 365. [Google Scholar] [CrossRef]
- Malrieu, J.-P.; Caballol, R.; Calzado, C.J.; de Graaf, C.; Guihéry, N. Magnetic Interactions in Molecules and Highly Correlated Materials: Physical Content, Analytical Derivation, and Rigorous Extraction of Magnetic Hamiltonians. Chem. Rev. 2014, 114, 429–492. [Google Scholar] [CrossRef]
- Neese, F.; Lang, L.; Chilkuri, V.G. Effective Hamiltonians in Chemistry. In Topology, Entanglement, and Strong Correlations, Modeling and Simulation; Pavarini, E., Koch, E., Eds.; Forschungszentrum Jülich: Julich, Germany, 2020; Volume 10, ISBN 978-3-95806-466-9. [Google Scholar]
- Maurice, R.; Bastardis, R.; de Graaf, C.; Suaud, N.; Mallah, T.; Guihéry, N. Universal Theoretical Approach to Extract Anisotropic Spin Hamiltonians. J. Chem. Theory Comput. 2009, 5, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Boča, R. Program Terms22. University of Ss. Cyril and Methodius in Trnava: Trnava, Slovak Republic, 2022. [Google Scholar]
- Inorganic Crystal Structure Database, Fachinformationszentrum Karlsruhe, Germany. Available online: http://www.fiz-informationsdienste.de/en/DB/icsd/index.html (accessed on 20 January 2023).
- Titiš, J.; Boča, R. Magnetostructural D Correlation in Nickel(II) Complexes: Reinvestigation of the Zero-Field Splitting. Inorg. Chem. 2011, 50, 11838–11845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Ng, S.W. Hexaaqua cobalt(II) bis (6-hydroxy pyridine-3-carboxylate). Acta Crystallogr. E 2005, E61, m1140–m1141. [Google Scholar] [CrossRef]
- Buvaylo, E.A.; Kokozay, V.N.; Vassilyeva, O.Y.; Skelton, B.W.; Ozarowski, A.; Titiš, J.; Vranovičová, B.; Boča, R. Field-Assisted Slow Magnetic Relaxation in a Six-Coordinate Co(II)–Co(III) Complex with Large Negative Anisotropy. Inorg. Chem. 2017, 56, 6999–7009. [Google Scholar] [CrossRef]
- Hudák, J.; Boča, R.; Dlháň, Ľ.; Kožíšek, J.; Moncoľ, J. Structure and magnetism of mono-, di-, and trinuclear benzoato cobalt(II) complexes. Polyhedron 2011, 30, 1367–1373. [Google Scholar] [CrossRef]
- Moseley, D.H.; Stavretis, S.E.; Thirunavukkuarasu, K.; Ozerov, M.; Cheng, Y.; Daemen, L.L.; Ludwig, J.; Lu, Z.; Smirnov, D.; Brown, C.M.; et al. Spin–phonon couplings in transition metal complexes with slow magnetic relaxation. Nat. Commun. 2018, 9, 2572. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Boča, R.; Moncol’, J.; Padělková, Z. Self-assembled cobalt(II) Schiff base complex: Synthesis, structure, and magnetic properties. Mon. Chem. 2011, 142, 789–795. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Moncoľ, J.; Renz, F.; Boča, R. Field-Supported Slow Magnetic Relaxation in Hexacoordinate Co(II) Complexes with Easy Plane Anisotropy. Eur. J. Inorg. Chem. 2017, 2017, 1520–1525. [Google Scholar] [CrossRef]
- Titiš, J.; Boča, R. Magnetostructural D Correlations in Hexacoordinated Cobalt(II) Complexes. Inorg. Chem. 2011, 50, 11838–11845. [Google Scholar] [CrossRef]
- Herchel, R.; Váhovská, L.; Potočňák, U.; Trávníček, Z. Slow Magnetic Relaxation in Octahedral Cobalt(II) Field-Induced Single-Ion Magnet with Positive Axial and Large Rhombic Anisotropy. Inorg. Chem. 2014, 53, 5896–5898. [Google Scholar] [CrossRef]
- Rajnák, C.; Varga, F.; Titiš, J.; Moncoľ, J.; Boča, R. Octahedral–Tetrahedral Systems [Co(dppmO,O)3]2+[CoX4]2− Showing Slow Magnetic Relaxation with Two Relaxation Modes. Inorg. Chem. 2018, 57, 4352–4358. [Google Scholar] [CrossRef]
- Varga, F.; Rajnák, C.; Titiš, J.; Moncoľ, J.; Boča, R. Slow magnetic relaxation in a Co(II) octahedral–tetrahedral system formed of a [CoL3]2+ core with L = bis(diphenylphosphanoxido) methane and tetrahedral [CoBr4]2− counter anions. Dalton Trans. 2017, 46, 4148–4151. [Google Scholar] [CrossRef]
- Lin, J.-L.; Fei, S.-T.; Lei, K.-W.; Zheng, Y.-Q. Hexakis(imidazole-κN3)cobalt(II) diformate. Acta Crystallogr. E 2006, E62, m2439–m24441. [Google Scholar] [CrossRef]
- Valigura, D.; Rajnák, C.; Moncoľ, J.; Titiš, J.; Boča, R. A mononuclear Co(II) complex formed from pyridinedimethanol with manifold slow relaxation channels. Dalton Trans. 2017, 46, 10950–10956. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Moncol, J.; Boča, R. Effect of the distant substituent on the slow magnetic relaxation of the mononuclear Co(ii) complex with pincer-type ligands. Dalton Trans. 2020, 49, 4206–4210. [Google Scholar] [CrossRef] [PubMed]
- Boča, R.; Rajnák, C.; Moncol, J.; Titiš, J.; Valigura, D. Breaking the Magic Border of One Second for Slow Magnetic Relaxation of Cobalt-Based Single Ion Magnets. Inorg. Chem. 2018, 57, 14314–14321. [Google Scholar] [CrossRef]
- Papánková, B.; Svoboda, I.; Fuess, H. Bis(acetato-[kappa]O)diaqua bis(1-methyl imidazole-[kappa]N3)cobalt(II). Acta Crystallogr. E 2006, E62, m1916–m1918. [Google Scholar] [CrossRef] [Green Version]
- Papánková, B.; Boča, R.; Dlháň, Ľ.; Nemec, I.; Titiš, J.; Svoboda, I.; Fuess, H. Magneto-structural relationships for a mononuclear Co(II) complex with large zero-field splitting. Inorg. Chim. Acta 2010, 363, 147–156. [Google Scholar] [CrossRef]
- Pike, R.D.; Lim, M.J.; Willcox, E.A.L.; Tronic, T.A. Nickel(II) and cobalt(II) nitrate and chloride networks with 2-aminopyrimidine. J. Chem. Cryst. 2006, 36, 781–791. [Google Scholar] [CrossRef] [Green Version]
- Idešicová, M.; Boča, R. Magnetism, IR and Raman spectra of a tetracoordinate and hexacoordinate Co(II) complexes derived from aminopyrimidine. Inorg. Chim. Acta 2013, 408, 162–171. [Google Scholar] [CrossRef]
- Váhovská, L.; Potočňák, I.; Dušek, M.; Vitushkina, S.; Titiš, J.; Boča, R. Low-dimensional compounds containing cyanido groups. XXVI. Crystal structure, spectroscopic and magnetic properties of Co(II) complexes with non-linear pseudohalide ligands. Polyhedron 2014, 81, 396–408. [Google Scholar] [CrossRef]
- Świtlicka, A.; Machura, B.; Penkala, M.; Bieńko, A.; Bieńko, D.C.; Titiš, J.; Rajnák, C.; Boča, R.; Ozarowski, A. Slow magnetic relaxation in hexacoordinated cobalt(II) field-induced single-ion magnets. Inorg. Chem. Front. 2020, 7, 2637–2650. [Google Scholar] [CrossRef]
- Statgraphics Centurion XV, HP Inc. © 1982 Statpoint, Inc. Available online: https://www.statgraphics.com/centurion-xvi (accessed on 19 February 2023).
- Rajnák, C.; Boča, R. Reciprocating thermal behaviour of single ion magnets. Coord. Chem. Rev. 2021, 436, 213808. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Boča, R. Reciprocating Thermal Behavior in Multichannel Relaxation of Cobalt(II) Based Single Ion Magnets. Magnetochemistry 2021, 7, 76. [Google Scholar] [CrossRef]
- Sahu, P.K.; Kharel, R.; Shome, S.; Goswami, S.; Konar, S. Understanding the unceasing evolution of Co(II) based single-ion magnets. Coord. Chem. Rev. 2023, 475, 214871. [Google Scholar] [CrossRef]
- Vallejo, J.; Castro, I.; Ruiz-García, R.; Cano, J.; Julve, M.; Lloret, F.; De Munno, G.; Wernsdorfer, W.; Pardo, E. Field-Induced Slow Magnetic Relaxation in a Six-Coordinate Mononuclear Cobalt(II) Complex with a Positive Anisotropy. J. Am. Chem. Soc. 2012, 134, 15704–15707. [Google Scholar] [CrossRef]
- Tripathi, S.; Vaidya, S.; Ahmed, N.; Klahn, E.A.; Cao, H.; Spillecke, L.; Koo, C.; Spachmann, S.; Klingeler, R.; Rajaraman, G.; et al. Structure-property correlation in stabilizing axial magnetic anisotropy in octahedral Co(II) complexes. Cell Rep. Phys. Sci. 2021, 2, 100404. [Google Scholar] [CrossRef]
- Craig, G.A.; Murrie, M. 3d single ion magnets. Chem. Soc. Rev. 2015, 44, 2135–2147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Coca, S.; Aravena, D.; Morales, R.; Ruiz, E. Large magnetic anisotropy in mononuclear metal complexes. Coord. Chem. Rev. 2015, 289, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Frost, J.M.; Harriman, K.L.M.; Murugesu, M. The rise of 3-d single-ion magnets in molecular magnetism: Towards materials from molecules. Chem. Sci. 2016, 7, 2470–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Coca, S.; Urtizberea, A.; Cremades, E.; Alonso, P.J.; Camón, A.; Ruiz, E.; Luis, F. Origin of slow magnetic relaxation in Kramers ions with non-uniaxial anisotropy. Nat. Commun. 2014, 5, 4300. [Google Scholar] [CrossRef] [Green Version]
- Juráková, J.; Šalitroš, I. Co(II) single-ion magnets: Synthesis, structure, and magnetic properties. Monatsh. Chem. 2022, 153, 1001–1036. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
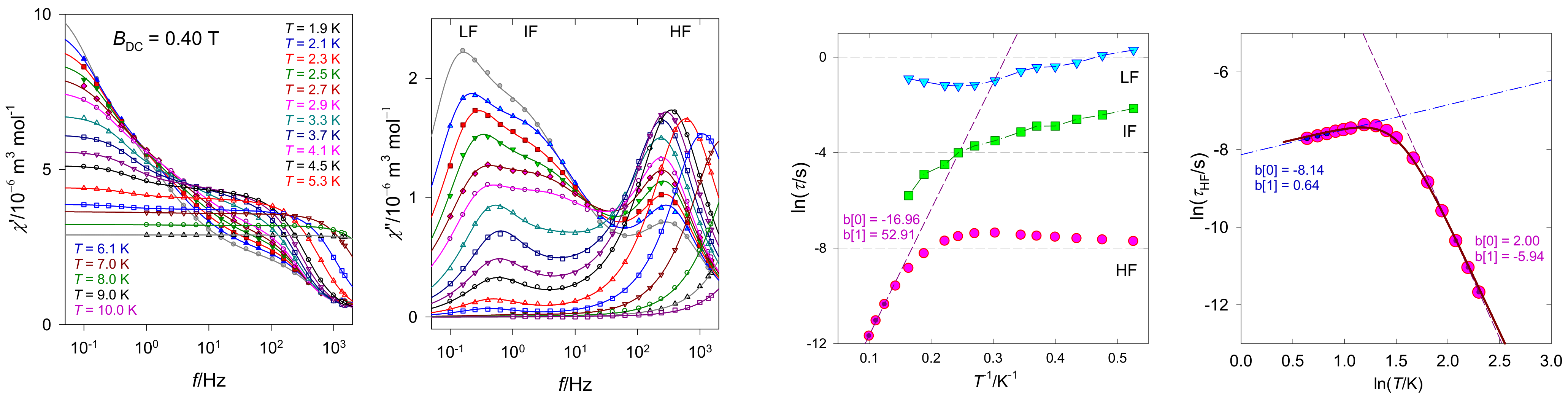{kind=link}
| Free Atom/Ion | Molecule/Complex | |||
|---|---|---|---|---|
| Operators | ||||
| Wave function | Atomic term | Atomic multiplet | Multielectron term | Spin–orbit multiplet |
| Notation | |dn: ν, L, ML, S, MS> | |(νLS), J, MJ> | |Γ, γ, a; S, MS> | |Γ′, γ′, a′> |
| Irreducible representations b | D(L)(2L + 1): S, P, D, F, G, H, I | 2S + 1DJ(2J + 1) | mA(1), mB(1), mE(2), mT(3) b | Γi(1, 2, 3, 4) |
| -for Kramers systems | S = 1/2, 3/2, 5/2, 7/2 | J = |L − S|,…L + S | m = 2S + 1 = 2, 4, 6, 8 | Γi(2), Γ8(4) |
| A, [Co(H2O)6]2+ (OHnic−)2, [CoH12O6]2+ 2(C6H4NO3)− | CAS Theory: Spin–Orbit Multiplets | ||||
|---|---|---|---|---|---|
| CCDC FONQUV, 295 K, Rgt = 0.054 [39,40]  | {CoO4O’2} Co-O’ 2.113 Å Co-O 2.042 Å Dstr = +7.1 pm Estr = 0 | KD1, 0.61 a | KD2, 0.78 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 209 | δ5,6 = 526 | δ7,8 = 814 | ||
| 41·| ± 1/2> + 57·| ± 3/2> | 58·| ± 1/2> + 40·| ± 3/2> | 55·| ± 1/2> + 42·| ± 3/2> | 42·| ± 1/2> +57·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: score S1 = 7, S2 = 4, classification 1–invalid | ||||
 | GF model λeff = −188 cm−1 gL = −1.10 Δax = −112 cm−1 | 4Δ0 = 0 4Δ1 = 199 4Δ2 = 2468 | D = −100.9 D1 = −119.9 D2 = +10.5 | E/D = 0.16 E1 = −0.01 E2 = −10.8 | g1 = 1.762g2 = 1.906g3 = 3.104giso = 2.258 |
| B, [CoIICoIII(L1H2)2(H2O)(ac)]·(H2O)3, [C26H35Co2N2O13] 3(H2O) | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1440294, 100 K, Rgt = 0.039 [41] | {CoO4O’2} Co-O’ 2.150 Å Co-O 2.061 Å Dstr = +8.9 pm Estr = 0 | KD1, 0.71 | KD2, 0.88 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 220 | δ5,6 = 736 | δ7,8 = 1006 | ||
| 56·| ± 1/2> + 43·| ± 3/2> | 38·| ± 1/2> + 58·| ± 3/2> | 36·| ± 1/2> + 62·| ± 3/2> | 64·| ± 1/2> +34·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 30, S2 = 17, classification 3–questionable | ||||
 | GF model λeff = −198 cm−1 gLz = −1.64 gLx = −1.11 Δax = −774 cm−1 | 4Δ0 = 0 4Δ1 = 444 4Δ2 = 1516 | D = −100.9 D1 = −114.8 D2 = +20.0 | E/D = 0.25 E1 = −0.01 E2 = −20.0 | g1 = 1.842g2 = 2.293g3 = 3.102giso = 2.412 |
| C, trans-[Co(bz)2(H2O)2(nca)2], [C26H26CoN4O8] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 804191, 293 K, Rgt = 0.038 [42] | {CoO2O’2N2} Co-N 2.147 Å Co-O 2.084 Å Co-O’w 2.143 Å Dstr = +7.75 pm Estr = 1.85 pm | KD1, 0.58 | KD2, 0.73 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 256 | δ5,6 = 525 | δ7,8 = 850 | ||
| 65·| ± 1/2> + 34·| ± 3/2> | 31·| ± 1/2> + 67·| ± 3/2> | 31·| ± 1/2> + 67·| ± 3/2> | 72·| ± 1/2> +27·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: score S1 = 3, S2 = 2, classification 1–invalid | ||||
 | GF model λeff = −172 cm−1 gLz = −2.06 gLx = −1.50 Δax = −739 cm−1 | 4Δ0 = 0 4Δ1 = 117 4Δ2 = 1138 | D = −113.3 D1 = −131.0 D2 = +26.9 | E/D = 0.31 E1 = −0.08 E2 = −27.0 | g1 = 1.507g2 = 2.042g3 = 3.160giso = 2.237 |
| D, [Co(acac)2(H2O)2], [C10H18CoO6] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1842364, 100 K, Rgt = 0.024 [43] | {CoO2O’2Ow} Co-O 2.040Å Co-O’ 2.034 Å Co-Ow 2.157 Å Dstr = +12.0 pm Estr = 0.30 pm | KD1, 0.81 | KD2, 0.93 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 155 | δ5,6 = 915 | δ7,8 = 1153 | ||
| 53·| ± 1/2> + 45·| ± 3/2> | 46·| ± 1/2> + 52·| ± 3/2> | 56·| ± 1/2> + 40·| ± 3/2> | 40·| ± 1/2> +57·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 91, S2 = 48, classification 5–fulfilled | ||||
 | SH-zfs model from ab initio calculations | 4Δ0 = 0 4Δ1 = 763 4Δ2 = 1398 | D = +72.0 D1 = +39.7 D2 = +22.7 | E/D = 0.23 E1 = −39.6 E2 = −22.8 | g1 = 1.943g2 = 2.462g3 = 2.804giso = 2.403 |
| E, [CoL22Cl2]·3.5H2O, [C40H36Cl2CoN4O2]·3.5H2O | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 796703, 150 K, Rgt = 0.045 [44] | {CoN2O2Cl2} Co-N 2.081 Å Co-O 2.034 Å Co-Cl 2.492 Å Dstr = +9.45 pm Estr = 2.65 pm Estr/Dstr = 0.28 | KD1, 0.91 | KD2, 0.96 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 94 | δ5,6 = 1238 | δ7,8 = 1441 | ||
| 88·| ± 1/2> + 9·| ± 3/2> | 8·| ± 1/2> + 89·| ± 3/2> | 11·| ± 1/2> + 86·| ± 3/2> | 87·| ± 1/2> +9·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: S1 = 257, S2 = 226, classification 5–fulfilled | ||||
 | SH-zfs model D = 75.1 cm−1 E = 4.8 cm−1 gz = 2 gx = 2.51 gy = 2.36 | 4Δ0 = 0 4Δ1 = 1217 4Δ2 = 2039 | D = +43.3 D1 = +21.5 D2 = +13.9 | E/D = 0.24 E1 = +13.6 E2 = −3.8 | g1 = 2.032g2 = 2.341g3 = 2.566giso = 2.313 |
| Fa, [Co(bzpy)4Cl2], [C48H44Cl2CoN4] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1497488, 120 K, Rgt = 0.027 [45] | {CoN4Cl2} Unit A Co-Cl 2.443 Å Co-N 2.235 Å Co-N 2.176 Å Dstr = +7.05 pm Estr = 1.15 pm | KD1, 0.69 | KD2, 0.89 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 179 | δ5,6 = 633 | δ7,8 = 911 | ||
| 69·| ± 1/2> + 29·| ± 3/2> | 24·| ± 1/2> + 73·| ± 3/2> | 36·| ± 1/2> + 62·| ± 3/2> | 73·| ± 1/2> +24·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 27, S2 = 19, classification 3–questionable | ||||
 | GF model/11 λeff = −175 cm−1 gLz = −1.02 gLx = −1.28 Δax = −424 cm−1 | 4Δ0 = 0 4Δ1 = 448 4Δ2 = 993 | D = +87.6 D1 = +43.2 D2 = +31.6 | E/D = 0.13 E1 = + 43.1 E2 = −31.4 | g1 = 1.948g2 = 2.498g3 = 2.779giso = 2.408 |
 | SH-zfs model D = +106 cm−1 gx = 2.53 gz = 2 | ||||
| Fb, [Co(bzpy)4Cl2], [C48H44Cl2CoN4] | CAS Theory: Spin–Orbit Multiplets | ||||
| Structure as above for Fa | {CoN4Cl2} Unit B Co-Cl 2.433 Å Co-N 2.187 Å Co-N 2.169 Å Dstr = −3.5 pm Estr = 0.9 pm E/|D| = 0.26 | KD1, 0.49 | KD2, 0.68 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 252 | δ5,6 = 455 | δ7,8 = 787 | ||
| 54·| ± 1/2> + 44·| ± 3/2> | 47·| ± 1/2> + 51·| ± 3/2> | 34·| ± 1/2> + 65·| ± 3/2> | 66·| ± 1/2> +33·| ± 3/2> | ||
| Magnetic data as above for Fa | SH theory: score S1 = 2, S2 = 1, classification 1–invalid | ||||
| 4Δ0 = 0 4Δ1 = 130 4Δ2 = 804 | D = +120.9 D1 = + 56.2 D2 = +34.8 | E/D = 0.17 E1 = + 56.2 E2 = −34.7 | g1 = 1.604g2 = 2.163g3 = 2.942giso = 2.237 | ||
| Ga, [Co(hfac)2(etpy)2], [C24H20CoF12N2O4] | CAS Theory: Spin–Orbit Multiplets | ||||
|---|---|---|---|---|---|
CCDC 2223471, 100 K, Rgt = 0.050  | A: {CoO4N2} Co-N 2.132 Å Co-O 2.056 Å Co-O 2.048 Å Dstr = −2.0 pm Estr = 0.4 pm | KD1, 0.50 | KD2, 0.73 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 237 | δ5,6 = 461 | δ7,8 = 804 | ||
| 49·| ± 1/2> + 50·| ± 3/2> | 50·| ± 1/2> + 49·| ± 3/2> | 52·| ± 1/2> + 46·| ± 3/2> | 46·| ± 1/2> + 52·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 2, S2 = 1, classification 1–invalid | ||||
 | GF model λeff = −159 cm−1 gLz = −1.96 gLx = −1.79 Δax = −771 cm−1 | 4Δ0 = 0 4Δ1 = 109 4Δ2 = 785 | D = +112 D1 = +59.8 D2 = +33.4 | E/D = 0.20 E1 = 58.8 E2 = −33.4 | g1 = 1.661 g2 = 2.043 g3 = 2.932 giso = 2.212 |
| Gb, [Co(hfac)2(etpy)2], [C24H20CoF12N2O4] | CAS Theory: Spin–Orbit Multiplets | ||||
| B: {CoO4N2} Co-N = 2.151Å Co-O 2.040 Å Co-O 2.058 Å Dstr = −1.45 pm Estr = 0.35 pm | KD1, 0.64 | KD2, 0.87 | KD3 | KD4 | |
| δ1,2 = 0 | δ3,4 = 196 | δ5,6 = 568 | δ7,8 = 873 | ||
| 37·| ± 1/2> + 63·| ± 3/2> | 45·| ± 1/2> + 55·| ± 3/2> | 55·| ± 1/2> + 45·| ± 3/2> | 40·| ± 1/2> + 60·| ± 3/2> | ||
| Magnetic data as above | SH theory: S1 = 16, S2 = 10, classification 2–problematic | ||||
| 4Δ0 = 0 4Δ1 = 359 4Δ2 = 901 | D = +94 D1 = +47.9 D2 = +29.7 | E/D = 0.18 E1 = −47.9 E2 = 29.7 | g1 = 1.931 g2 = 2.351 g3 = 2.808 giso = 2.364 | ||
| H, [Co(hfac)2(bzpyCl)2], [C34H22Cl2CoF12N2O4] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 2223472, 100 K, Rgt = 0.036  | {CoO4N2}* Co-N 2.137 Å Co-O 2.061 Å Co-O 2.062 Å Dstr = −2.45 pm Estr = 0.05 pm | KD1, 0.58 | KD2, 0.83 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 188 | δ5,6 = 582 | δ7,8 = 883 | ||
| 24·| ± 1/2> + 74·| ± 3/2> | 78·| ± 1/2> + 20·| ± 3/2> | 79·| ± 1/2> + 20·| ± 3/2> | 6·| ± 1/2> + 90·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 17, S2 = 13, classification 2–problematic | ||||
 | GF model λeff = −170 cm−1 gLz = −1.83 gLx = −1.11 Δax = −643 cm−1 | 4Δ0 = 0 4Δ1 = 392 4Δ2 = 905 | D = +91 D1 = +45.9 D2 = +29.6 | E/D = 0.16 E1 = 45.7 E2 = −29.0 | g1 = 1.954 g2 = 2.372 g3 = 2.781 giso = 2.369 |
| I, [Co(abpt)2(tcm)2], [C32H20CoN18] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 997721, 173 K, Rgt = 0.036 [47] | {CoN4N’2}* Co-N’ 2.133 Å Co-N 2.109 Å Co-N 2.125 Å Dstr = −2.0 pm Estr = 0.4 pm | KD1, 0.86 | KD2, 0.96 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 131 | δ5,6 = 862 | δ7,8 = 1066 | ||
| 20·| ± 1/2> + 79·| ± 3/2> | 76·| ± 1/2> + 17·| ± 3/2> | 85·| ± 1/2> + 13·| ± 3/2> | 5·| ± 1/2> + 92·| ± 3/2> | ||
| Magnetic data, SMR–yes [47] | SH theory: S1 = 115, S2 = 91, classification 5–fulfilled | ||||
 | SH-zfs model D = +55 cm−1 E = 14.6 cm−1 gx = 2.53 gz = 2 | 4Δ0 = 0 4Δ1 = 900 4Δ2 = 1878 | D = +50.3 D1 = +28.5 D2 = +17.6 | E/D = 0.29 E1 = +28.5 E2 = −17.6 | g1 = 2.037 g2 = 2.333 g3 = 2.636 giso = 2.335 |
| J, [Co(dppmO,O)3][Co(NCS)4], [C75H66CoO6P6]2+ Co(NCS)42− | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1526142, 100 K, Rgt = 0.041 [48] | {CoO2O’2O”2} Co-O 2.094 Å Co-O’ 2.089 Å Co-O” 2.074 Å Dstr = −1.65 pm Estr = 0.35 pm | KD1, 0.61 | KD2, 0.86 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 211 | δ5,6 = 562 | δ7,8 = 966 | ||
| 51·| ± 1/2> + 48·| ± 3/2> | 47·| ± 1/2> + 51·| ± 3/2> | 47·| ± 1/2> + 51·| ± 3/2> | 57·| ± 1/2> + 41·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 21, S2 = 11, classification 2–problematic | ||||
 | SH-zfs model D = +93 cm−1 gx = 2.76 gz = 2 | 4Δ0 = 0 4Δ1 = 445 4Δ2 = 539 | D = +105.5 D1 = +46.3 D2 = +42.0 | E/D = 0.03 E1 = +45.7 E2 = −41.9 | g1 = 1.972 g2 = 2.592 g3 = 2.688 giso = 2.417 |
| K, [Co(dppmO,O)3][CoBr4], [C75H66CoO6P6]2+ CoBr42− | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1526141, 100 K, Rgt = 0.044 [49] | {CoO2O’2O”2} Co-O 2.109 Å Co-O’ 2.102 Å Co-O” 2.091 Å Dstr = −1.45 pm Estr = 0.35 pm | KD1, 0.61 | KD2, 0.86 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 211 | δ5,6 = 562 | δ7,8 = 966 | ||
| 51·| ± 1/2> + 47·| ± 3/2> | 46·| ± 1/2> + 53·| ± 3/2> | 47·| ± 1/2> + 52·| ± 3/2> | 59·| ± 1/2> + 39·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 19, S2 = 10, classification 2–problematic | ||||
 | SH-zfs model D = +122 cm−1 gx = 2.68 gz = 2 | 4Δ0 = 0 4Δ1 = 445 4Δ2 = 539 | D = +105.5 D1 = +46.3 D2 = +42.53 | E/D = 0.03 E1 = +45.7 E2 = −41.9 | g1 = 1.972 g2 = 2.592 g3 = 2.688 giso = 2.417 |
| L, [Co(dppmO,O)3][CoI4], [C75H66CoO6P6]2+ CoI42− | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1526143, 100 K, Rgt = 0.028 [48] | {CoO2O’2O”2} Co-O 2.092 Å Co-O’ 2.076 Å Co-O” 2.065 Å Dstr = +2.15 pm Estr = 0.55 pm | KD1, 0.57 | KD2, 0.80 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 223 | δ5,6 = 508 | δ7,8 = 874 | ||
| 45·| ± 1/2> + 54·| ± 3/2> | 56·| ± 1/2> + 41·| ± 3/2> | 49·| ± 1/2> + 49·| ± 3/2> | 49·| ± 1/2> + 48·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 8, S2 = 4, classification 1–invalid | ||||
 | SH-zfs model D = +99 cm−1 gx = 2.70 gz = 2 | 4Δ0 = 0 4Δ1 = 258 4Δ2 = 732 | D = +107.9 D1 = +54.6 D2 = +34.1 | E/D = 0.15 E1 = +54.6 E2 = −34.1 | g1 = 1.860 g2 = 2.319 g3 = 2.868 giso = 2.349 |
| M [Co(iz)6]2+(fm−)2, [C18H24CoN12]2+ 2(CHO2)− | CAS Theory: Spin–Orbit Multiplets | ||||
|---|---|---|---|---|---|
CCDC 624939, 296 K, Rgt = 0.034 [39,50] | {CoN4N’2} Co-N’ 2.211 Å Co-N 2.197 Å Co-N’ 2.143 Å Dstr = −6.10 pm Estr = 0.71 pm | KD1, 0.46 | KD2, 0.54 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 256 | δ5,6 = 450 | δ7,8 = 836 | ||
| 60·| ± 1/2> + 38·| ± 3/2> | 34·| ± 1/2> + 64·| ± 3/2> | 35·| ± 1/2> + 64·| ± 3/2> | 72·| ± 1/2> + 26·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: S1 = 0.4, S2 = 0.2, classification 1–invalid | ||||
 | SH-zfs model D = +69.2 cm−1 gx = 2.75 gz = 2 | 4Δ0 = 0 4Δ1 = 35 4Δ2 = 591 | D = +124.0 D1 = +62.4 D2 = +39.0 | E/D = 0.15 E1 = +61.9 E2 = −37.0 | g1 = 1.302 g2 = 1.829 g3 = 2.974 giso = 2.035 |
| Na, [Co(bzpy)4(NCS)2], [C50H44CoN6S2] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1497489, 120 K, Rgt = 0.036 [45] | {CoN4N’2} Unit A Co-N’ 2.086 Å Co-N 2.217 Å Co-N 2.180 Å Dstr = −11.7 pm Estr = 1.35 pm | KD1, 0.68 | KD2, 0.88 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 187 | δ5,6 = 646 | δ7,8 = 965 | ||
| 79·| ± 1/2> + 21·| ± 3/2> | 13·| ± 1/2> + 85·| ± 3/2> | 33·| ± 1/2> + 67·| ± 3/2> | 78·| ± 1/2> + 20·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 28, S2 = 22, classification 3–questionable | ||||
 | SH-zfs model D = +90.5 cm−1 gx = 2.52 gz = 2 | 4Δ0 = 0 4Δ1 = 473 4Δ2 = 838 | D = +88.9 D1 = +47.2 D2 = +30.7 | E/D = 0.17 E1 = +47.0 E2 = −30.4 | g1 = 1.932 g2 = 2.446 g3 = 2.823 giso = 2.400 |
| Nb, [Co(bzpy)4(NCS)2], [C50H44CoN6S2] | CAS Theory: Spin–Orbit Multiplets | ||||
| Unit B Co-N’ 2.094 Å Co-N 2.213 Å Co-N 2.196 Å Dstr = −11.0 pm Estr = 0.85 pm | KD1, 0.67 | KD2, 0.88 | KD3 | KD4 | |
| δ1,2 = 0 | δ3,4 = 189 | δ5,6 = 638 | δδ7,8 = 975 | ||
| 79·| ± 1/2> + 21·| ± 3/2> | 12·| ± 1/2> + 85·| ± 3/2> | 33·| ± 1/2> + 66·| ± 3/2> | 80·| ± 1/2> + 18·| ± 3/2> | ||
| Magnetic data as above | SH theory: S1 = 28, S2 = 22, classification 3–questionable | ||||
| 4Δ0 = 0 4Δ1 = 481 4Δ2 = 776 | D = +91.7 D1 =+47.0 D2 = +32.1 | E/D = 0.15 E1 = +46.6 E2 = −31.6 | g1 = 1.938 g2 = 2.466 g3 = 2.806 giso = 2.403 | ||
| O, [Co(pydm)2]2+(dnbz)−2, [C14H18CoN2O4]2+·2(C7H3N2O6)− pincer type | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1533249, 100 K, Rgt = 0.037 [51] | {CoO4N2} Co-N 2.039 Å Co-O 2.110 Å Co-O 2.171 Å Dstr* = −20.15 Estr* = 3.05 | KD1, 0.71 | KD2, 0.89 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 188 | δ5,6 = 864 | δ7,8 = 1099 | ||
| 22·| ± 1/2> + 75·| ± 3/2> | 75·| ± 1/2> + 21| ± 3/2> | 59·| ± 1/2> + 36·| ± 3/2> | 37·| ± 1/2> + 61·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 58, S2 = 44, classification 5–fulfilled | ||||
 | SH-zfs model D = −62 cm−1 gz = 2.13 gx = 2 | 4Δ0 = 0 4Δ1 = 615 4Δ2 = 2199 | D = −91.8 D1 = −103.0 D2 = +8.9 | E/D = 0.13 E1 = −0.3 E2 = −11.4 | g1 = 1.983 g2 = 2.169 g3 = 3.058 giso = 2.403 |
| P, [Co(pydm)2]2+(dmnbz)−2, [C14H18CoN2O4]2+·2(C8H5N2O6)−; pincer type | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1945478, 100 K, Rgt = 0.042 [52] | {CoO4N2} Co-N 2.038 Å Co-O 2.120 Å Co-O 2.114 Å Dstr* = −17.9 pm Estr* = 0.30 pm | KD1, 0.68 | KD2, 0.88 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 145 | δ5,6 = 870 | δ7,8 = 1099 | ||
| 40·| ± 1/2> + 57·| ± 3/2> | 57·| ± 1/2> + 38·| ± 3/2> | 63·| ± 1/2> + 35·| ± 3/2> | 32·| ± 1/2> + 65·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 72, S2 = 41, classification 5–fulfilled | ||||
 | SH-zfs model D = −50.0 cm−1 gz = 2.30 gx = 2 | 4Δ0 = 0 4Δ1 = 708 4Δ2 = 1831 | D = −69.0 D1 = −86.9 D2 = +11.7 | E/D = 0.19 E1 = −0.02 E2 = −11.8 | g1 = 2.047 g2 = 2.213 g3 = 2.878 giso = 2.379 |
| Qa, [Co(pydca)(dmpy)], [C14H12CoN2O6]; pincer type | CAS Theory: Spin–Orbit Multiplets | ||||
| [Co(pydca)(dmpy)]·0.5 H2 O CCDC 1585697, 100 K, Rgt = 0.041 [53]  | A: {CoO4N2} Co-N 2.031 Å Co-O 2.152 Å Co-O 2.163 Å Dstr* = −22.6 pm Estr* = 0.55 pm | KD1, 0.78 | KD2, 0.93 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 162 | δ5,6 = 813 | δ7,8 = 1046 | ||
| 64·| ± 1/2> + 34·| ± 3/2> | 37·| ± 1/2> + 61·| ± 3/2> | 20·| ± 1/2> + 77·| ± 3/2 > 2 | 77·| ± 1/2> + 21·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 62, S2 = 40, classification 5–fulfilled | ||||
 | SH-zfs model D = −89.5 cm−1 gx = 2.42 gz = 2.50 | 4Δ0 = 0 4Δ1 = 614 4Δ2 = 2228 | D = −77.2 D1 = −93.6 D2 = +11.0 | E/D = 0.18 E1 = −0.01 E2 = −11.4 | g1 = 1.992 g2 = 2.226 g3 = 2.945 giso = 2.388 |
 | GF model λεff = −141 cm−1 gL = −1.13 Δax = −811 cm−1 | ||||
| Qb, [Co(pydca)(dmpy)], [C14H12CoN2O6]; pincer | CAS Theory: Spin–Orbit Multiplets | ||||
| B: {CoO4N2} Co-N 2.028 Å Co-O 2.133 Å Co-O 2.176 Å Dstr* = −22.6 pm Estr* = 2.15 pm | KD1, 0.82 | KD2, 0.95 | KD3 | KD4 | |
| δ1,2 = 0 | δ3,4 = 147 | δ5,6 = 968 | δ7,8 = 1179 | ||
| 8·| ± 1/2> + 90·| ± 3/2> | 90·| ± 1/2> + 7·| ± 3/2> | 83·| ± 1/2> + 12·| ± 3/2> | 14·| ± 1/2> + 85·| ± 3/2> | ||
| Magnetic data as above | SH theory: S1 = 107, S2 = 96, classification 5–fulfilled | ||||
| 4Δ0 = 0 4Δ1 = 786 4Δ2 = 2692 | D = −97.1 D1 = −112.0 D2 = +9.6 | E/D = 0.10 E1 = −0.08 E2 = −6.9 | g1 = 2.022 g2 = 2.112 g3 = 2.898 giso = 2.377 | ||
| R, [Co(ac)2(H2O)2(MeIm)2], [C12H22CoN4O6] | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 618142, 100 K, Rgt = 0.033 [54,55] | {CoO2O’2N2} Co-N 2.127 Å Co-O’ 2.122 Å Co-Ow 2.170 Å Dstr = −11.9 pm Estr = 2.4 pm | KD1, 0.84 | KD2, 0.93 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 156 | δ5,6 = 1030 | δ7,8 = 1230 | ||
| 41·| ± 1/2> + 57·| ± 3/2> | 58·| ± 1/2> + 40·| ± 3/2> | 54·| ± 1/2> + 44·| ± 3/2> | 41·| ± 1/2> + 57·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 123, S2 = 70, classification–5 fulfilled | ||||
 | GF model λeff = −217 cm−1 gLz = −1.23 gLz = −1.37 Δax = +568 cm−1 | 4Δ0 = 0 4Δ1 = 878 4Δ2 = 1593 | D = +75.1 D1 = +35.0 D2 = +23.2 | E/D = 0.16 E1 = +35.0 E2 = −23.0 | g1 = 1.910 g2 = 2.508 g3 = 2.764 giso = 2.394 |
 | SH-zfs model D = +82 cm−1 gx = 2.54 gz = 2 | ||||
| S, [Co(ampyd)2Cl2], [C16H20Cl2CoN12] | CAS Theory: Spin–Orbit Multiplets | ||||
| CCDC SEQFUQ, 293 K, Rgt = 0.023 [56,57]  | {CoN4Cl2} Co-Cl 2.450 Å Co-N 2.233 Å Co-N 2.233 Å Dstr = −7.03 pm Estr = 0 | KD1, 0.50 | KD2, 0.63 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 262 | δ5,6 = 461 | δ7,8 = 800 | ||
| 54·| ± 1/2> + 45·| ± 3/2> | 41·| ± 1/2> + 57·| ± 3/2> | 50·| ± 1/2> + 49·| ± 3/2> | 53·| ± 1/2> + 46·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: S1 = 1, S2 = 0.6, classification 1–invalid | ||||
 | GF model λeff = −181 cm−1 gLz = −1.5 gLx = −1.3 Δax = +377 cm−1 | 4Δ0 = 0 4Δ1 = 77 4Δ2 = 869 | D = +121 D1 = +60.9 D2 = +30.9 | E/D = 0.23 E1 = +60.9 E2 = −30.9 | g1 = 1.434 g2 = 1.900 g3 = 3.066 giso = 2.133 |
 | SH-zfs model D = +146 cm−1 gx = 2.91 gz = 2 | ||||
| T, [Co(dppmO,O)3]2+·CoCl42−, [C75H66CoO6P6]2+ CoCl42− | CAS Theory: Spin–Orbit Multiplets | ||||
|---|---|---|---|---|---|
CCDC 296003, 100 K, Rgt = 0.066 [48] | {CoO3O’3} Co-O 2.112 Å Co-O’ 2.074 Å | KD1, 0.40 | KD2, 0.72 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 317 | δ5,6 = 389 | δ7,8 = 925 | ||
| 50·| ± 1/2> + 49·| ± 3/2> | 49·| ± 1/2> + 49·| ± 3/2> | 49·| ± 1/2> + 50·| ± 3/2> | 48·| ± 1/2> + 50·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 0.7, S2 = 0.4, classification 1–invalid | ||||
 | SH-zfs model D = +77 cm−1 gx = 2.55 gz = 2 | 4Δ0 = 0 4Δ1 = 142 4Δ2 = 142 | D = +158.3 D1 = +59.1 D2 = +59.1 | E/D = 0.00 E1 = +59.1 E2 = −59.1 | g1 = 1.781 g2 = 2.505 g3 = 2.505 giso = 2.263 |
| Ua, cis-[Co(phen)2(dca)2], [C28H16CoN10] α-polymorph | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 997503, 193 K, Rgt = 0.029 [58] | {CoN4N’2} Co-N 2.153 Å Co-N’dca 2.076 Å | KD1, 0.54 | KD2, 0.65 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 243 | δ5,6 = 495 | δ7,8 = 838 | ||
| 19·| ± 1/2> + 79·| ± 3/2> | 82·| ± 1/2> + 17·| ± 3/2> | 82·| ± 1/2> + 15·| ± 3/2> | 8·| ± 1/2> + 90·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: S1 = 2, S2 = 2, classification 1–invalid | ||||
 | SH-zfs model D = +91 cm−1 gx = 2.66 gz = 2 | 4Δ0 = 0 4Δ1 = 110 4Δ2 = 961 | D = 108.2 D1 = 63.7 D2 = 27.3 | E/D = 0.30 E1 = 63.7 E2 = −27.2 | g1 = 1.487 g2 = 1.956 g3 = 3.085 giso = 2.176 |
| Ub, cis-[Co(phen)2(dca)2], [C28H16CoN10] β-polymorph | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 997504, 293 K, Rgt = 0.040 [58] | {CoN4N’2} Co-N 2.153 Å Co-N’dca 2.071 Å | KD1, 0.76 | KD2, 0.91 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 168 | δ5,6 = 737 | δ7,8 = 1029 | ||
| 51·| ± 1/2> + 48·| ± 3/2> | 46·| ± 1/2> + 51·| ± 3/2> | 52·| ± 1/2> + 46·| ± 3/2> | 47·| ± 1/2> + 50·| ± 3/2> | ||
| Magnetic data, SMR–n.a. | SH theory: S1 = 51, S2 = 26, classification 5–fulfilled | ||||
 | SH-zfs model D = +85 cm−1 gx = 2.60 gz = 2 | 4Δ0 = 0 4Δ1 = 618 4Δ2 = 1041 | D = 81.2 D1 = 40.3 D2 = 25.7 | E/D = 0.16 E1 = 40.1 E2 = −25.7 | g1 = 1.923 g2 = 2.460 g3 = 2.768 giso = 2.383 |
| V, [μ-(dca)Co(pypz)(H2O)] dca a | |||||
CCDC 1973544, 295 K, Rgt = 0.033 [59] | {CoN3N’2O} Co-N 2.155 Å Co-N’ 2.076 Å Co-O 2.134 Å | ||||
| Magnetic data, SMR–yes | |||||
 | GF model λeff = −131 cm−1 gL = −2.00 Δax = −2000 cm−1 | ||||
| W, [Co(pypz)2]2+(tcm)−2, [C22H18CoN10]2+·2(C3N4)− a | CAS Theory: Spin–Orbit Multiplets | ||||
CCDC 1973546, 295 K, Rgt = 0.037 [59] | {CoN4N’2} Co-N’ 2.082 Å Co-N 2.164 Å Co-N 2.164 Å Dstr* = −8.2 pm Estr = 0 | KD1, 0.74 | KD2, 0.91 | KD3 | KD4 |
| δ1,2 = 0 | δ3,4 = 159 | δ5,6 = 717 | δ7,8 = 1003 | ||
| 14·| ± 1/2> + 85·| ± 3/2> | 85·| ± 1/2> + 11·| ± 3/2> | 88·| ± 1/2> + 11·| ± 3/2> | 3·| ± 1/2> + 95·| ± 3/2> | ||
| Magnetic data, SMR–yes | SH theory: S1 = 47, S2 = 40, classification 4–acceptable | ||||
 | GF model λeff = −87 cm−1 gL = −2.77 Δax = −4000 cm−1 | 4Δ0 = 0 4Δ1 = 571 4Δ2 = 1179 | D = +72.2 D1 = +43.4 D2 = +21.0 | E/D = 0.26 E1 = +43.6 E2 = −21.0 | g1 = 1.990 g2 = 2.349 g3 = 2.818 giso = 2.384 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boča, R.; Rajnák, C.; Titiš, J. Zero-Field Splitting in Hexacoordinate Co(II) Complexes. Magnetochemistry 2023, 9, 100. https://doi.org/10.3390/magnetochemistry9040100
Boča R, Rajnák C, Titiš J. Zero-Field Splitting in Hexacoordinate Co(II) Complexes. Magnetochemistry. 2023; 9(4):100. https://doi.org/10.3390/magnetochemistry9040100
Chicago/Turabian StyleBoča, Roman, Cyril Rajnák, and Ján Titiš. 2023. "Zero-Field Splitting in Hexacoordinate Co(II) Complexes" Magnetochemistry 9, no. 4: 100. https://doi.org/10.3390/magnetochemistry9040100