

Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 3: Fe, Co, Ni, Pd, and Pt Glycinates



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
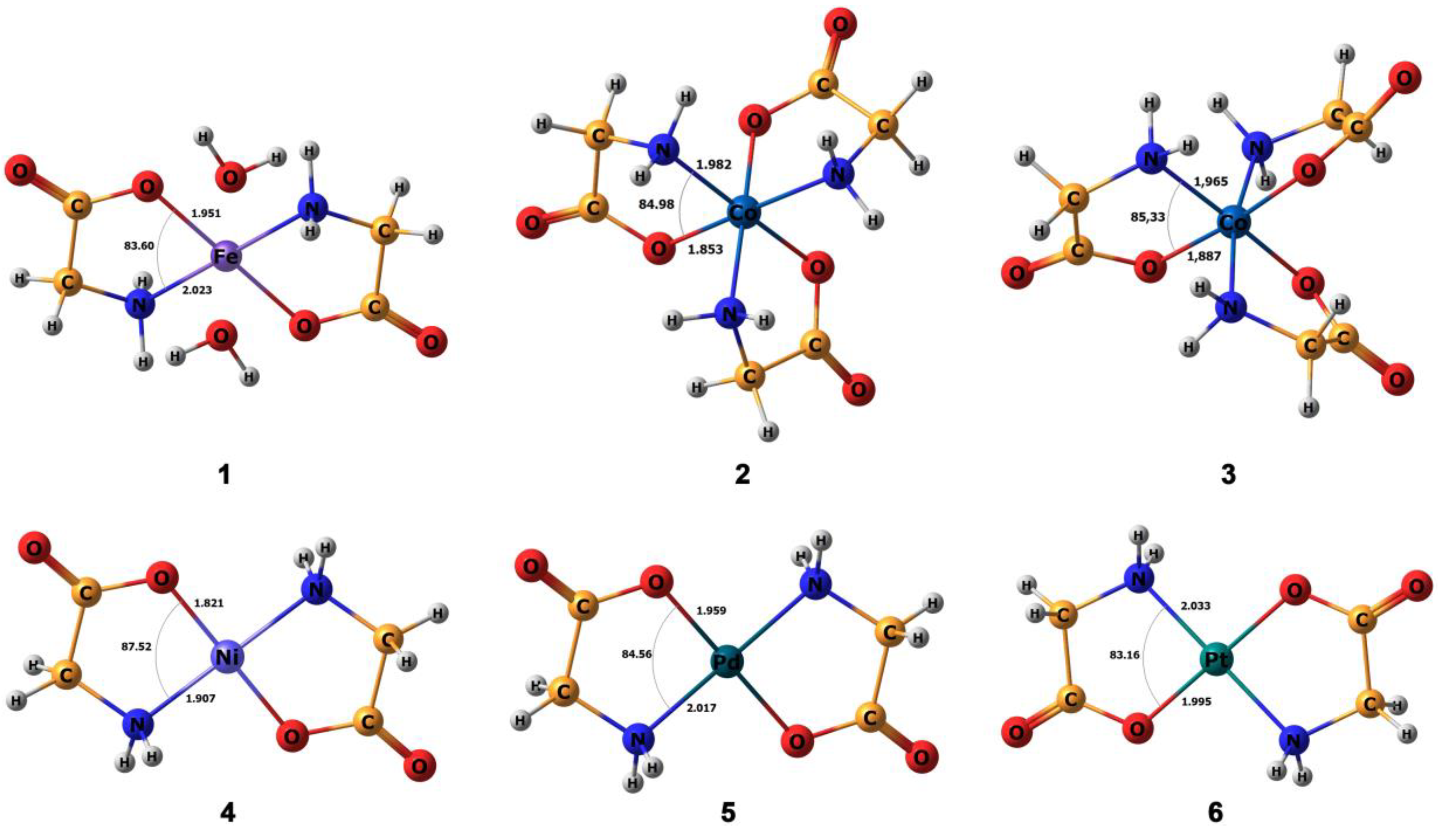
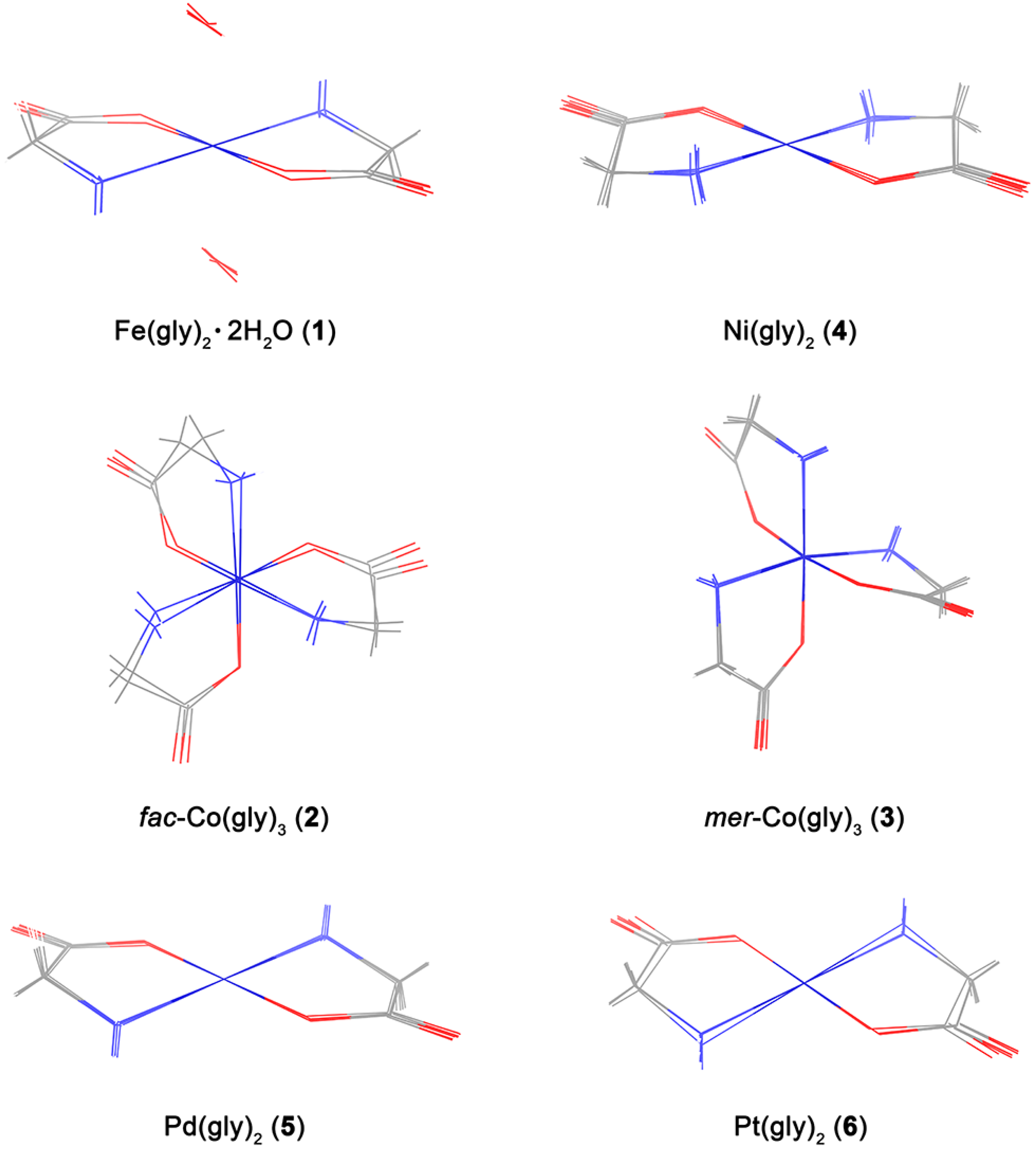
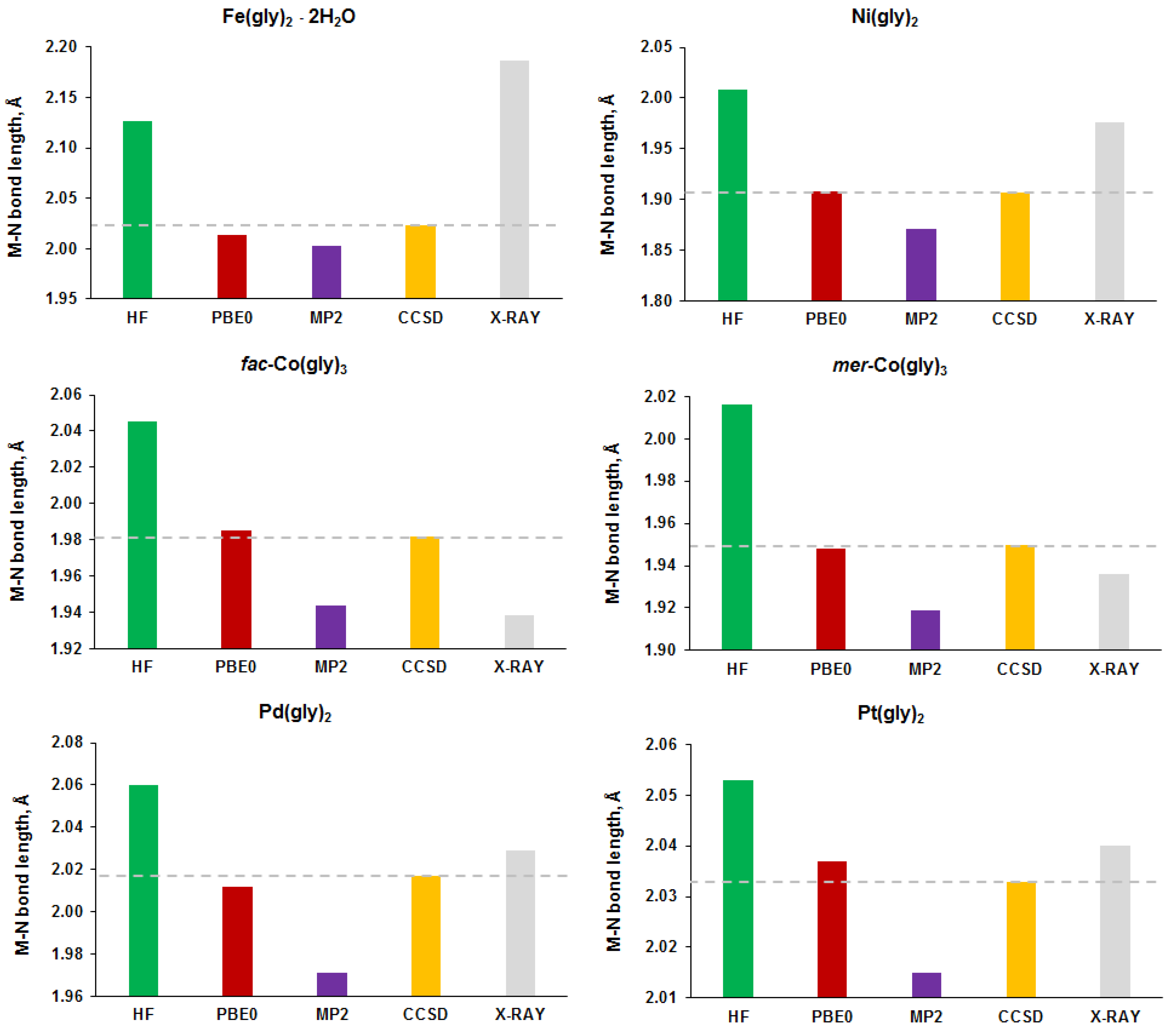
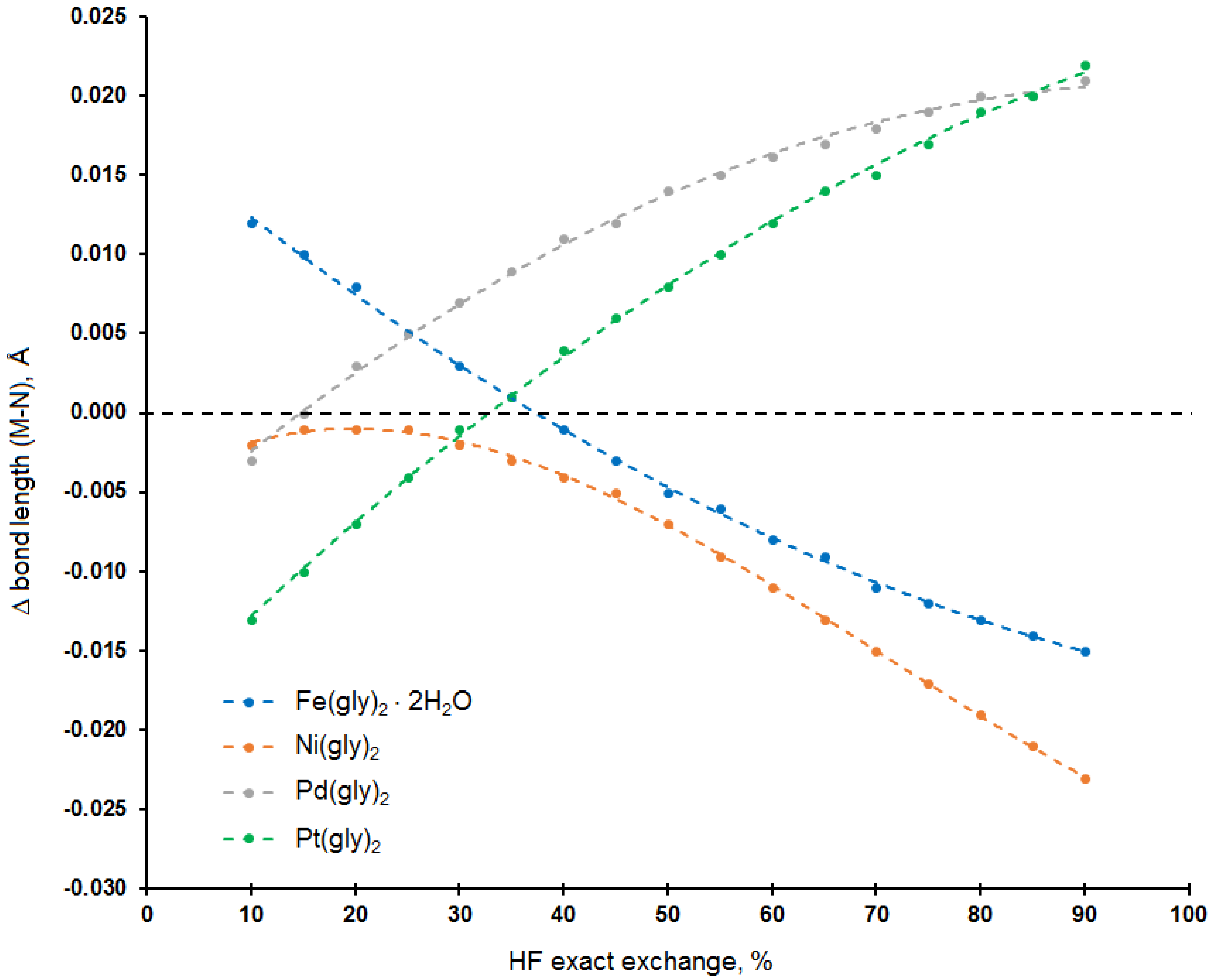
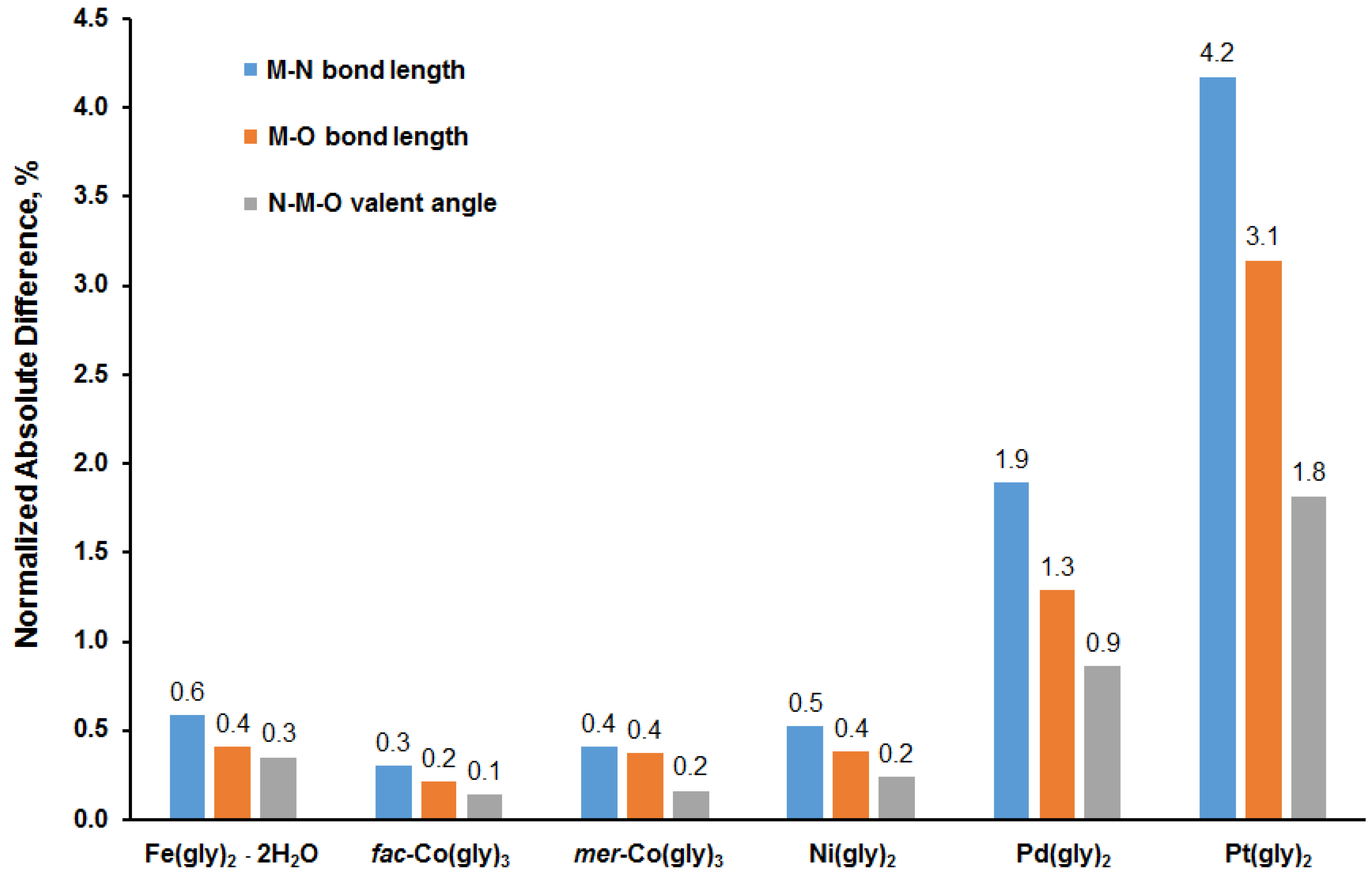
3.1. Calculation of Molecular Geometry
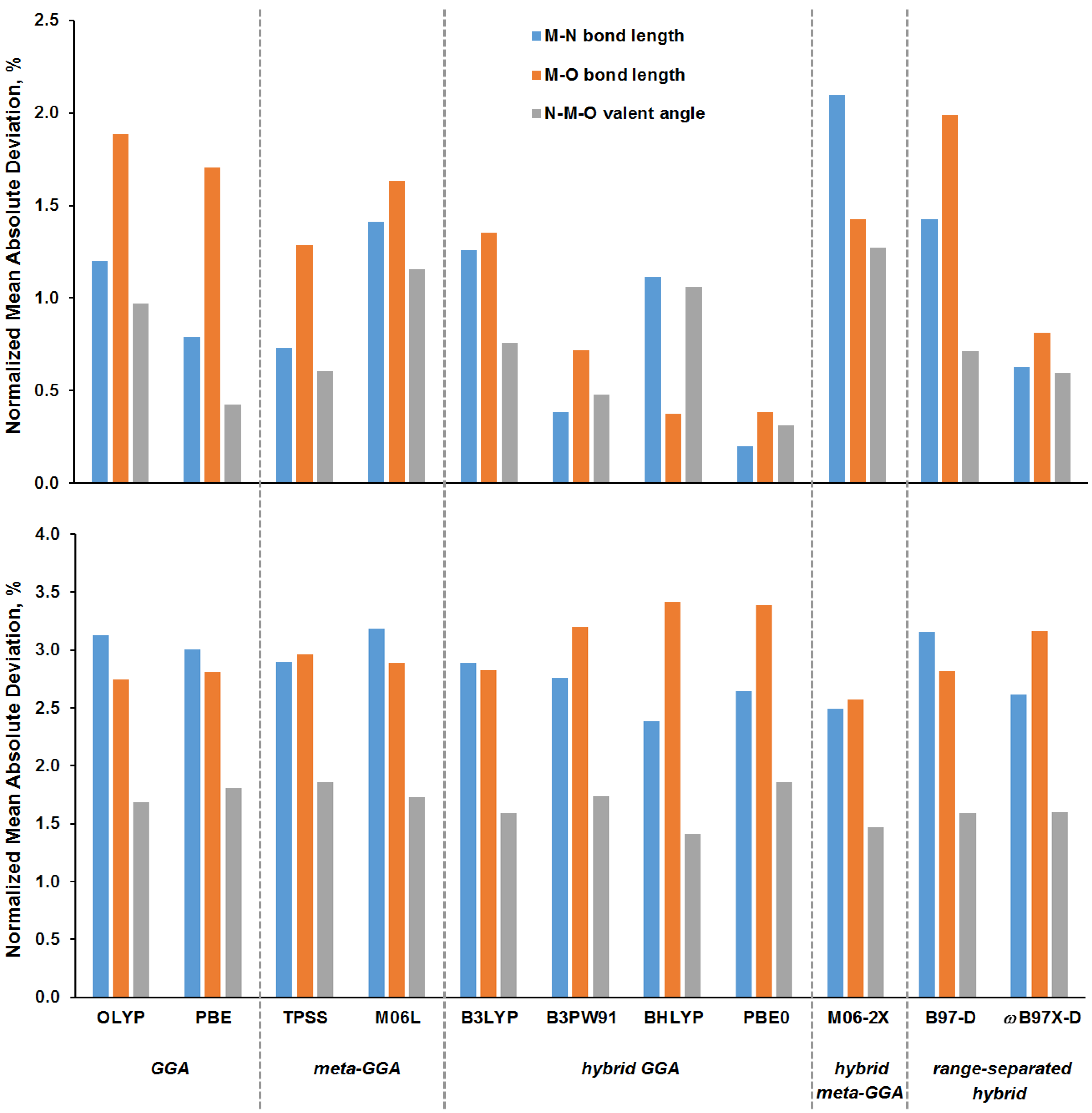
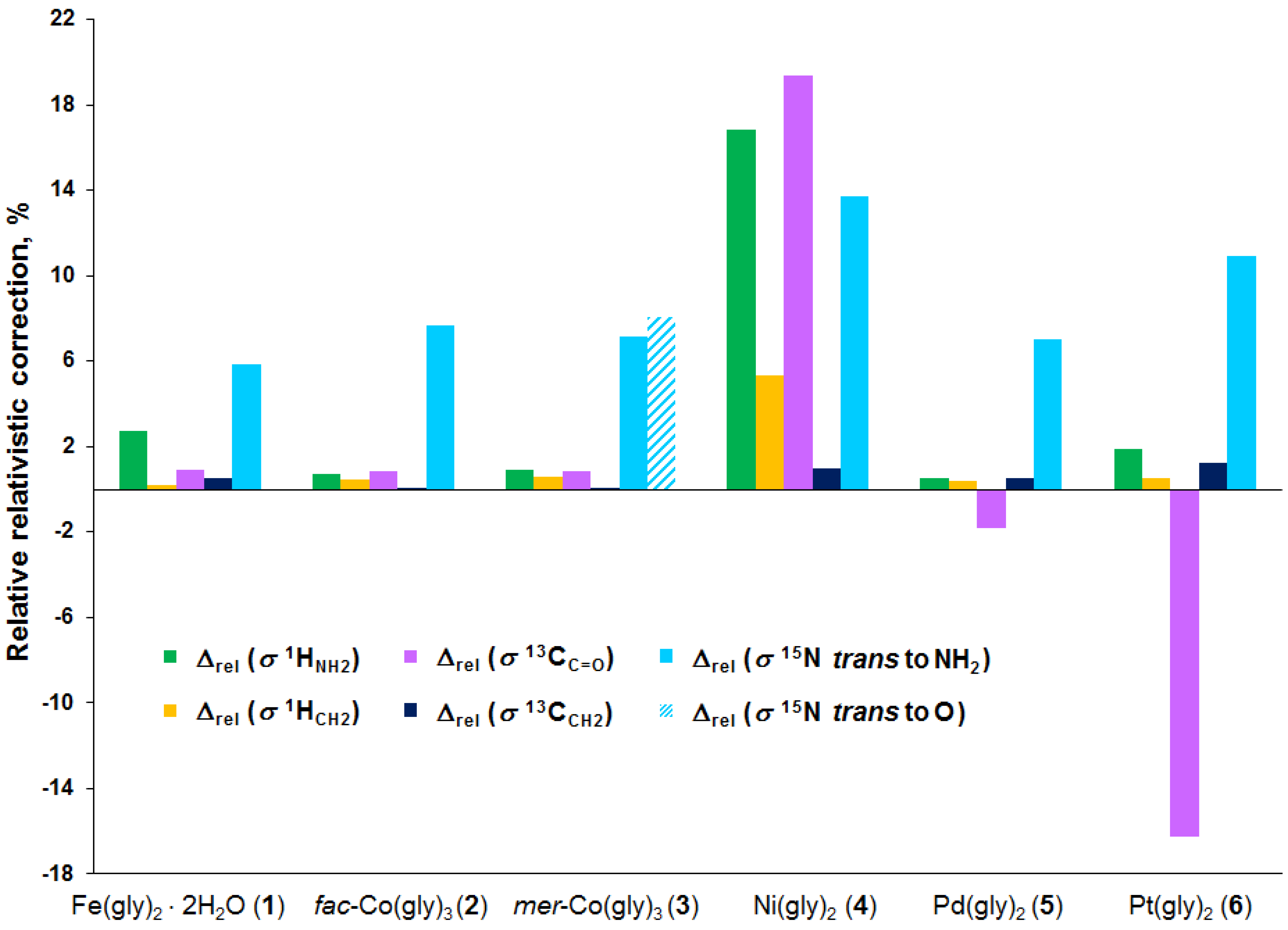
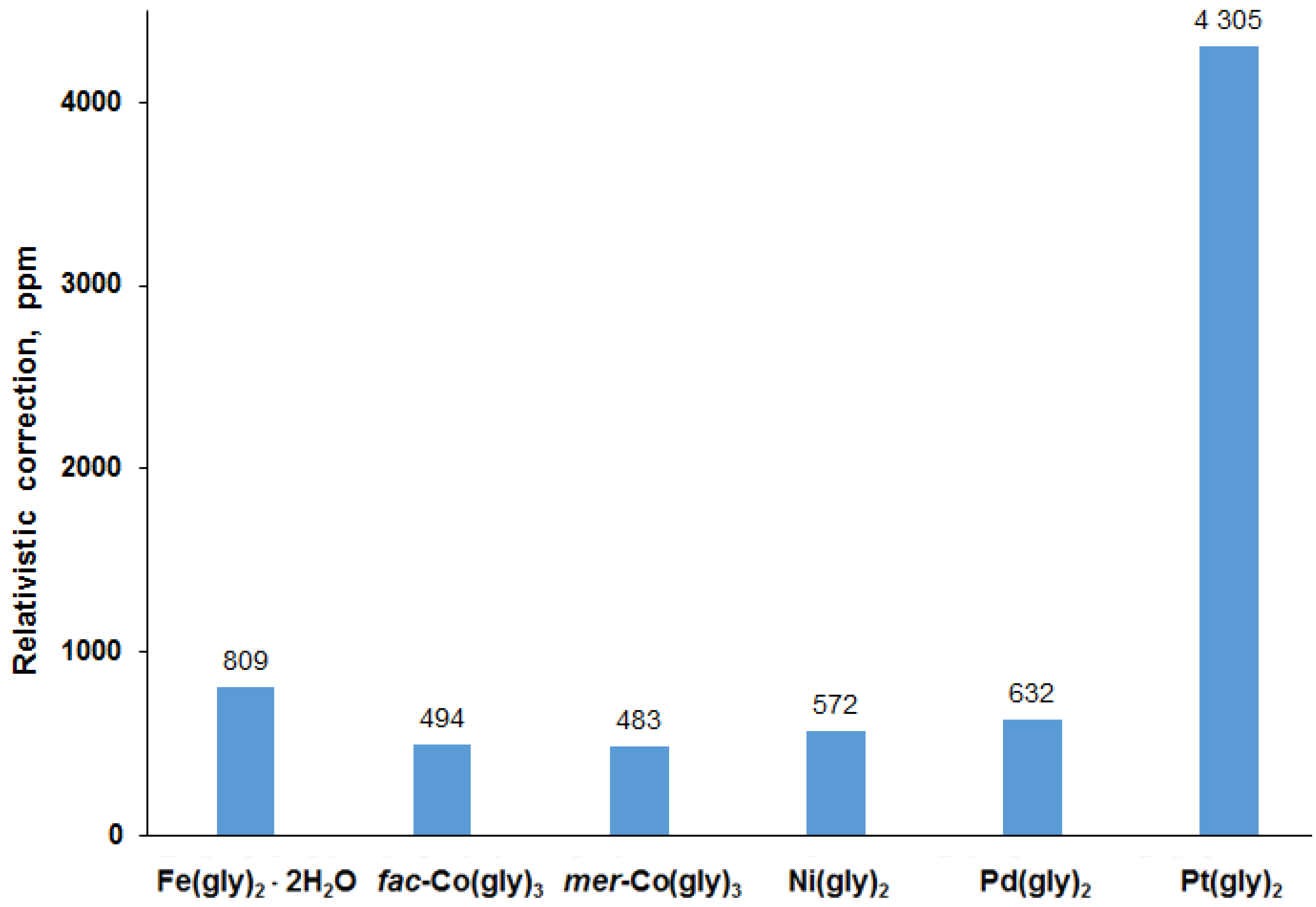
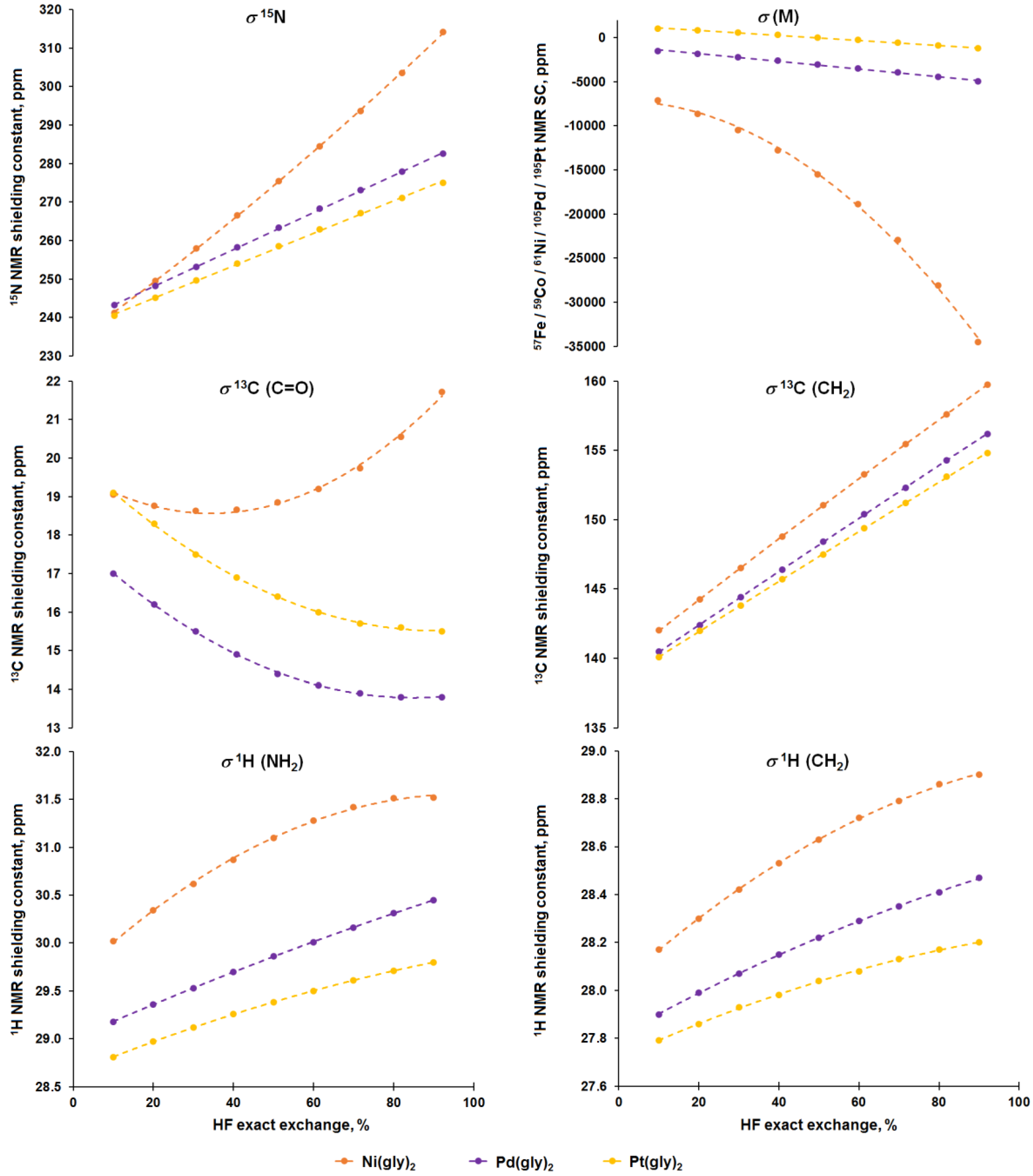
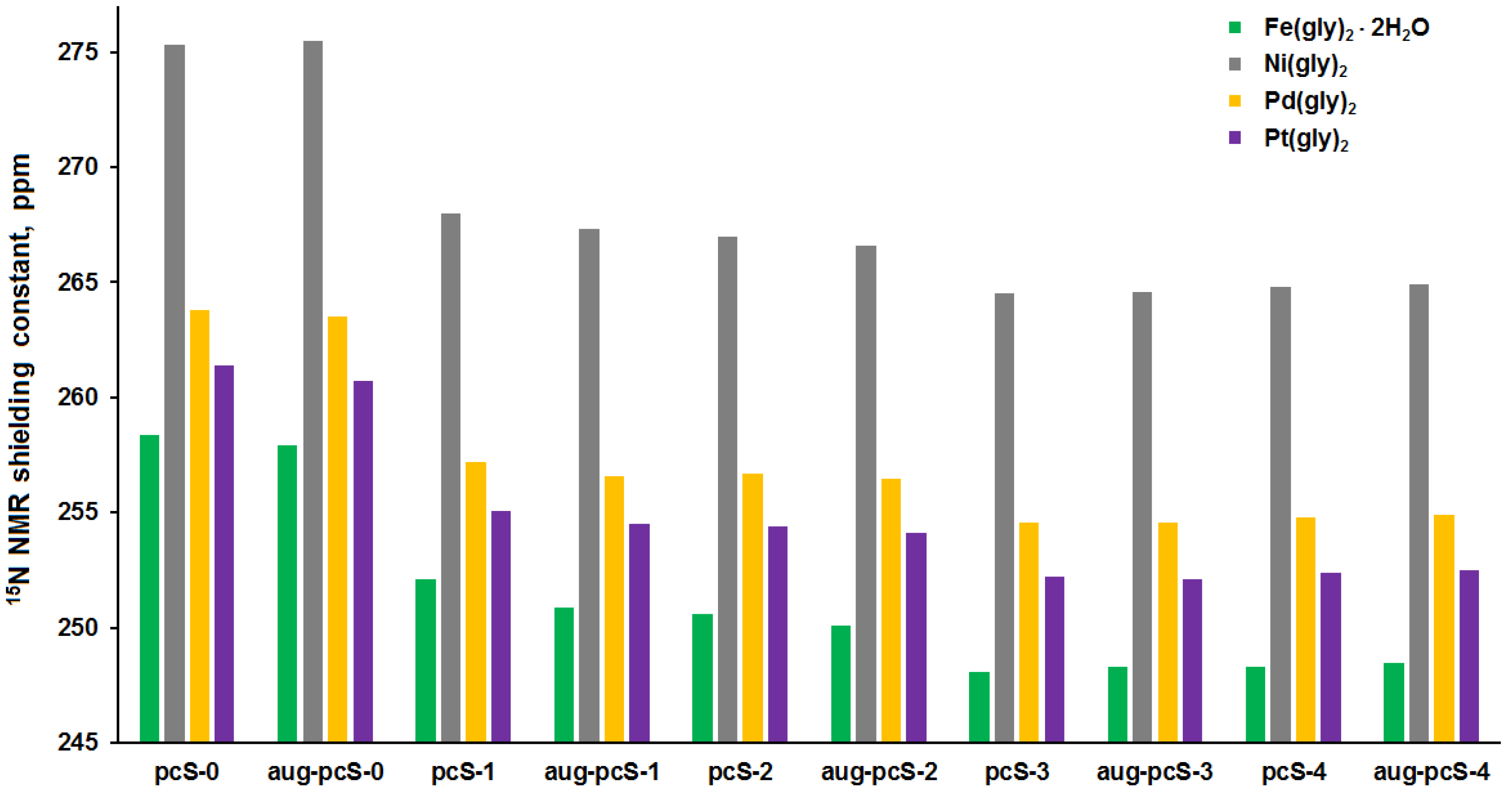
3.2. Analysis of Calculated Shielding Constants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nomura, Y.; Takeuchi, Y. Substituent effects in aromatic proton NMR spectra. III substituent effects caused by halogens. Tetrahedron Lett. 1969, 10, 639–642. [Google Scholar] [CrossRef]
- Vícha, J.; Novotný, J.; Komorovsky, S.; Straka, M.; Kaupp, M.; Marek, M. Relativistic heavy-neighbor-atom effects on NMR shifts: Concepts and trends across the Periodic Table. Chem. Rev. 2020, 120, 7065–7103. [Google Scholar] [CrossRef] [PubMed]
- Rusakova, I.L.; Rusakov, Y.Y. Relativistic effects from heavy main group p-elements on the NMR chemical shifts of light atoms: From pioneering studies to recent advances. Magnetochemistry 2023, 9, 24. [Google Scholar] [CrossRef]
- Rusakova, I.L. Quantum Chemical Approaches to the Calculation of NMR Parameters: From Fundamentals to Recent Advances. Magnetochemistry 2022, 8, 50. [Google Scholar] [CrossRef]
- Samultsev, D.O.; Semenov, V.A.; Krivdin, L.B. Four-component relativistic calculations of NMR shielding constants of the transition metal complexes. Part 1: Pentaammines of cobalt, rhodium, and iridium. Magn. Reson. Chem. 2022, 60, 463–468. [Google Scholar] [CrossRef]
- Samultsev, D.O.; Semenov, V.A.; Rusakova, I.L.; Krivdin, L.B. Four-component relativistic calculations of NMR shielding constants of the transition metal complexes. Part 2: Nitrogen-coordinated complexes of cobalt. Int. J. Mol. Sci. 2022, 23, 13178. [Google Scholar] [CrossRef]
- Jorge, F.E.; Canal Neto, A.; Camiletti, G.G.; Machado, S.F. Contracted Gaussian basis sets for Douglas–Kroll–Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B. GAUSSIAN 09, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009; Available online: http://www.gaussian.com (accessed on 15 March 2023).
- Hess, B.A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742–3748. [Google Scholar] [CrossRef] [Green Version]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Emiliano Deustua, J.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [Green Version]
- Jensen, H.J.A.; Bast, R.; Saue, T.; Visscher, L. DIRAC16, Revision 2016. Available online: http://www.diracprogram.org (accessed on 15 March 2023).
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1998, 298, 113–119. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic and nonrelativistic finite nucleus optimized double zeta basis sets for the 4p, 5p and 6p elements. Theor. Chem. Acc. 1998, 99, 366–371. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic and nonrelativistic finite nucleus optimized triple-zeta basis sets for the 4p, 5p and 6p elements. Theor. Chem. Acc. 2002, 108, 335–340. [Google Scholar] [CrossRef]
- Jensen, F. Basis set convergence of nuclear magnetic shielding constants calculated by density functional methods. J. Chem. Theory Comp. 2008, 4, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Bernal, I.; Somoza, F. Experimental Crystal Structure Determination; CCDC 645775; Cambridge Crystallographic Data Centre: Cambridge, UK, 2009. [Google Scholar] [CrossRef]
- Gu, K.-Q.; Sun, Y.X.; Zhang, R.; Zhang, N.W.; Che, H.W. Tris(glycinato-κ2N,O)cobalt(III). Acta Cryst. Sect. E Struct. Rep. Online 2007, 63, m740. [Google Scholar] [CrossRef]
- Jungen, S.; Chen, P. Alkyl Radical Generation by an Intramolecular Homolytic Substitution Reaction between Iron(II) and Trialkylsulfonium Groups. Chem. Eur. J. 2018, 24, 11008. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Zhang, Z.-B. A monoclinic polymorph of bis(2-amino acetato)nickel(II) monohydrate. Acta Cryst. 2006, E62, m2546. [Google Scholar] [CrossRef]
- Baidina, I.A.; Podberezskaya, N.V.; Borisov, S.V.; Golubovskaya, E.V. Crystal structure of the β-modification of palladium(II)-trans-diglycinate. J. Struct. Chem. 1982, 23, 241–244. [Google Scholar] [CrossRef]
- Freeman, H.C.; Golomb, M.L. The crystal structure of trans-bisglycinatoplatinum(II), Pt(NH2CH2COO)2. Acta Cryst. 1969, B25, 1203. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1. [Google Scholar] [CrossRef]
- Manninen, P.; Lantto, P.; Vaara, J.; Ruud, K. Perturbational ab initio calculations of relativistic contributions to nuclear magnetic resonance shielding tensors. J. Chem. Phys. 2003, 119, 2623–2637. [Google Scholar] [CrossRef]
- Semenov, V.A.; Samultsev, D.O.; Rusakova, I.L.; Krivdin, L.B. Computational multinuclear NMR of platinum complexes: A relativistic four-component study. J. Phys. Chem. A 2019, 123, 4908–4920. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samultsev, D.O.; Semenov, V.A.; Krivdin, L.B. Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 3: Fe, Co, Ni, Pd, and Pt Glycinates. Magnetochemistry 2023, 9, 83. https://doi.org/10.3390/magnetochemistry9030083
Samultsev DO, Semenov VA, Krivdin LB. Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 3: Fe, Co, Ni, Pd, and Pt Glycinates. Magnetochemistry. 2023; 9(3):83. https://doi.org/10.3390/magnetochemistry9030083
Chicago/Turabian StyleSamultsev, Dmitry O., Valentin A. Semenov, and Leonid B. Krivdin. 2023. "Four-Component Relativistic Calculations of NMR Shielding Constants of the Transition Metal Complexes—Part 3: Fe, Co, Ni, Pd, and Pt Glycinates" Magnetochemistry 9, no. 3: 83. https://doi.org/10.3390/magnetochemistry9030083