
Improved Annotation of the Peach (Prunus persica) Genome and Identification of Tissue- or Development Stage-Specific Alternative Splicing through the Integration of Iso-Seq and RNA-Seq Data
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Illumina RNA-Seq Library Construction
2.3. PacBio Technology-Based Full-Length cDNA Library Preparation and Sequencing
2.4. Preprocessing of PacBio Reads
2.5. Gene Fusion Characterization
2.6. Identification of Novel Genes
2.7. Identification of Alternative Splicing (AS) Isoforms and Poly(A) Sites
2.8. Prediction of lncRNAs
2.9. Illumina Sequencing Data Analysis
2.10. Real-Time Quantitative PCR (RT–qPCR)
3. Results
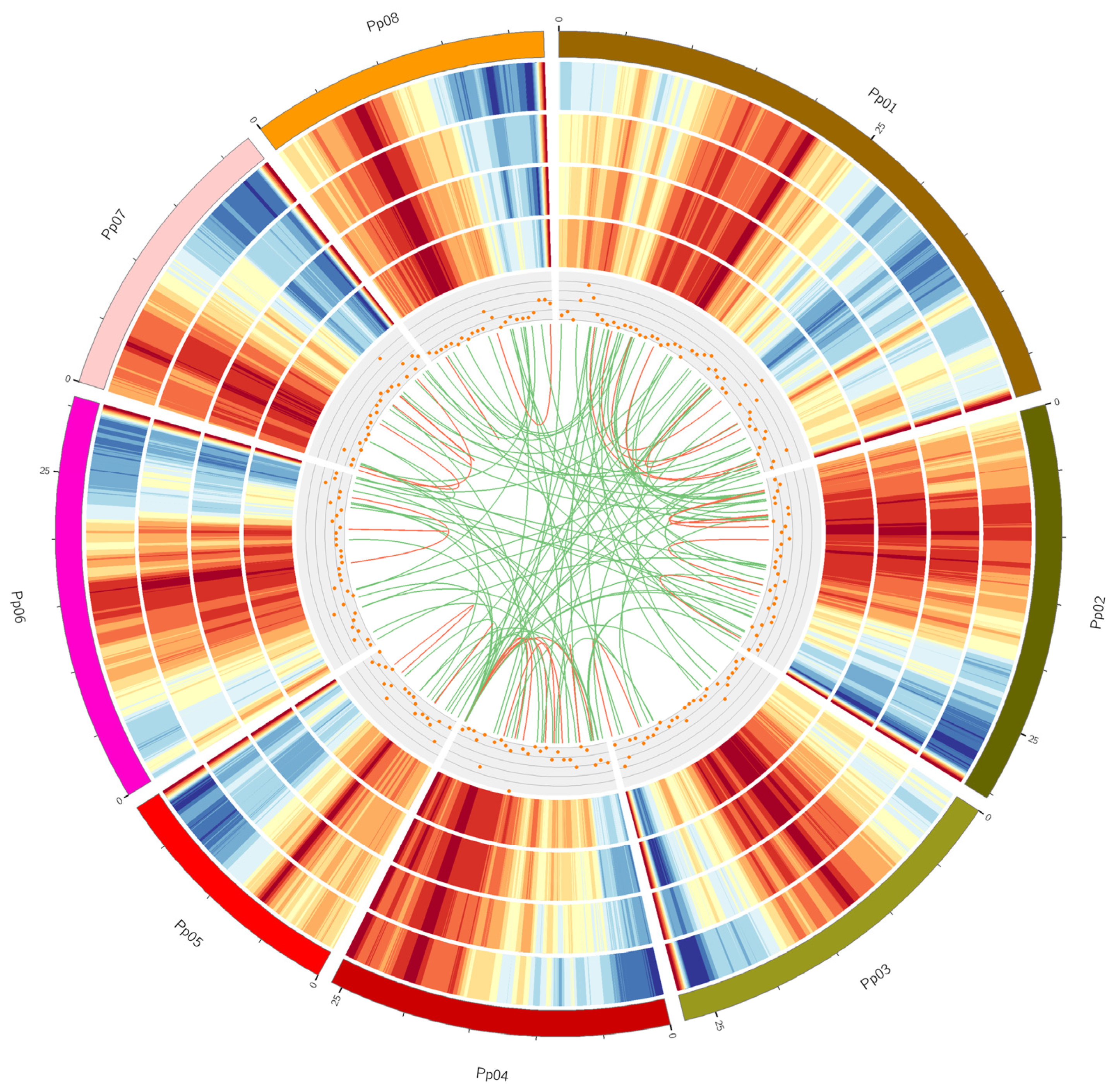
3.1. Peach PacBio Iso-Seq
3.2. Discovery of Novel Transcripts
3.3. Identification of Fusion Transcripts
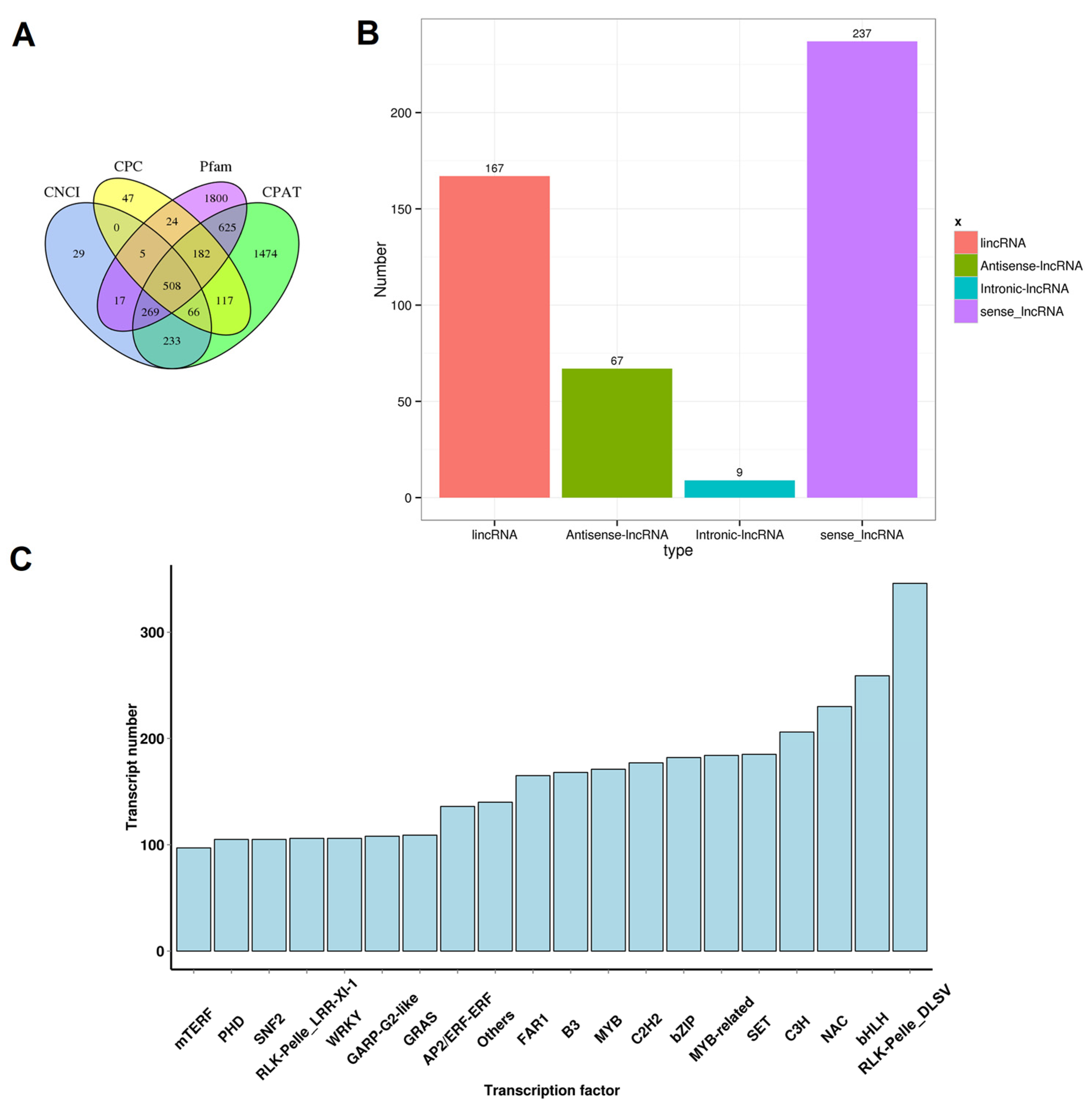
3.4. Prediction of lncRNAs and Determination of TFs
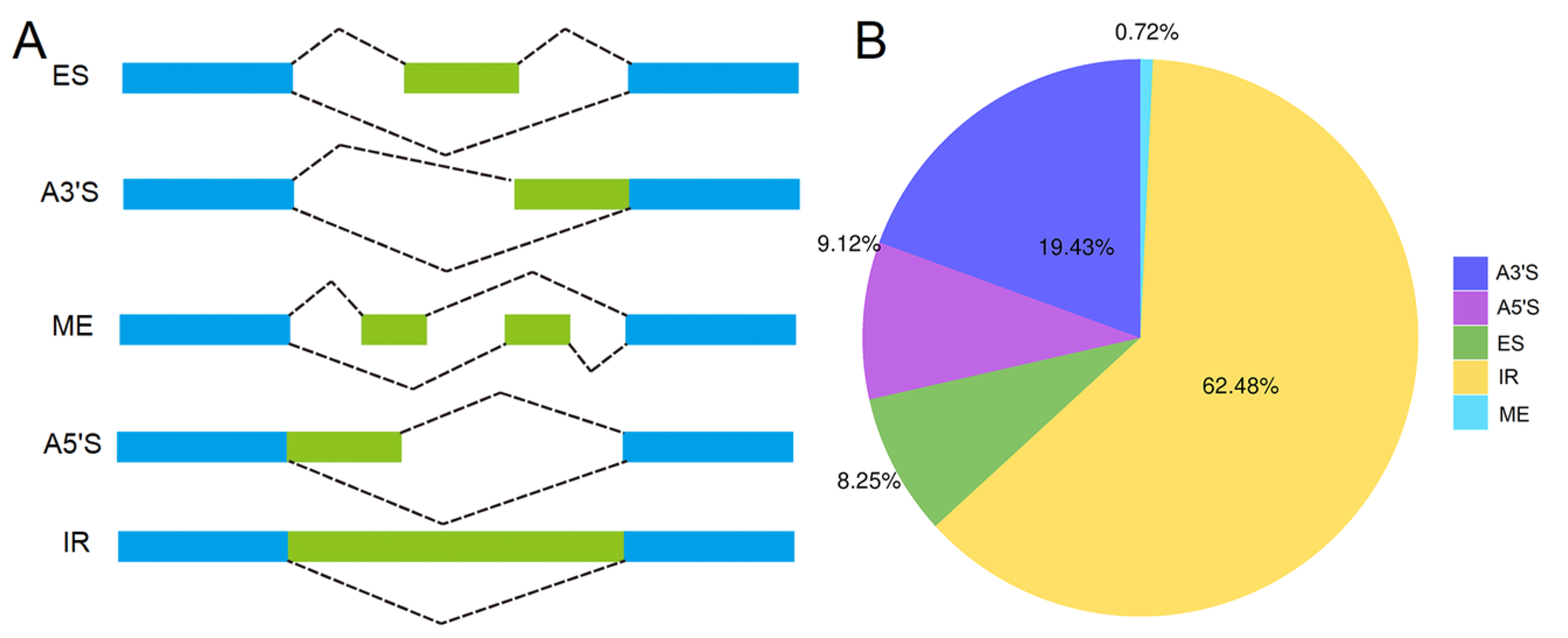
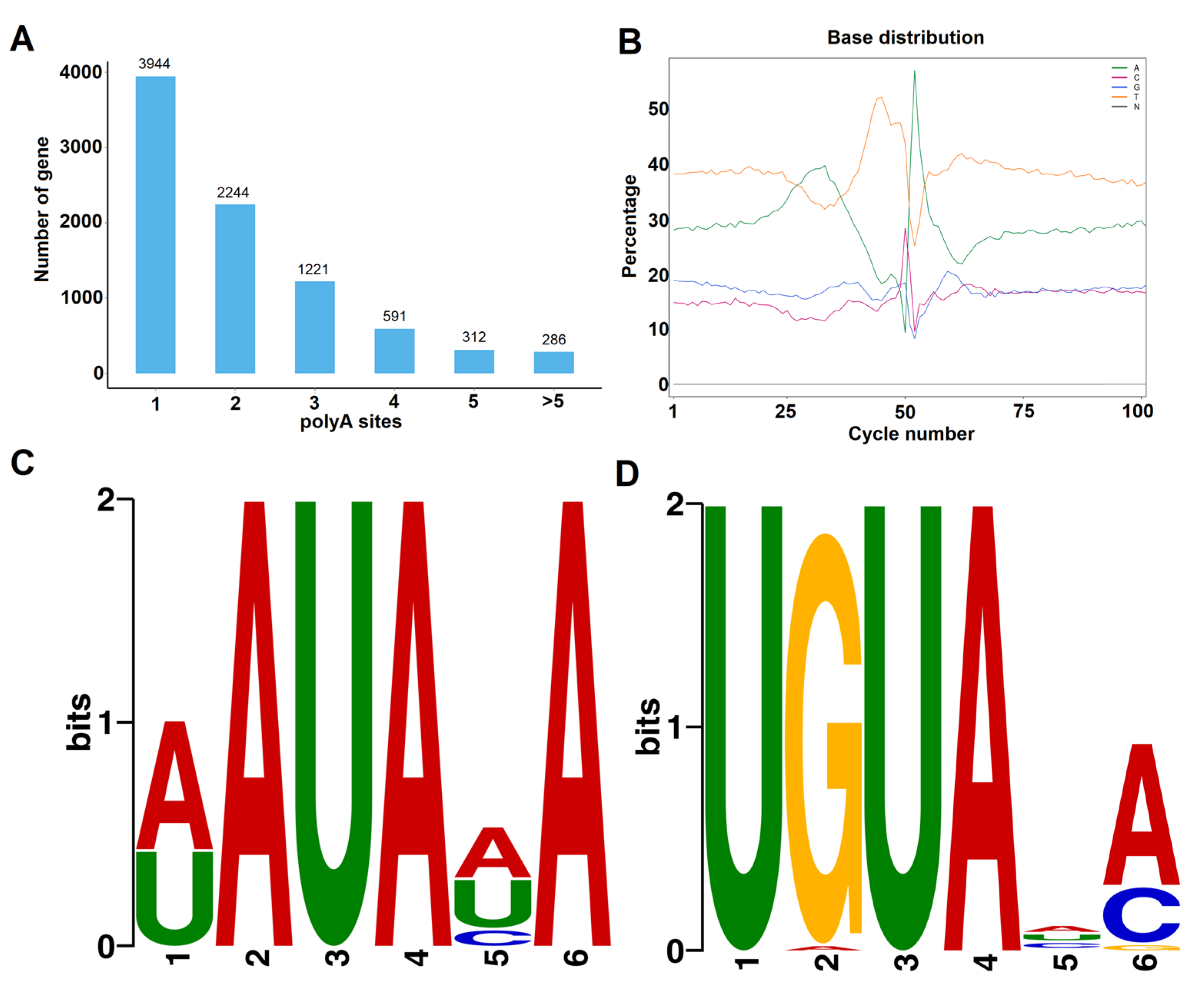
3.5. Analysis of AS and APA Events
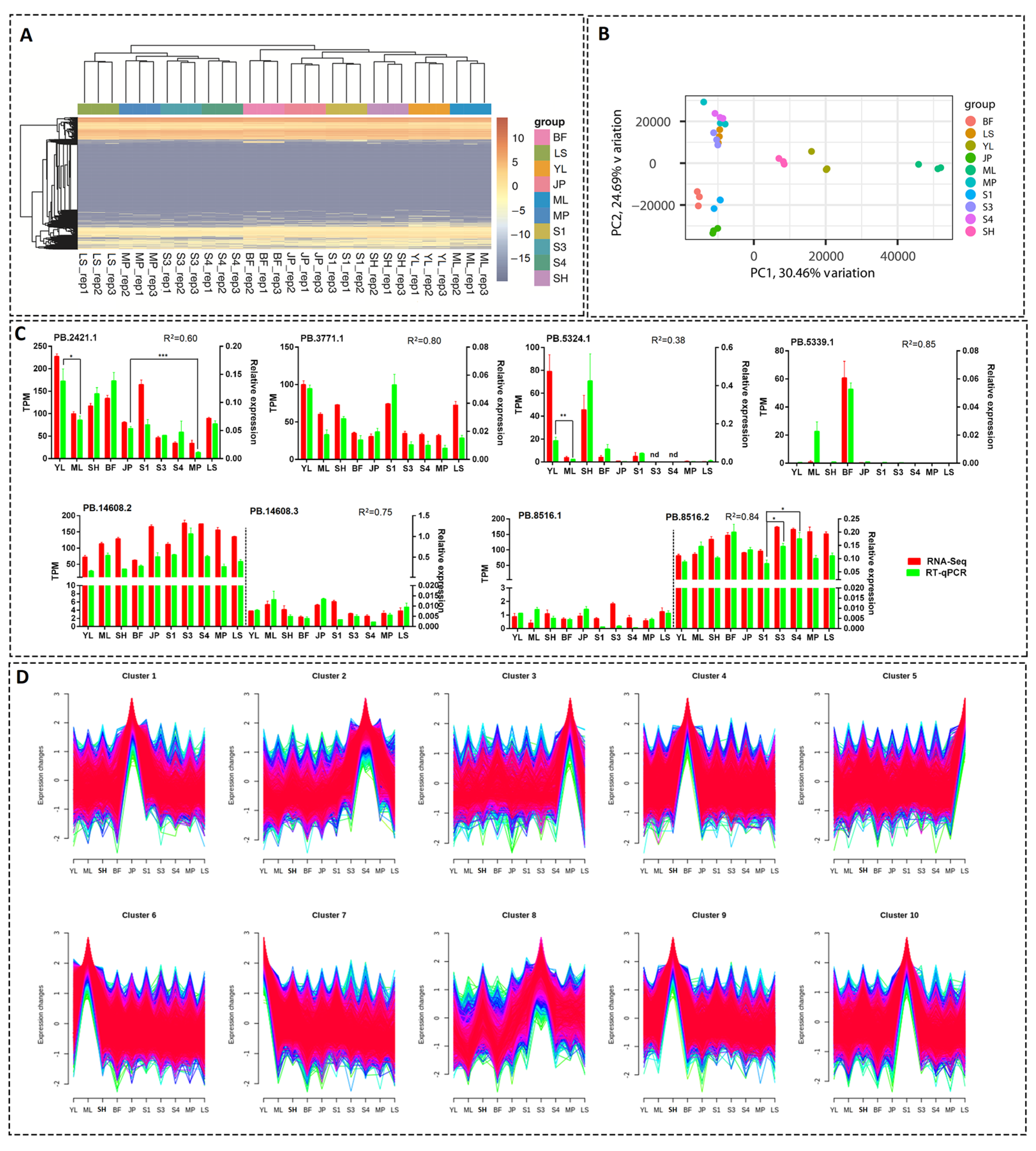
3.6. Estimation of Expression Levels of the Transcripts
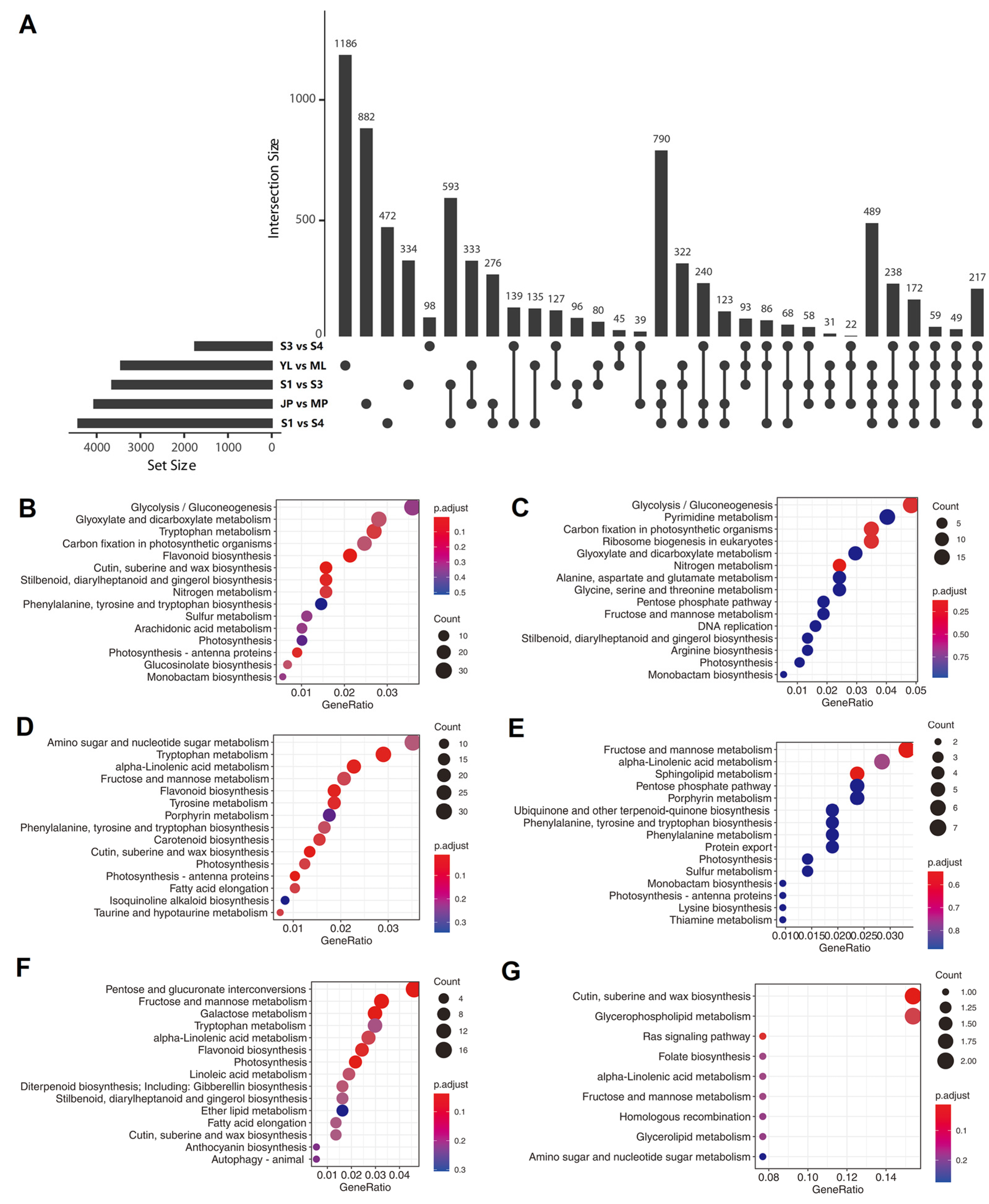
3.7. Analysis of Tissue- or Development Stage-Specific Differentially Expressed Transcripts (DETs)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Khan, M.; Rahim, T.; Naeem, M.; Shah, M.; Bakhtiar, Y.; Tahir, M. Post harvest economic losses in peach produce in district Swat. Sarhad J. Agric. 2008, 24, 705–711. [Google Scholar]
- Luo, C.-X.; Schnabel, G.; Hu, M.; De Cal, A. Global distribution and management of peach diseases. Phytopathol. Res. 2022, 4, 30. [Google Scholar] [CrossRef]
- Minas, I.S.; Tanou, G.; Molassiotis, A. Environmental and orchard bases of peach fruit quality. Sci. Hortic.-Amst. 2018, 235, 307–322. [Google Scholar] [CrossRef]
- Arumuganathan, K.; Earle, E. Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Verde, I.; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; Cattonaro, F.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verde, I.; Jenkins, J.; Dondini, L.; Micali, S.; Pagliarani, G.; Vendramin, E.; Paris, R.; Aramini, V.; Gazza, L.; Rossini, L.; et al. The Peach v2.0 release: High-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Shen, N.; Lu, Z.; Xu, G.; Wang, Y.; Jin, B. Analysis and comprehensive comparison of PacBio and nanopore-based RNA sequencing of the Arabidopsis transcriptome. Plant Methods 2020, 16, 85. [Google Scholar] [CrossRef]
- Parker, M.T.; Knop, K.; Sherwood, A.V.; Schurch, N.J.; Mackinnon, K.; Gould, P.D.; Hall, A.J.; Barton, G.J.; Simpson, G.G. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m(6)A modification. Elife 2020, 9, e49658. [Google Scholar] [CrossRef]
- Yu, Y.; Guan, J.; Xu, Y.; Ren, F.; Zhang, Z.; Yan, J.; Fu, J.; Guo, J.; Shen, Z.; Zhao, J.; et al. Population-scale peach genome analyses unravel selection patterns and biochemical basis underlying fruit flavor. Nat. Commun. 2021, 12, 3604. [Google Scholar] [CrossRef]
- Cao, K.; Yang, X.; Li, Y.; Zhu, G.; Fang, W.; Chen, C.; Wang, X.; Wu, J.; Wang, L. New high-quality peach (Prunus persica L. Batsch) genome assembly to analyze the molecular evolutionary mechanism of volatile compounds in peach fruits. Plant J. 2021, 108, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Wang, Z. Next-generation transcriptome assembly. Nat. Rev. Genet. 2011, 12, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.; Hirst, M.; Marra, M.A. Applications of new sequencing technologies for transcriptome analysis. Annu. Rev. Genom. Hum. Genet. 2009, 10, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, A.; Cole, C.; Volden, R.; Vollmers, C. Realizing the potential of full-length transcriptome sequencing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20190097. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinf. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayega, A.; Wang, Y.C.; Oikonomopoulos, S.; Djambazian, H.; Fahiminiya, S.; Ragoussis, J. Transcript profiling using long-read sequencing technologies. Methods Mol. Biol. 2018, 1783, 121–147. [Google Scholar]
- McCarthy, A. Third generation DNA sequencing: Pacific biosciences’ single molecule real time technology. Chem. Biol. 2010, 17, 675–676. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhang, X.; Lv, P.; Wang, W.; Wang, J.; He, Y.; Song, Z.; Cai, D. Full-length transcriptome reconstruction reveals genetic differences in hybrids of Oryza sativa and Oryza punctata with different ploidy and genome compositions. BMC Plant Biol. 2022, 22, 131. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Feng, S.; Xu, M.; Liu, F.; Cui, C.; Zhou, B. Reconstruction of the full-length transcriptome atlas using PacBio Iso-Seq provides insight into the alternative splicing in Gossypium australe. BMC Plant Biol. 2019, 19, 365. [Google Scholar] [CrossRef] [Green Version]
- Hackl, T.; Hedrich, R.; Schultz, J.; Forster, F. proovread: Large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 2014, 30, 3004–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35 (Suppl. S2), W297–W299. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35 (Suppl. S2), W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernandez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Zahra, S.; Das, D.; Kumar, S. AtFusionDB: A database of fusion transcripts in Arabidopsis thaliana. Database 2019, 2019, bay135. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zheng, H.; Sui, N. Regulation mechanism of long non-coding RNA in plant response to stress. Biochem. Biophys. Res. Commun. 2018, 503, 402–407. [Google Scholar] [CrossRef]
- Zhou, H.; Ren, F.; Wang, X.; Qiu, K.; Sheng, Y.; Xie, Q.; Shi, P.; Zhang, J.; Pan, H. Genome-wide identification and characterization of long noncoding RNAs during peach (Prunus persica) fruit development and ripening. Sci. Rep. 2022, 12, 11044. [Google Scholar] [CrossRef]
- Hu, H.; Yang, W.; Zheng, Z.; Niu, Z.; Yang, Y.; Wan, D.; Liu, J.; Ma, T. Analysis of alternative splicing and alternative polyadenylation in Populus alba var. pyramidalis by single-molecular long-read sequencing. Front Genet. 2020, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.M.; Schmieder, R.; Highnam, G.; Mittelman, D. Big data challenges and opportunities in high-throughput sequencing. Syst. Biomed. 2013, 1, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef]
- Martin, G.; Marquez, Y.; Mantica, F.; Duque, P.; Irimia, M. Alternative splicing landscapes in Arabidopsis thaliana across tissues and stress conditions highlight major functional differences with animals. Genome Biol. 2021, 22, 35. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Chao, Q.; Gao, Z.-F.; Zhang, D.; Zhao, B.-G.; Dong, F.-Q.; Fu, C.-X.; Liu, L.-J.; Wang, B.-C. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2019, 17, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Kuo, R.; Coulter, M.; Calixto, C.P.G.; Entizne, J.C.; Guo, W.; Marquez, Y.; Milne, L.; Riegler, S.; Matsui, A.; et al. A high-resolution single-molecule sequencing-based Arabidopsis transcriptome using novel methods of Iso-seq analysis. Genome Biol. 2022, 23, 149. [Google Scholar] [CrossRef] [PubMed]
- Tapial, J.; Ha, K.C.H.; Sterne-Weiler, T.; Gohr, A.; Braunschweig, U.; Hermoso-Pulido, A.; Quesnel-Vallieres, M.; Permanyer, J.; Sodaei, R.; Marquez, Y.; et al. An atlas of alternative splicing profiles and functional associations reveals new regulatory programs and genes that simultaneously express multiple major isoforms. Genome Res. 2017, 27, 1759–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grau-Bove, X.; Ruiz-Trillo, I.; Irimia, M. Origin of exon skipping-rich transcriptomes in animals driven by evolution of gene architecture. Genome Biol. 2018, 19, 135. [Google Scholar] [CrossRef]
- Wang, B.-B.; Brendel, V. Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. USA 2006, 103, 7175–7180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Gschwend, A.R.; Ouyang, Y.; Long, M. Evolution of gene structural complexity: An alternative-splicing-based model accounts for intron-containing retrogenes. Plant Physiol. 2014, 165, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Barbazuk, W.B.; Fu, Y.; McGinnis, K.M. Genome-wide analyses of alternative splicing in plants: Opportunities and challenges. Genome Res. 2008, 18, 1381–1392. [Google Scholar] [CrossRef] [Green Version]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, M.; Downie, B.; Liang, C.; Ji, G.; Li, Q.Q.; Hunt, A.G. Genome-wide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. USA 2011, 108, 12533–12538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Zhang, N.; Wang, Q.; Fu, Y.; Zhao, H.; Wang, J.; Wu, G.; Wang, F.; Li, X.; Liao, H. Full-length transcriptome sequencing reveals the molecular mechanism of potato seedlings responding to low-temperature. BMC Plant Biol. 2022, 22, 125. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, X.; Wen, X.; Zhang, Y.; Ding, Y.; Zhang, Y.; Gao, B.; Zhang, D. PacBio full-length transcriptome of wild apple (Malus sieversii) provides insights into canker disease dynamic response. BMC Genom. 2021, 22, 52. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, Q.Q. Coupling epigenetics and RNA polyadenylation: Missing links. Trends Plant Sci. 2023, 28, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Kumar, S.; Zeng, J.; McEwan, R.; Wright, T.R.; Gupta, M. Transcription terminator-mediated enhancement in transgene expression in maize: Preponderance of the AUGAAU motif overlapping with poly(A) signals. Front Plant Sci. 2020, 11, 570778. [Google Scholar] [CrossRef]
- Bennett, C.L.; Brunkow, M.E.; Ramsdell, F.; O’Briant, K.C.; Zhu, Q.; Fuleihan, R.L.; Shigeoka, A.O.; Ochs, H.D.; Chance, P.F. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 2001, 53, 435–439. [Google Scholar] [CrossRef]
- Staiger, D.; Brown, J.W. Alternative splicing at the intersection of biological timing, development, and stress responses. Plant Cell 2013, 25, 3640–3656. [Google Scholar] [CrossRef] [Green Version]
- Kriechbaumer, V.; Wang, P.; Hawes, C.; Abell, B.M. Alternative splicing of the auxin biosynthesis gene YUCCA4 determines its subcellular compartmentation. Plant J. 2012, 70, 292–302. [Google Scholar] [CrossRef]
- Remy, E.; Cabrito, T.R.; Baster, P.; Batista, R.A.; Teixeira, M.C.; Friml, J.; Sá-Correia, I.; Duque, P. A major facilitator superfamily transporter plays a dual role in polar auxin transport and drought stress tolerance in Arabidopsis. Plant Cell 2013, 25, 901–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naftaly, A.S.; Pau, S.; White, M.A. Long-read RNA sequencing reveals widespread sex-specific alternative splicing in threespine stickleback fish. Genome Res. 2021, 31, 1486–1497. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ci, D.; Li, T.; Li, P.; Song, Y.; Chen, J.; Quan, M.; Zhou, D.; Zhang, D. The role of DNA methylation in xylogenesis in different tissues of poplar. Front Plant Sci. 2016, 7, 1003. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peach Iso-Seq Data | |
|---|---|
| cDNA size | 1–6K |
| CCS Number | 250,803 |
| Read Bases of CCS | 552,197,124 |
| Mean Read Length of CCS | 2201 |
| Number of FLNC reads | 223,186 (88.99%) |
| Number of consensus isoforms | 72,175 |
| Average consensus isoforms read length | 2097 |
| Number of polished high-quality isoforms | 70,436 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.; Sheng, Y.; Qiu, K.; Ren, F.; Shi, P.; Xie, Q.; Guo, J.; Pan, H.; Zhang, J. Improved Annotation of the Peach (Prunus persica) Genome and Identification of Tissue- or Development Stage-Specific Alternative Splicing through the Integration of Iso-Seq and RNA-Seq Data. Horticulturae 2023, 9, 175. https://doi.org/10.3390/horticulturae9020175
Zhou H, Sheng Y, Qiu K, Ren F, Shi P, Xie Q, Guo J, Pan H, Zhang J. Improved Annotation of the Peach (Prunus persica) Genome and Identification of Tissue- or Development Stage-Specific Alternative Splicing through the Integration of Iso-Seq and RNA-Seq Data. Horticulturae. 2023; 9(2):175. https://doi.org/10.3390/horticulturae9020175
Chicago/Turabian StyleZhou, Hui, Yu Sheng, Keli Qiu, Fei Ren, Pei Shi, Qingmei Xie, Jiying Guo, Haifa Pan, and Jinyun Zhang. 2023. "Improved Annotation of the Peach (Prunus persica) Genome and Identification of Tissue- or Development Stage-Specific Alternative Splicing through the Integration of Iso-Seq and RNA-Seq Data" Horticulturae 9, no. 2: 175. https://doi.org/10.3390/horticulturae9020175