
Identification and Characterization of CCD Gene Family in Rose (Rosa chinensis Jacq. ‘Old Blush’) and Gene Co-Expression Network in Biosynthesis of Flower Scent

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification and Characteristics of the CCD Gene Family in Rose Genome
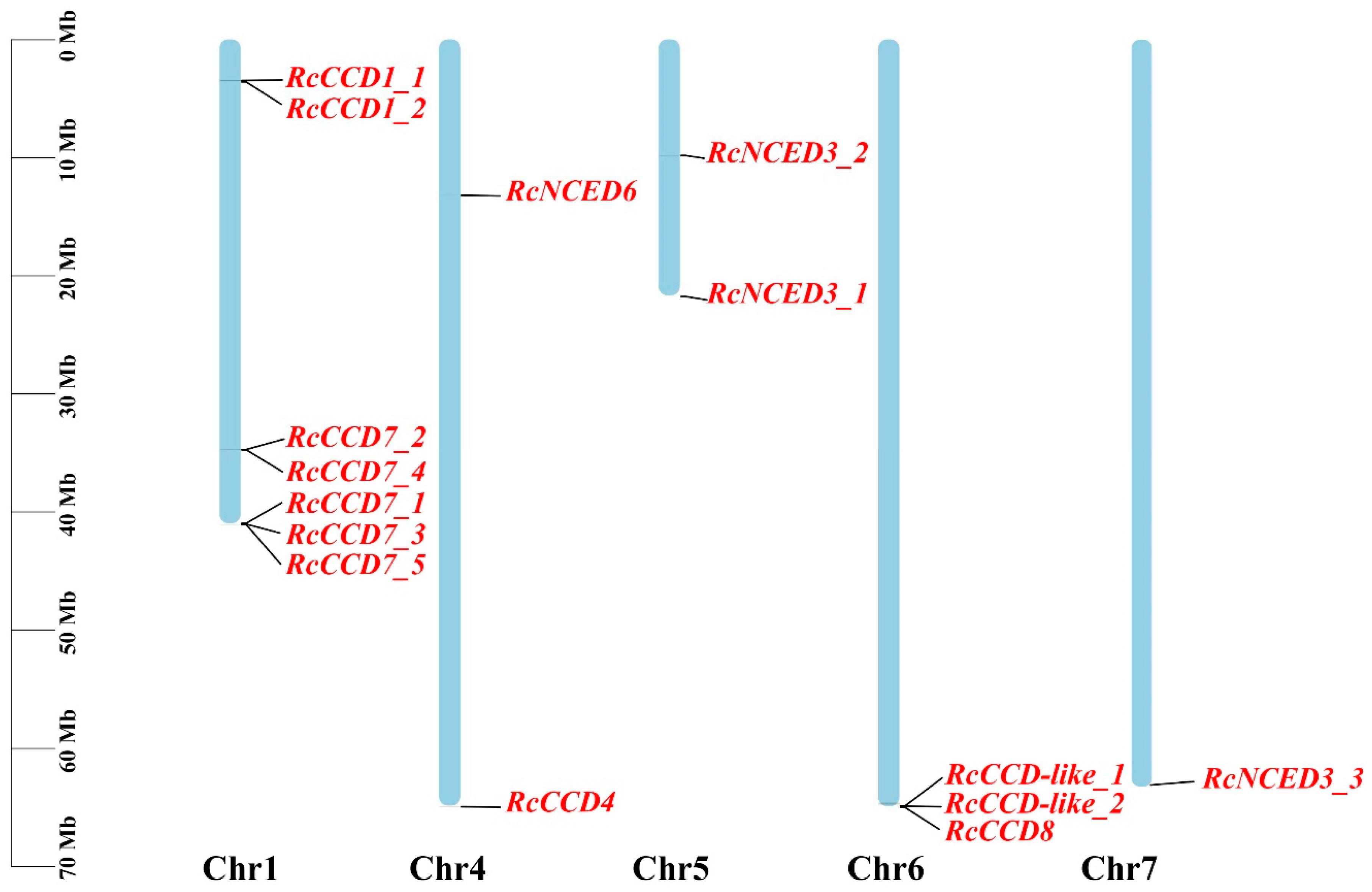
2.2. Chromosome Distribution and Collinearity Analysis
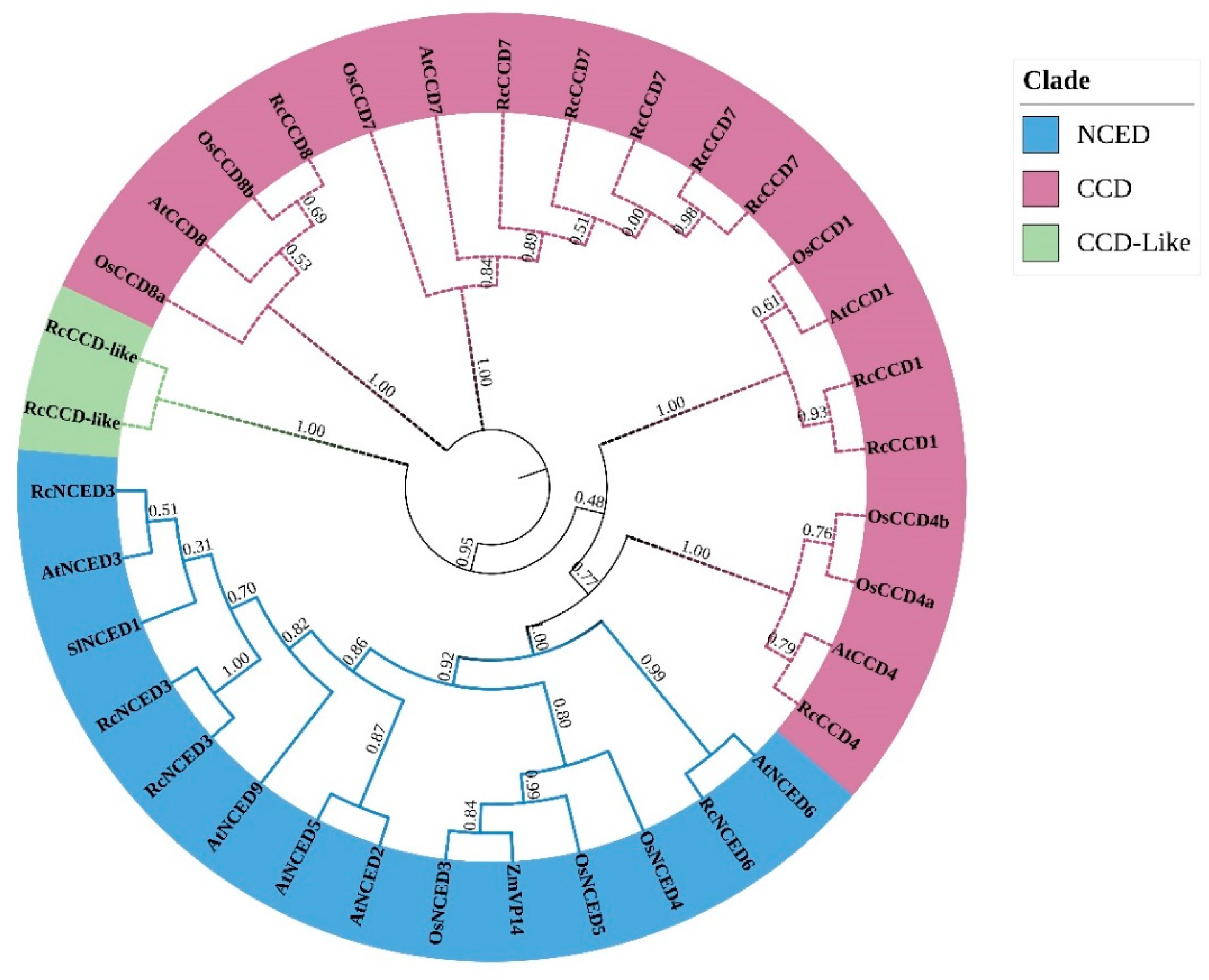
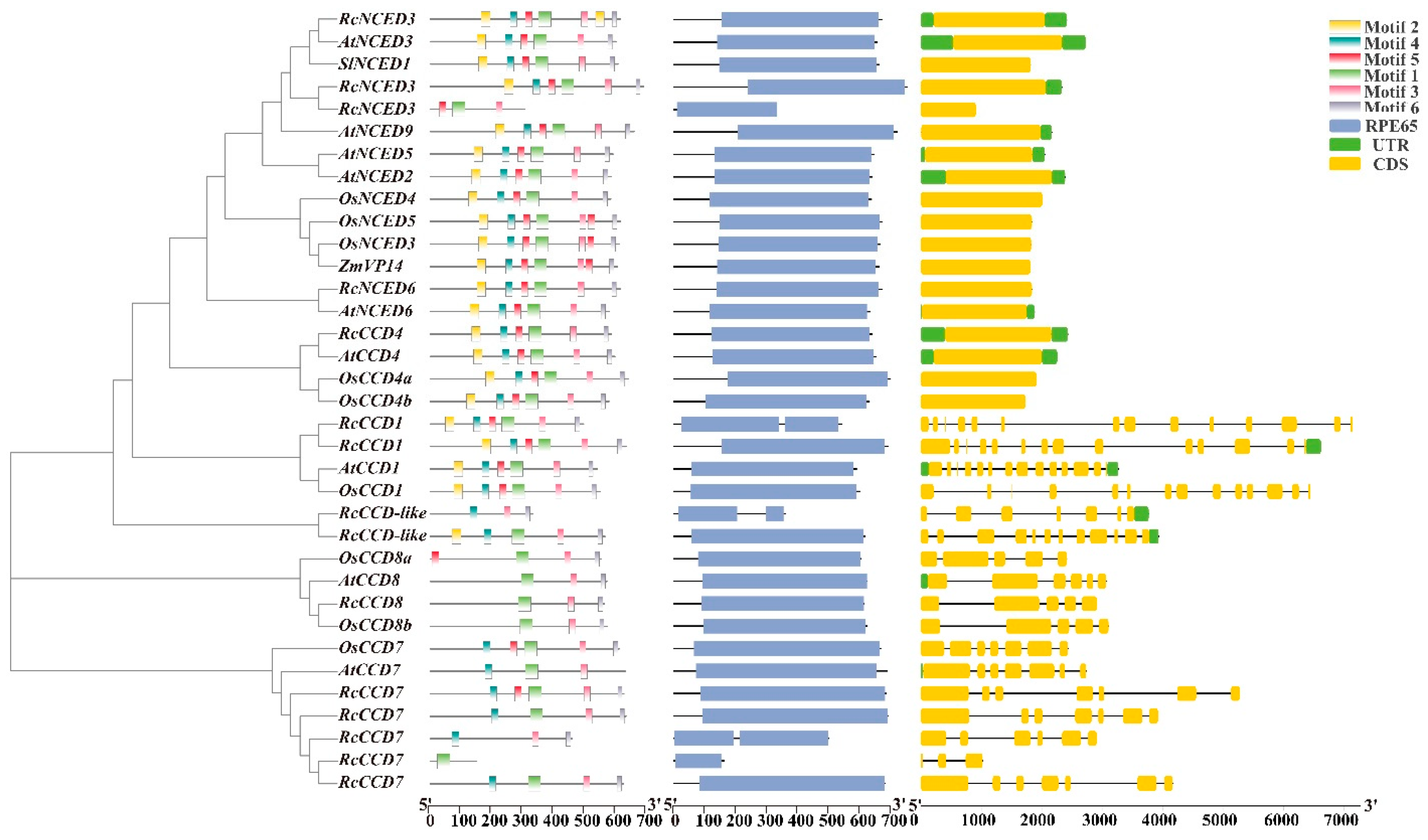
2.3. Phylogenetic and CCD Gene Family Structure Analysis
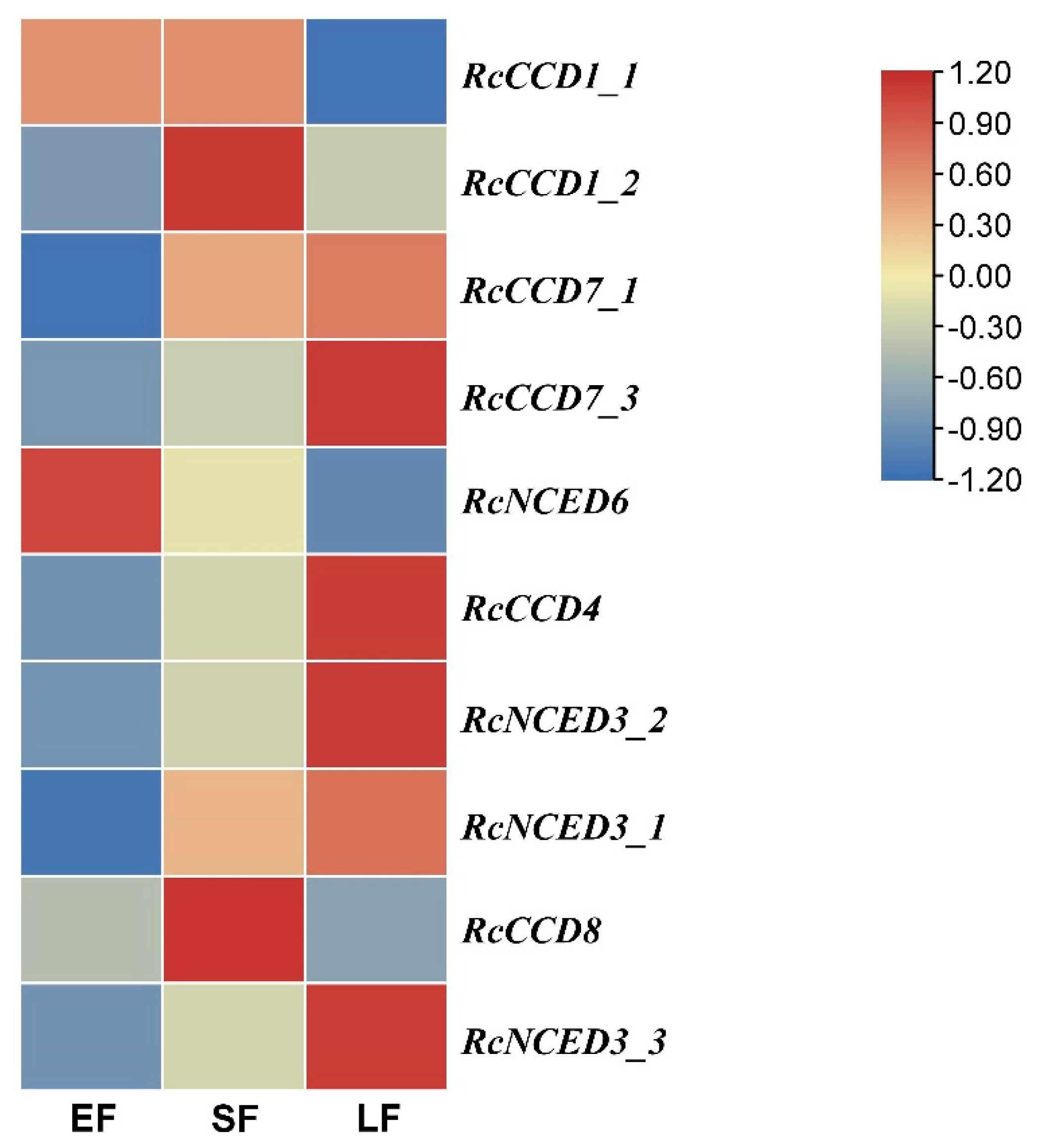
2.4. Expression of RcCCD Genes in Floral Organs of R. hybrida at Three Flower Developmentstages
2.5. Weighted Gene Co-Expression Network Analysis, WGCNA
3. Results
3.1. Identification and Characterization of the CCD Gene Family in Rose
3.2. Chromosomal Locations and Microsynteny
3.3. Phylogenetic Tree and Sequence Structure Analysis of Rose CCD Genes
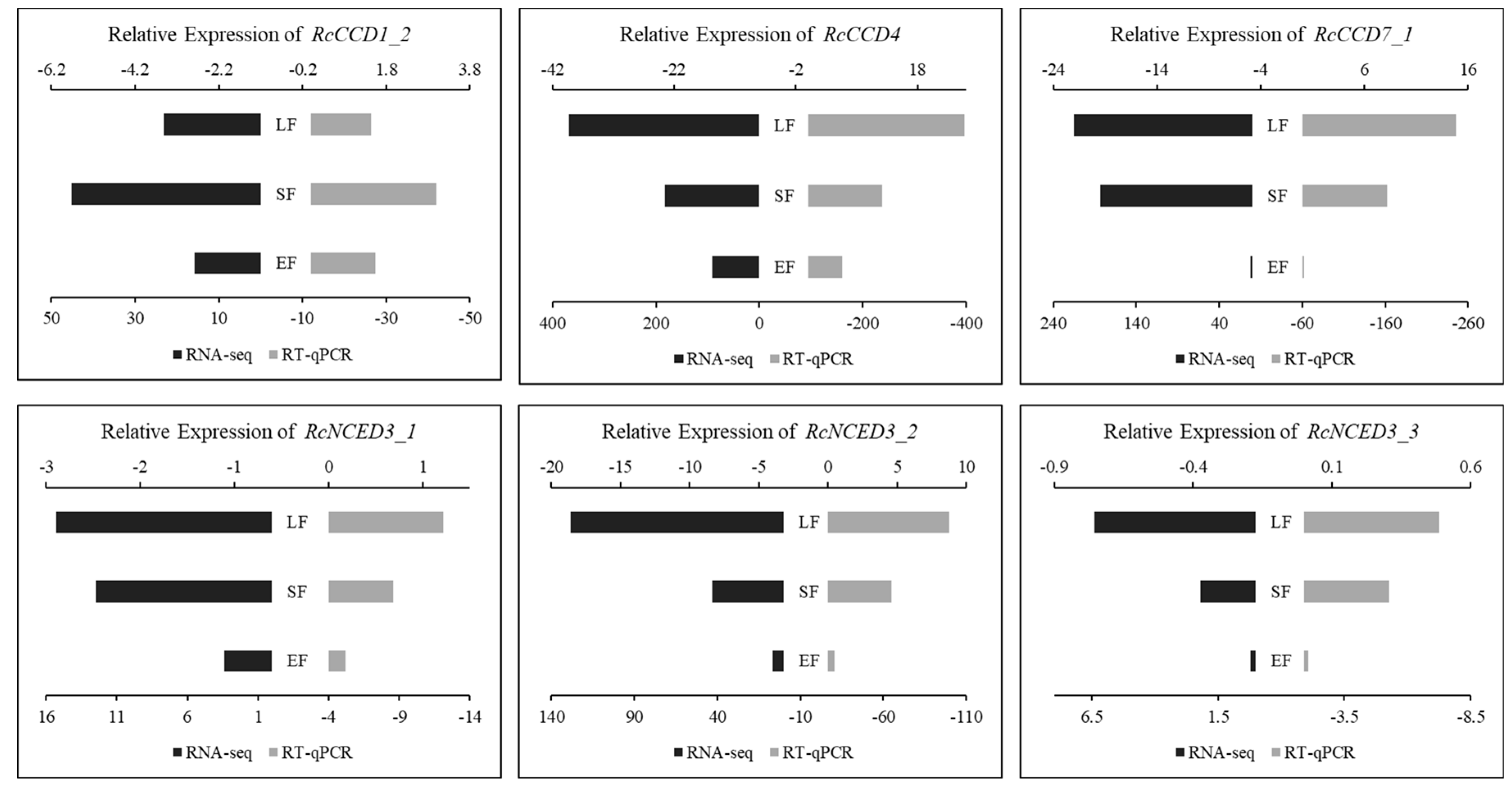
3.4. Expression of RcCCD genes in Floral Organs of R. hybrida ‘Tianmidemeng’ at Three Flower Development Stages
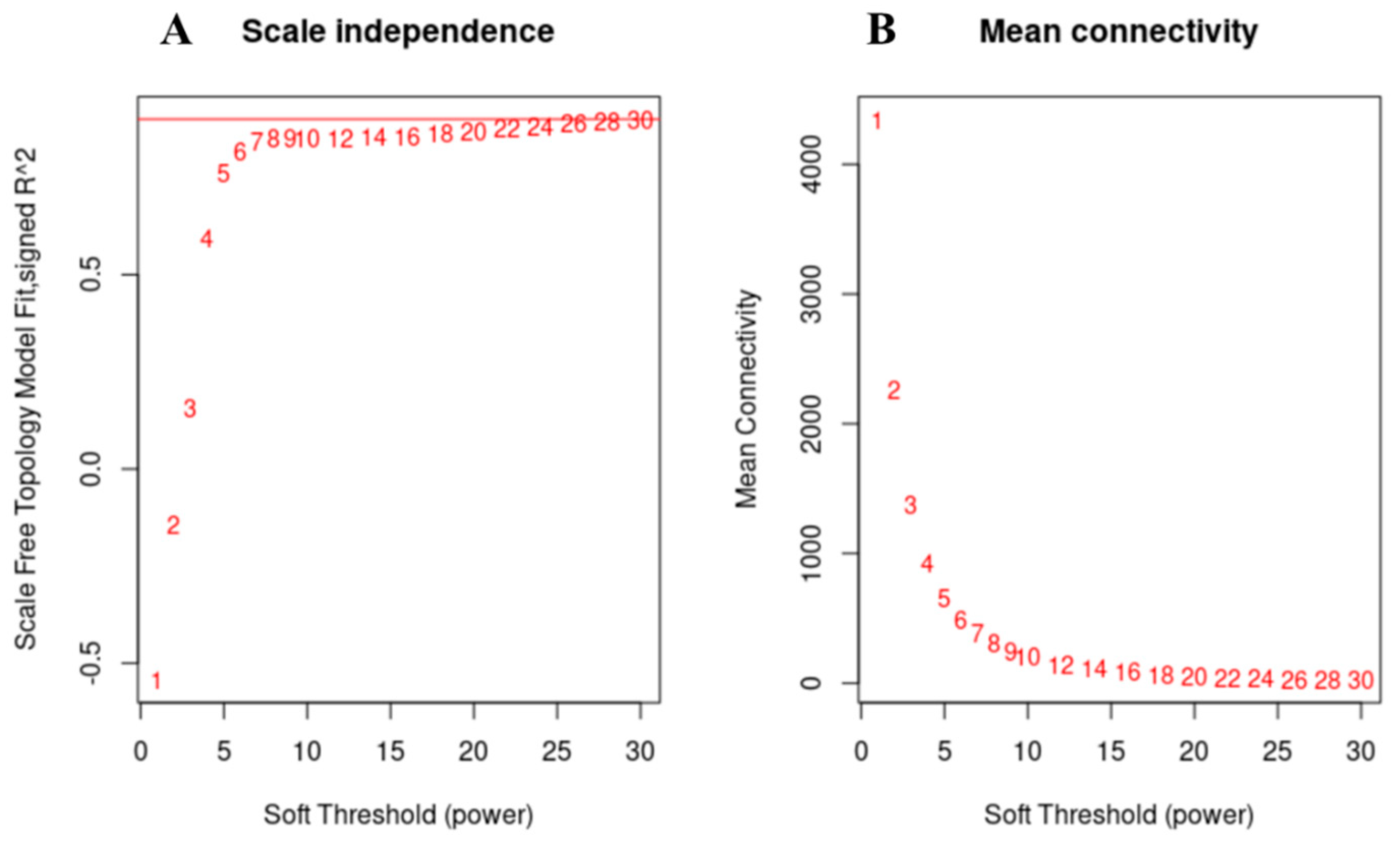
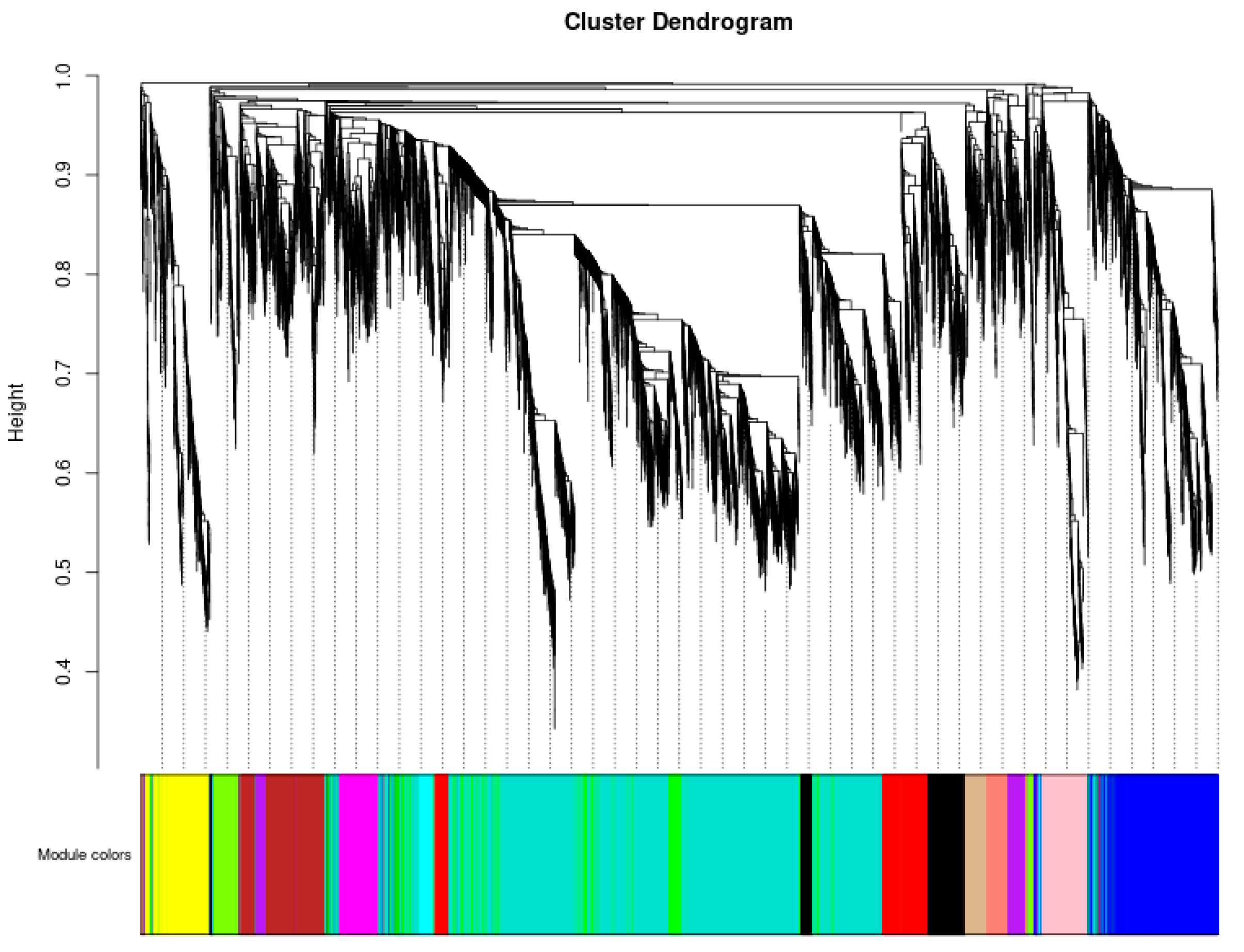
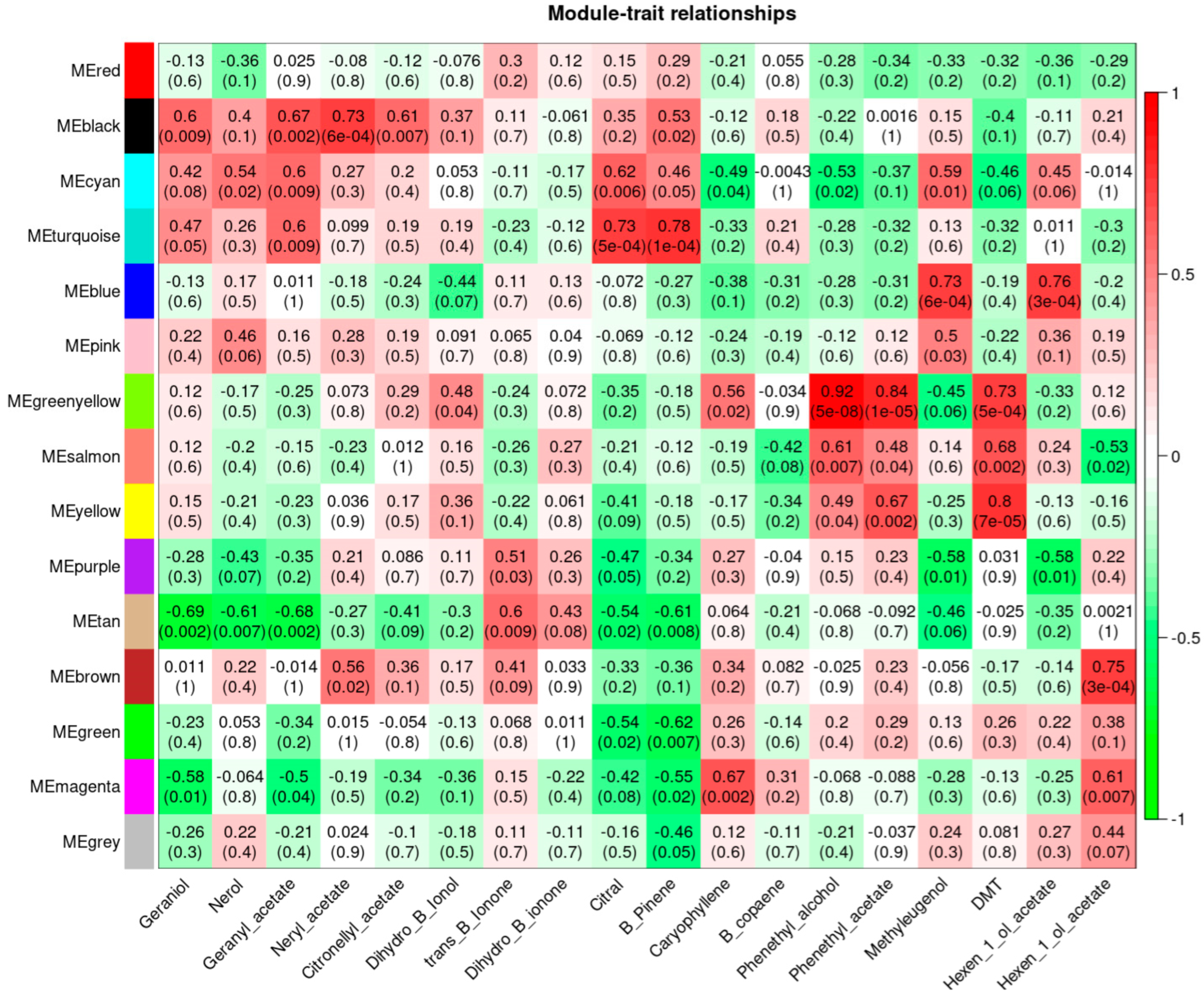
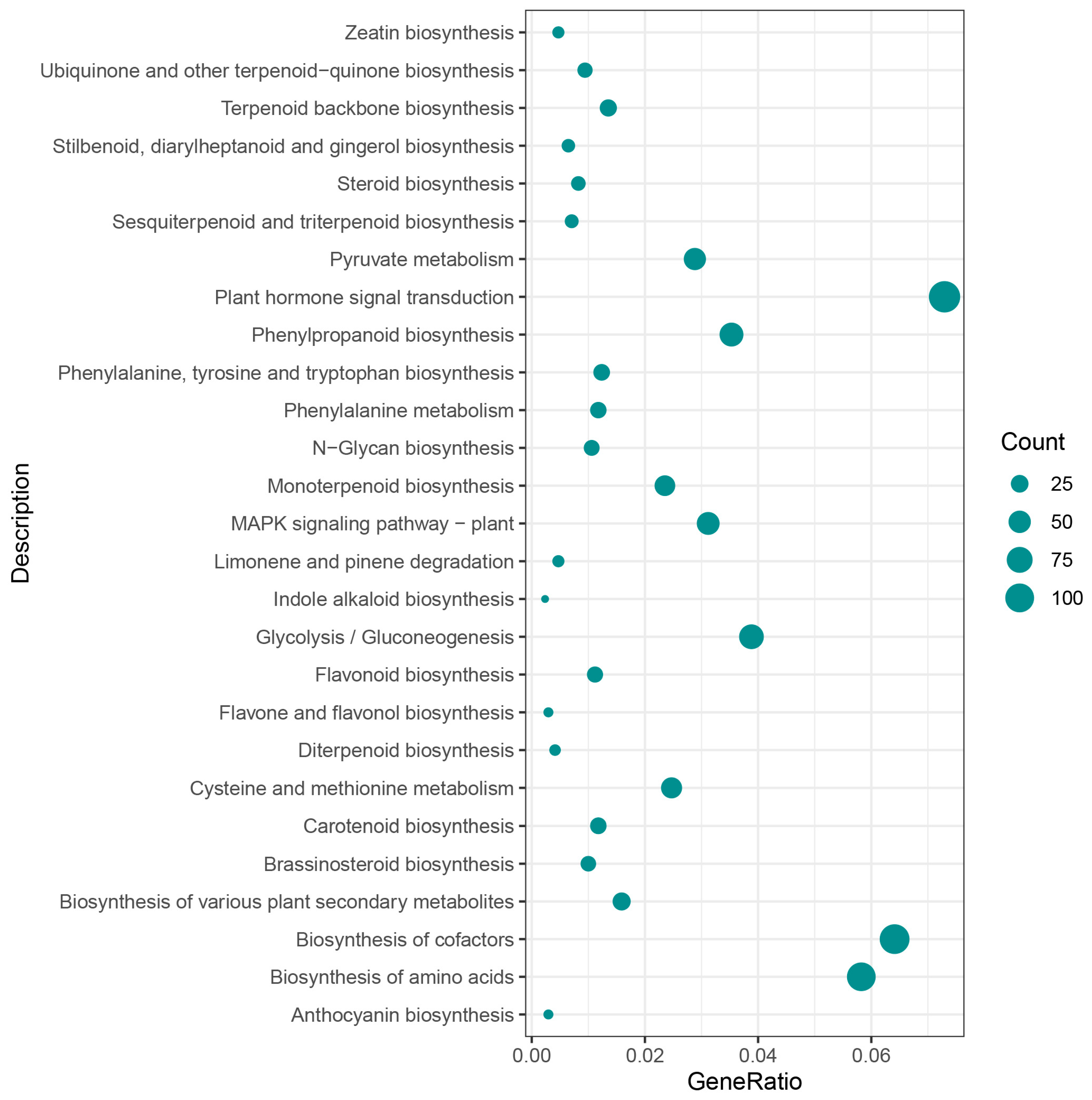
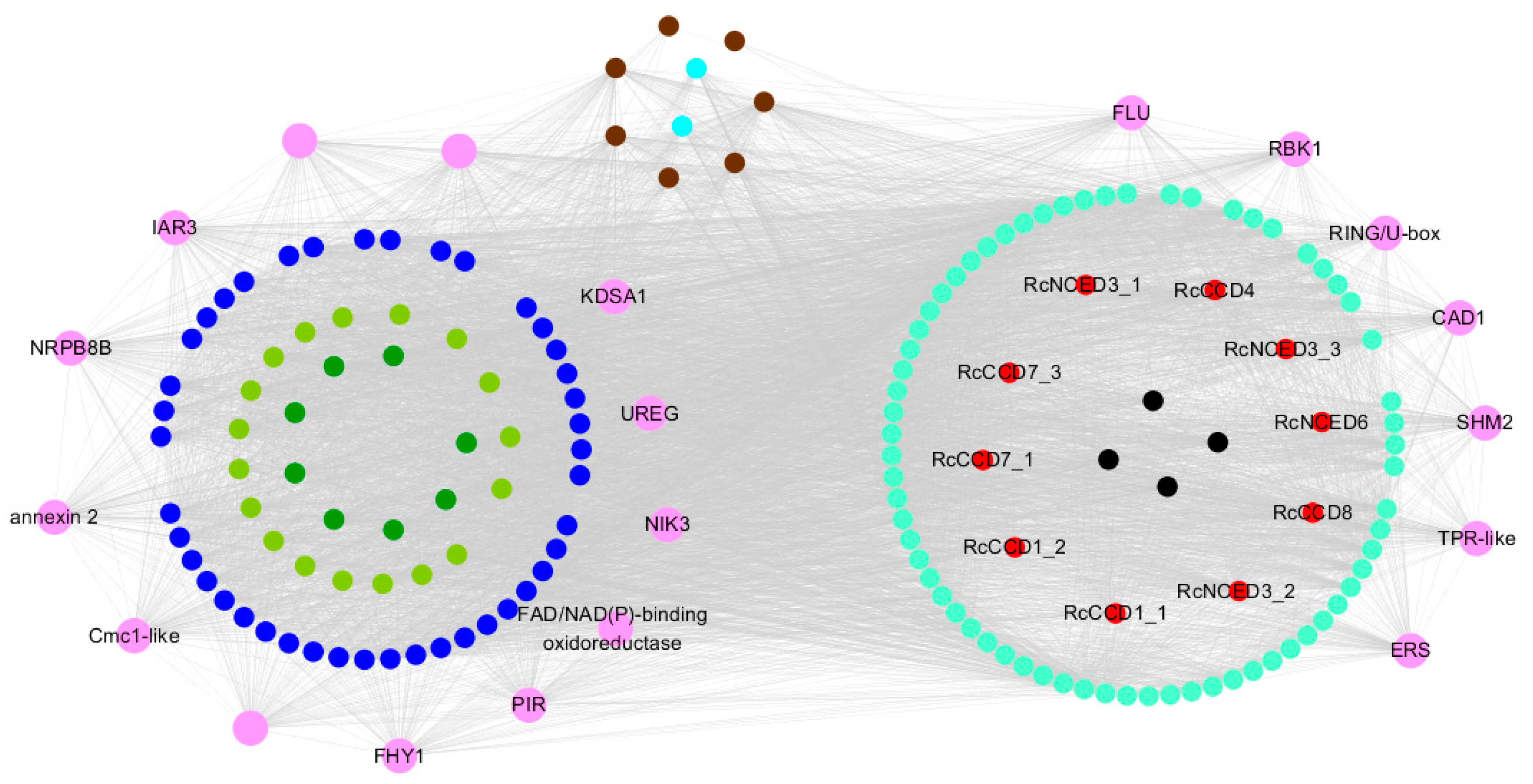
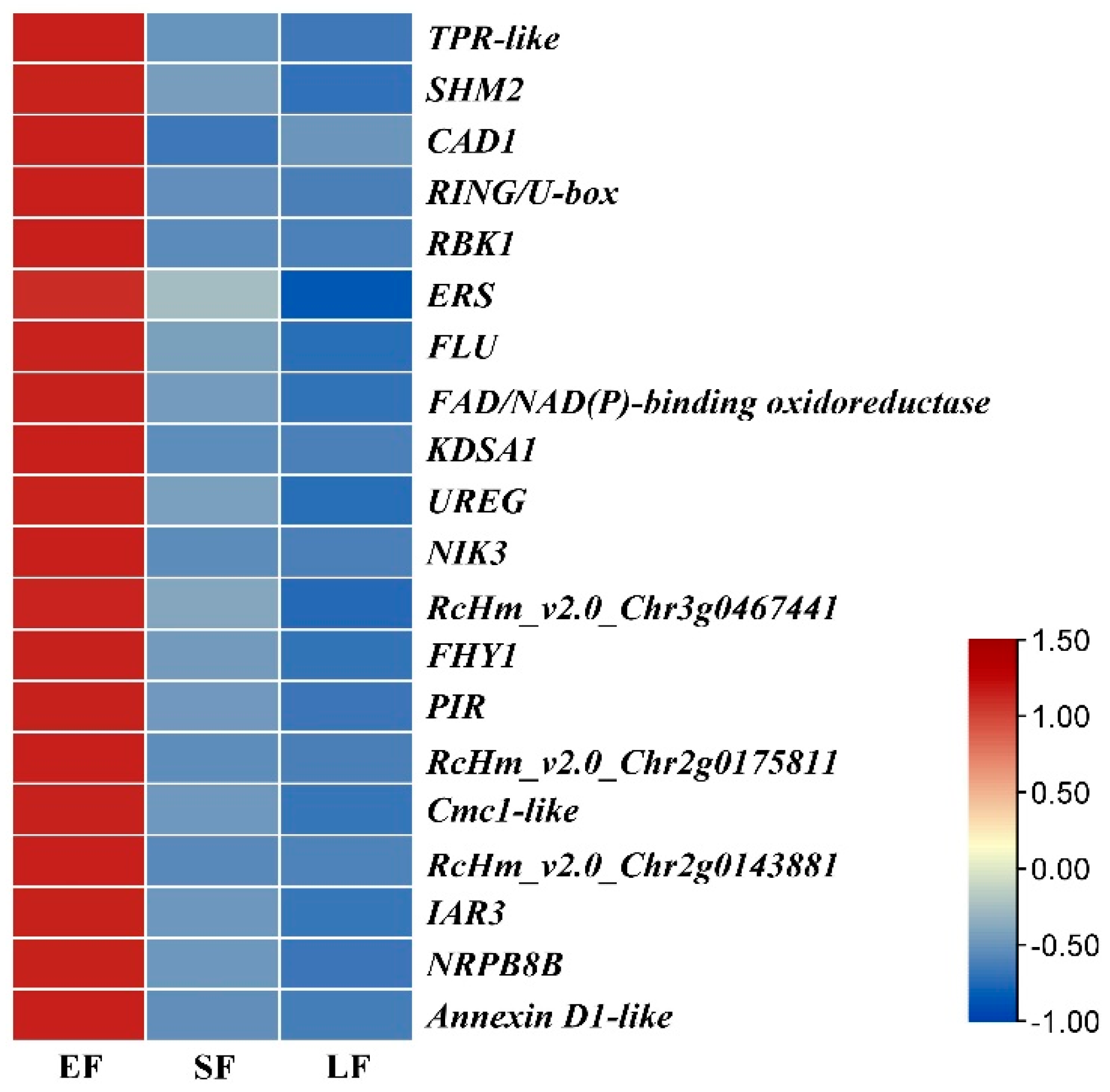
3.5. Weighted Gene Co-Expression Network Analysis, WGCNA


4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Venkatesha, K.T.; Gupta, A.; Rai, A.N.; Jambhulkar, S.J.; Bisht, R.; Padalia, R.C. Recent developments, challenges, and opportunities in genetic improvement of essential oil-bearing rose (Rosa damascena): A review. Ind. Crops Prod. 2022, 184, 471–479. [Google Scholar] [CrossRef]
- Dudareva, N.; Pichersky, E.; Gershenzon, J. Biochemistry of plant Volatiles. Plant Physiol. 2004, 135, 1893–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, M.H.; Chang, C.Y.; Hsu, S.J.; Chen, J.J. Chloroplast localization of methylerythritol 4-phosphate pathway enzymes and regulation of mitochondrial genes in ispD and ispE albino mutants in Arabidopsis. Plant Mol. Biol. 2008, 66, 663–673. [Google Scholar] [CrossRef]
- Knudsen, J.T.; Eriksson, R.; Gershenzon, J.; Stahl, B. Diversity and distribution of floral scent. Bot. Rev. 2006, 72, 1–120. [Google Scholar] [CrossRef]
- Simkin, A.J.; Guirimand, G.; Papon, N.; Courdavault, V.; Thabet, I.; Ginis, O.; Bouzid, S.; Giglioli-Guivarc’h, N.; Clastre, M. Peroxisomal localisation of the final steps of the mevalonic acid pathway in planta. Planta 2011, 234, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Pulido, P.; Perello, C.; Rodriguez-Concepcion, M. New Insights into Plant Isoprenoid Metabolism. Mol. Plant 2012, 5, 964–967. [Google Scholar] [CrossRef] [Green Version]
- Boatright, J.; Negre, F.; Chen, X.L.; Kish, C.M.; Wood, B.; Peel, G.; Orlova, I.; Gang, D.; Rhodes, D.; Dudareva, N. Understanding in vivo benzenoid metabolism in petunia petal tissue. Plant Physiol. 2004, 135, 1993–2011. [Google Scholar] [CrossRef] [Green Version]
- Bouvier, F.; Isner, J.C.; Dogbo, O.; Camara, B. Oxidative tailoring of carotenoids: A prospect towards novel functions in plants. Trends Plant Sci. 2005, 10, 187–194. [Google Scholar] [CrossRef]
- Auldridge, M.E.; Block, A.; Vogel, J.T.; Dabney-Smith, C.; Mila, I.; Bouzayen, M.; Magallanes-Lundback, M.; DellaPenna, D.; McCarty, D.R.; Klee, H.J. Characterization of three members of the Arabidopsis carotenoid cleavage dioxygenase family demonstrates the divergent roles of this multifunctional enzyme family. Plant J. 2006, 45, 982–993. [Google Scholar] [CrossRef]
- Wei, Y.P.; Wan, H.J.; Wu, Z.M.; Wang, R.Q.; Ruan, M.Y.; Ye, Q.J.; Li, Z.M.; Zhou, G.Z.; Yao, Z.P.; Yang, Y.J. A Comprehensive Analysis of Carotenoid Cleavage Dioxygenases Genes in Solanum lycopersicum. Plant Mol. Biol. Rep. 2016, 34, 512–523. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, G.Q.; Gu, T.T.; Ding, J.; Li, Y. Bioinformatic and expression analyses on carotenoid dioxygenase genes in fruit development and abiotic stress responses in Fragaria vesca. Mol. Genet. Genom. 2017, 292, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Zuo, X.Y.; Shao, H.X.; Fan, S.; Ma, J.J.; Zhang, D.; Zhao, C.P.; Yan, X.Y.; Liu, X.J.; Han, M.Y. Genome-wide analysis of carotenoid cleavage oxygenase genes and their responses to various phytohormones and abiotic stresses in apple (Malus domestica). Plant Physiol. Biochem. 2018, 123, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.C.; Joseph, L.M.; Deng, W.T.; Liu, L.J.; Li, Q.B.; Cline, K.; McCarty, D.R. Molecular characterization of the Arabidopsis 9-cis epoxycarotenoid dioxygenase gene family. Plant J. 2003, 35, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Kloer, D.P.; Ruch, S.; Al-Babili, S.; Beyer, P.; Schulz, G.E. The structure of a retinal-forming carotenoid oxygenase. Science 2005, 308, 267–269. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, S.H.; Tan, B.C.; Gage, D.A.; Zeevaart, J.A.D.; McCarty, D.R. Specific oxidative cleavage of carotenoids by VP14 of maize. Science 1997, 276, 1872–1874. [Google Scholar] [CrossRef] [Green Version]
- Vallabhaneni, R.; Bradbury, L.M.T.; Wurtzel, E.T. The carotenoid dioxygenase gene family in maize, sorghum, and rice. Arch. Biochem. Biophys. 2010, 504, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Lashbrooke, J.G.; Young, P.R.; Dockrall, S.J.; Vasanth, K.; Vivier, M.A. Functional characterisation of three members of the Vitis vinifera L. carotenoid cleavage dioxygenase gene family. BMC Plant Biol. 2013, 13, 156. [Google Scholar] [CrossRef] [Green Version]
- Schwab, W.; Huang, F.C.; Molnar, P. Carotenoid Cleavage Dioxygenase Genes from Fruit. In Proceedings of the American-Chemical-Society Symposium on Carotenoid Cleavage Products/243rd American-Chemical-Society National Meeting, San Diego, CA, USA, 25–26 March 2012; pp. 11–19. [Google Scholar]
- He, Y.J.; Wang, Z.W.; Ge, H.Y.; Liu, Y.; Chen, H.Y. Weighted gene co-expression network analysis identifies genes related to anthocyanin biosynthesis and functional verification of hub gene SmWRKY44. Plant Sci. 2021, 309, 110935. [Google Scholar] [CrossRef]
- Li, M.; Zhang, D.; Gao, Q.; Luo, Y.; Zhang, H.; Ma, B.; Chen, C.; Whibley, A.; Zhang, Y.; Cao, Y.; et al. Genome structure and evolution of Antirrhinum majus L. Nat. Plants 2019, 5, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Shi, S.; Zhang, S.; Wu, J.; Liu, X.; Zhang, Z. Identification of long non-coding RNAs involved in floral scent of Rosa hybrida. Front. Plant Sci. 2022, 13, 996474. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1265. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Verdejo, C.I.; Obrero, A.; Roman, B.; Gomez, P. Expression Profile of Carotenoid Cleavage Dioxygenase Genes in Summer Squash (Cucurbita pepo L.). Plant Foods Hum. Nutr. 2015, 70, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.C.; Horvath, G.; Molnar, P.; Turcsi, E.; Deli, J.; Schrader, J.; Sandmann, G.; Schmidt, H.; Schwab, W. Substrate promiscuity of RdCCD1, a carotenoid cleavage oxygenase from Rosa damascena. Phytochemistry 2009, 70, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Hwang, I.; Jung, H.J.; Park, J.I.; Kang, J.G.; Nou, I.S. Genome-Wide Classification and Abiotic Stress-Responsive Expression Profiling of Carotenoid Oxygenase Genes in Brassica rapa and Brassica oleracea. J. Plant Growth Regul. 2016, 35, 202–214. [Google Scholar] [CrossRef]
- Walter, M.H.; Strack, D. Carotenoids and their cleavage products: Biosynthesis and functions. Nat. Prod. Rep. 2011, 28, 663–692. [Google Scholar] [CrossRef]
- Zhou, Q.Q.; Li, Q.C.; Li, P.; Zhang, S.T.; Liu, C.; Jin, J.J.; Cao, P.J.; Yang, Y.X. Carotenoid Cleavage Dioxygenases: Identification, Expression, and Evolutionary Analysis of This Gene Family in Tobacco. Int. J. Mol. Sci. 2019, 20, 5796. [Google Scholar] [CrossRef] [Green Version]
- Faraji, S.; Heidari, P.; Amouei, H.; Filiz, E.; Abdullah; Poczai, P. Investigation and Computational Analysis of the Sulfotransferase (SOT) Gene Family in Potato (Solanum tuberosum): Insights into Sulfur Adjustment for Proper Development and Stimuli Responses. Plants 2021, 10, 2597. [Google Scholar] [CrossRef]
- Heidari, P.; Abdullah; Faraji, S.; Poczai, P. Magnesium transporter Gene Family: Genome-Wide Identification and Characterization in Theobroma cacao, Corchorus capsularis, and Gossypium hirsutum of Family Malvaceae. Agronomy 2021, 11, 1651. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, F.; Wu, J.; Zhang, Z. Identification and Characterization of CCD Gene Family in Rose (Rosa chinensis Jacq. ‘Old Blush’) and Gene Co-Expression Network in Biosynthesis of Flower Scent. Horticulturae 2023, 9, 115. https://doi.org/10.3390/horticulturae9010115
Ji F, Wu J, Zhang Z. Identification and Characterization of CCD Gene Family in Rose (Rosa chinensis Jacq. ‘Old Blush’) and Gene Co-Expression Network in Biosynthesis of Flower Scent. Horticulturae. 2023; 9(1):115. https://doi.org/10.3390/horticulturae9010115
Chicago/Turabian StyleJi, Fangfang, Jie Wu, and Zhao Zhang. 2023. "Identification and Characterization of CCD Gene Family in Rose (Rosa chinensis Jacq. ‘Old Blush’) and Gene Co-Expression Network in Biosynthesis of Flower Scent" Horticulturae 9, no. 1: 115. https://doi.org/10.3390/horticulturae9010115