
Transcriptomic Analysis of Sex-Associated DEGs in Female and Male Flowers of Kiwifruit (Actinidia deliciosa [A. Chev] C. F. Liang & A. R. Ferguson)

 , , ,
, , ,
Abstract
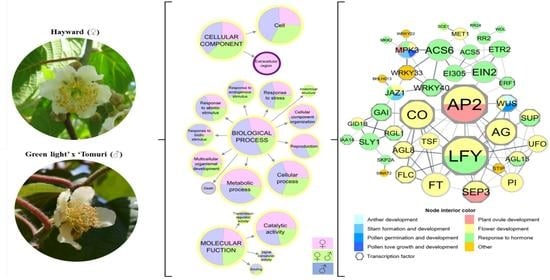
:
1. Introduction
2. Materials and Methods
2.1. Plant Material and RNA Extraction
2.2. Sequencing, Assembly and Gene Expression
2.3. Gene Ontology and Network Analysis
2.4. RT-qPCR Validation
3. Results
3.1. General Data of the RNA-seq
3.2. DEGs Function Annotation
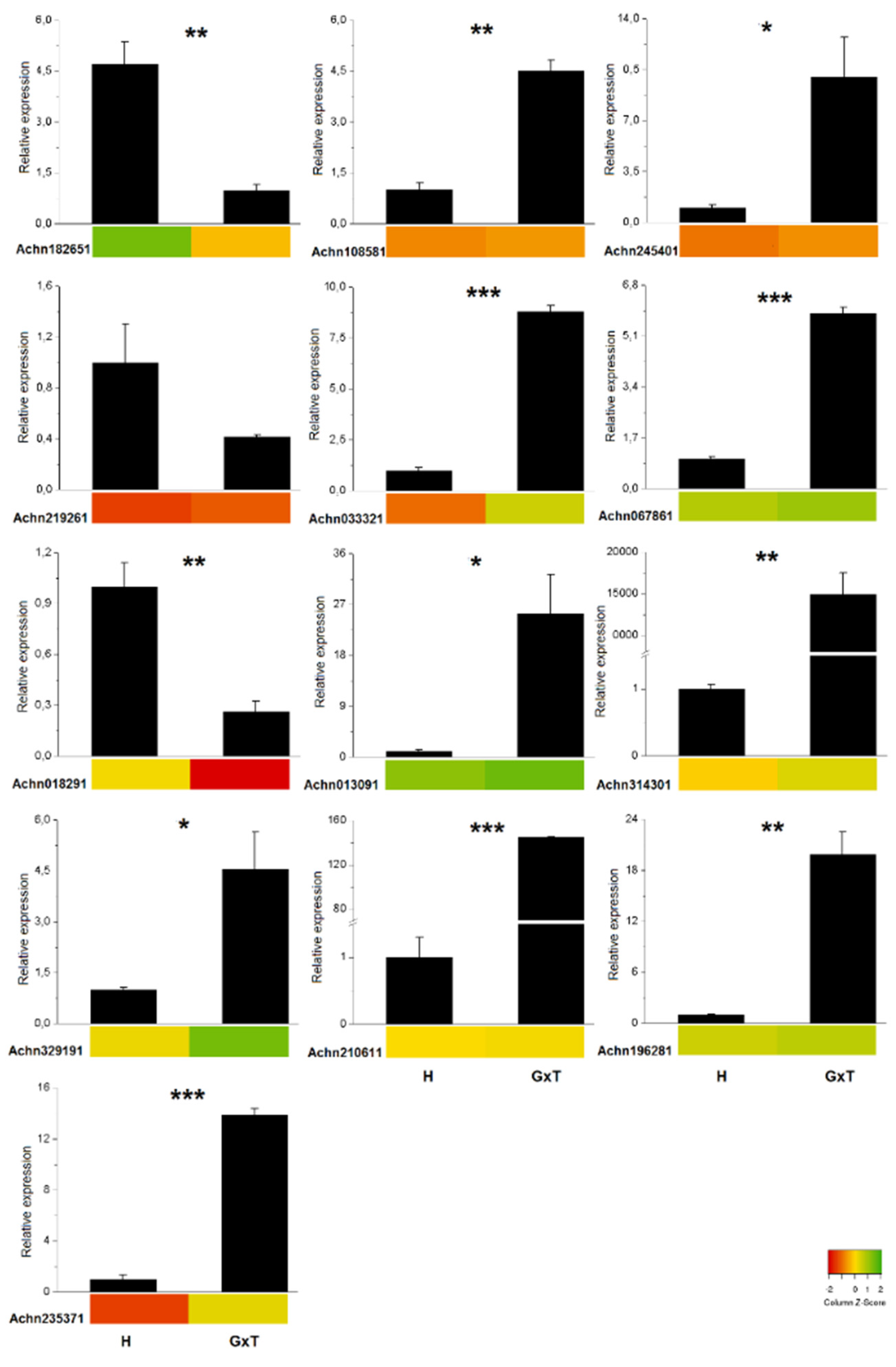
3.3. RT-qPCR Validation
4. Discussion
4.1. TFs Differentially Expressed in Male and Female Flowers
4.2. Methyltransferase Enzymes Expressed in Male and Female Flowers
4.3. Hormone-Related Genes Differentially Expressed in Male and Female Flowers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Liu, C.; Liu, Y.; VanBuren, R.; Yao, X.; Zhong, C.; Huang, H. High-density interspecific genetic maps of kiwifruit and the identification of sex-specific markers. DNA Res. 2015, 22, 367–375. [Google Scholar] [CrossRef] [Green Version]
- López-Sobaler, A.M.; Vizuete, A.A.; Anta, R.M.O. Beneficios nutricionales y sanitarios asociados al consumo de kiwi. Nutr. Hosp. 2016, 33, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinero, C.; Martino, J. Actinidia deliciosa (A. Chevalier, C.V. Liang et al., A. R. Ferguson) Kiwi. Edt. Excma. Diput Prov. Pontevedra 1997, 1, 1–3. [Google Scholar]
- Liu, Z.; Larsson, S. The Relationship between a Flower Gall Midge Pseudas-Phondylia SP. (Diptera: Cecidomyiidae) and its Host Plant Actinidia Valvata. Insect Sci. 1996, 3, 271–282. [Google Scholar] [CrossRef]
- Testolin, R.; Messina, R.; Lain, O.; Cipriani, G. A natural sex mutant in kiwifruit (Actinidia deliciosa). N. Z. J. Crop. Hortic. Sci. 2004, 32, 179–183. [Google Scholar] [CrossRef]
- Akagi, T.; Henry, I.M.; Ohtani, H.; Morimoto, T.; Beppu, K.; Kataoka, I.; Tao, R. A Y-Encoded Suppressor of Feminization Arose via Lineage-Specific Duplication of a Cytokinin Response Regulator in Kiwifruit. Plant Cell 2018, 30, 780–795. [Google Scholar] [CrossRef]
- Akagi, T.; Pilkington, S.M.; Varkonyi-Gasic, E.; Henry, I.M.; Sugano, S.S.; Sonoda, M.; Firl, A.; Mcneilage, M.A.; Douglas, M.J.; Wang, T.; et al. Two Y-chromosome-encoded genes determine sex in kiwifruit. Nat. Plants 2019, 5, 801–809. [Google Scholar] [CrossRef]
- Harkess, A.; Zhou, J.; Xu, C.; Bowers, J.E.; Van Der Hulst, R.; Ayyampalayam, S.; Mercati, F.; Riccardi, P.; McKain, M.R.; Kakrana, A.; et al. The asparagus genome sheds light on the origin and evolution of a young Y chromosome. Nat. Commun. 2017, 8, 1279. [Google Scholar] [CrossRef]
- Martin, A.; Troadec, C.; Boualem, A.; Rajab, M.; Fernandez, R.; Morin, H.; Pitrat, M.; Dogimont, C.; Bendahmane, A. A transposon-induced epigenetic change leads to sex determination in melon. Nat. Cell Biol. 2009, 461, 1135–1138. [Google Scholar] [CrossRef]
- Sebastian, P.; Schaefer, H.; Telford, I.R.H.; Renner, S.S. Cucumber (Cucumis sativus) and melon (C. melo) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc. Natl. Acad. Sci. USA 2010, 107, 14269–14273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, R.; Bendahmane, A.; Renner, S.S. Sex Chromosomes in Land Plants. Annu. Rev. Plant Biol. 2011, 62, 485–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, C.V.; Quek, S.-Y.; Stevenson, R.J.; Winz, R.A. Characterisation of bound volatile compounds of a low flavour kiwifruit species: Actinidia eriantha. Food Chem. 2012, 134, 655–661. [Google Scholar] [CrossRef]
- Garcia, C.V.; Stevenson, R.J.; Atkinson, R.G.; Winz, R.A.; Quek, S.-Y. Changes in the bound aroma profiles of ‘Hayward’ and ‘Hort16A’ kiwifruit (Actinidia spp.) during ripening and GC-olfactometry analysis. Food Chem. 2013, 137, 45–54. [Google Scholar] [CrossRef]
- Barrett, D.M.; Beaulieu, J.; Shewfelt, R. Color, Flavor, Texture, and Nutritional Quality of Fresh-Cut Fruits and Vegetables: Desirable Levels, Instrumental and Sensory Measurement, and the Effects of Processing. Crit. Rev. Food Sci. Nutr. 2010, 50, 369–389. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-Y.; Zhong, Y.; Zheng, J.; Ali, M.; Liu, G.-D.; Zheng, X.-L. L-ascorbic acid metabolism in an ascorbate-rich kiwifruit (Actinidia. Eriantha Benth.) cv. ‘White’ during postharvest. Plant Physiol. Biochem. 2018, 124, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Jorquera, C.; Campos-Vargas, R.; Jorgensen, C.; Zapata, P.; Infante, R. Effect of the application timing of 1-MCP on postharvest traits and sensory quality of a yellow-fleshed kiwifruit. Sci. Hortic. 2019, 244, 82–87. [Google Scholar] [CrossRef]
- Atak, A.; Aydın, B.; Kahraman, K.A. Sex determination of kiwifruit seedlings with molecular markers. Acta Hortic. 2014, 197–203. [Google Scholar] [CrossRef]
- Fraser, L.G.; Tsang, G.K.; Datson, P.M.; De Silva, H.N.; Harvey, C.F.; Gill, G.P.; Crowhurst, R.N.; Mcneilage, M.A. A gene-rich linkage map in the dioecious species Actinidia chinensis (kiwifruit) reveals putative X/Y sex-determining chromosomes. BMC Genom. 2009, 10, 102. [Google Scholar] [CrossRef] [Green Version]
- Scaglione, D.; Fornasiero, A.; Pinto, C.; Cattonaro, F.; Spadotto, A.; Infante, R.; Meneses, C.; Messina, R.; Lain, O.; Cipriani, G.; et al. A RAD-based linkage map of kiwifruit (Actinidia chinensis Pl.) as a tool to improve the genome assembly and to scan the genomic region of the gender determinant for the marker-assisted breeding. Tree Genet. Genomes 2015, 11, 115. [Google Scholar] [CrossRef]
- Harvey, C.F.; Gill, G.P.; Fraser, L.G.; Mcneilage, M.A. Sex determination in Actinidia. 1. Sex-linked markers and progeny sex ratio in diploid A. chinensis. Sex. Plant Reprod. 1997, 10, 149–154. [Google Scholar] [CrossRef]
- Jiménez-Durán, K.; Cruz-García, F. Incompatibilidad sexual, un mecanismo genético que evita la autofecundación y con-tribuye a la diversidad vegetal. Rev. Fitotec. Mex. 2011, 34, 1–9. [Google Scholar]
- Li, X.; Qin, H.; Wang, Z.; Zhang, Q.; Liu, Y.; Xu, P.; Zhao, Y.; Fan, S.; Yang, Y.; Ai, J. Differentially expressed genes in male and female flower buds of hardy kiwifruit (Actinidia Arguta (Sieb. et ZUCC.) Planch. Ex MIQ.). Bangladesh J. Bot. 2015, 44, 909–915. [Google Scholar]
- Tang, P.; Zhang, Q.; Yao, X. Comparative transcript profiling explores differentially expressed genes associated with sexual phenotype in kiwifruit. PLoS ONE 2017, 12, e0180542. [Google Scholar] [CrossRef]
- Zuccherelli, G.; Zuccherelli, G. La actinidia (KIWI), 2nd ed.; Ediciones Mundi-Prensa: Madrid, Spain, 1990; ISBN 8471141167. [Google Scholar]
- Salinero, M.; Vela, P.; Sainz, M. Phenological growth stages of kiwifruit (Actinidia deliciosa ‘Hayward’). Sci. Hortic. 2009, 121, 27–31. [Google Scholar] [CrossRef]
- Caporali, E.; Testolin, R.; Pierce, S.; Spada, A. Sex change in kiwifruit (Actinidia chinensis Planch.): A developmental framework for the bisexual to unisexual floral transition. Plant Reprod. 2019, 32, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [Green Version]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482–517. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Jun, S.S.; Choe, S.; Cho, J.Y.; Choi, S.B.; Kim, S.C. Identification of differentially expressed genes from male and female flowers of kiwifruit. Afr. J. Biotechnol. 2010, 9, 6684–6694. [Google Scholar]
- Varkonyi-Gasic, E.; Lough, R.H.; Moss, S.; Wu, R.; Hellens, R. Kiwifruit floral gene APETALA2 is alternatively spliced and accumulates in aberrant indeterminate flowers in the absence of miR172. Plant Mol. Biol. 2012, 78, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; Chen, H.-W.; Li, Q.; Tao, J.-J.; Bian, X.-H.; Ma, B.; Zhang, W.-K.; Chen, S.-Y.; Zhang, J.-S. Three SAUR proteins SAUR76, SAUR77 and SAUR78 promote plant growth in Arabidopsis. Sci. Rep. 2015, 5, 12477. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Cong, P.; Tian, Y.; Zhu, Y. Using RNA-seq data to select reference genes for normalizing gene expression in apple roots. PLoS ONE 2017, 12, e0185288. [Google Scholar] [CrossRef]
- Satyanarayana, P.; Guzdar, P.N.; Huba, J.D.; Ossakow, S.L. Rayleigh-Taylor instability in the presence of a stratified shear layer. J. Geophys. Res. Space Phys. 1984, 89, 2945. [Google Scholar] [CrossRef]
- Tao, Q.; Niu, H.; Wang, Z.; Zhang, W.; Wang, H.; Wang, S.; Zhang, X.; Li, Z. Ethylene responsive factor ERF110 mediates ethylene-regulated transcription of a sex determination-related orthologous gene in two Cucumis species. J. Exp. Bot. 2018, 69, 2953–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizek, B.A.; Fletcher, J.C. Molecular mechanisms of flower development: An armchair guide. Nat. Rev. Genet. 2005, 6, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Krizek, A.B. Arabidopsis: Flower Development and Patterning. In Encyclopedia of Life Sciences (ELS); John Wiley & Sons, Ltd.: Chichester, UK, 2020. [Google Scholar] [CrossRef]
- Sharma, P.; Singh, R.; Sehrawat, N. A critical review on: Significance of floral homeotic APETALA2 gene in plant system. J. Appl. Pharm. Sci. 2020, 10, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Wollmann, H.; Mica, E.; Todesco, M.; Long, J.A.; Weigel, D. On reconciling the interactions between APETALA2, miR172 and AGAMOUS with the ABC model of flower development. Development 2010, 137, 3633–3642. [Google Scholar] [CrossRef] [Green Version]
- Maes, T.; Van De Steene, N.; Zethof, J.; Karimi, M.; D’Hauw, M.; Mares, G.; Van Montagu, M.; Gerats, T. Petunia Ap2-like Genes and Their Role in Flower and Seed Development. Plant Cell 2001, 13, 229–244. [Google Scholar] [CrossRef] [Green Version]
- Byzova, M.V.; Franken, J.; Aarts, M.G.; De Almeida-Engler, J.; Engler, G.; Mariani, C.; Campagne, M.M.V.L.; Angenent, G.C. Arabidopsis Sterile Apetala, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 1999, 13, 1002–1014. [Google Scholar] [CrossRef] [Green Version]
- Pan, I.L.; McQuinn, R.; Giovannoni, J.J.; Irish, V.F. Functional diversification of AGAMOUS lineage genes in regulating tomato flower and fruit development. J. Exp. Bot. 2010, 61, 1795–1806. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Shi, T.; Zheng, B.; Yumul, R.E.; Liu, X.; You, C.; Gao, Z.; Xiao, L.; Chen, X. APETALA 2 antagonizes the transcriptional activity of AGAMOUS in regulating floral stem cells in Arabidopsis thaliana. New Phytol. 2017, 215, 1197–1209. [Google Scholar] [CrossRef]
- Ori, N.; Eshed, Y.; Chuck, G.; Bowman, J.; Hake, S. Mechanisms that control knox gene expression in the Arabidopsis shoot. Development 2000, 127, 5523–5532. [Google Scholar] [CrossRef]
- Song, Y.H.; Lee, I.; Lee, S.Y.; Imaizumi, T.; Hong, J.C. Constans and Asymmetric Leaves 1 complex is involved in the induction of Flowering Locus T in photoperiodic flowering in Arabidopsis. Plant J. 2012, 69, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.S.; Kubota, A.; Imaizumi, T. Circadian Clock and Photoperiodic Flowering in Arabidopsis: Constans is a Hub for Signal Integration. Plant Physiol. 2017, 173, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xiang, Y.; Xu, E.; Ge, X.; Li, Z. Major Co-localized QTL for Plant Height, Branch Initiation Height, Stem Diameter, and Flowering Time in an Alien Introgression Derived Brassica napus DH Population. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.B.; Shen, Y.; Chang, H.; Hou, Y.; Harris, A.; Ma, S.F.; McPartland, M.; Hymus, G.J.; Adam, L.; Marion, C.; et al. The flowering time regulator CONSTANS is recruited to the FLOWERING LOCUS T promoter via a unique cis-element. New Phytol. 2010, 187, 57–66. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Sawikowska, A.; Neumann, M.; Posé, D.; Capovilla, G.; Langenecker, T.; Neher, R.A.; Krajewski, P.; Schmid, M. Temporal dynamics of gene expression and histone marks at the Arabidopsis shoot meristem during flowering. Nat. Commun. 2017, 8, 15120. [Google Scholar] [CrossRef] [PubMed]
- Jue, D.; Sang, X.; Liu, L.; Shu, B.; Wang, Y.; Liu, C.; Xie, J.; Shi, S. Identification of WRKY Gene Family from Dimocarpus longan and Its Expression Analysis during Flower Induction and Abiotic Stress Responses. Int. J. Mol. Sci. 2018, 19, 2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY Transcription Factor Family in Model Plants and Crops. Crit. Rev. Plant Sci. 2017, 36, 311–335. [Google Scholar] [CrossRef]
- Amini, S.; Rosli, K.; Abu-Bakar, M.-F.; Alias, H.; Mat-Isa, M.-N.; Juhari, M.-A.-A.; Haji-Adam, J.; Goh, H.-H.; Wan, K.-L. Transcriptome landscape of Rafflesia cantleyi floral buds reveals insights into the roles of transcription factors and phytohormones in flower development. PLoS ONE 2019, 14, e0226338. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Liu, D.; Ma, J.; Li, M.; Sui, S. CpWRKY71, a WRKY Transcription Factor Gene of Wintersweet (Chimonanthus praecox), Promotes Flowering and Leaf Senescence in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.; Meng, X.; Khanna, R.; Lamontagne, E.; Liu, Y.; Zhang, S. Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in Arabidopsis. PLoS Genet. 2014, 10, e1004384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Abu Qamar, S.; Chen, Z.; Mengiste, T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. Plant J. 2006, 48, 592–605. [Google Scholar] [CrossRef]
- Gregis, V.; Sessa, A.; Colombo, L.; Kater, M.M. AGL24, Short Vegetative Phase, and Apetala1redundantly Control Agamous during Early Stages of Flower Development in Arabidopsis. Plant Cell 2006, 18, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Gregis, V.; Sessa, A.; Dorca-Fornell, C.; Kater, M.M. The Arabidopsis floral meristem identity genes AP1, AGL24 and SVP directly repress class B and C floral homeotic genes. Plant J. 2009, 60, 626–637. [Google Scholar] [CrossRef]
- Dorca-Fornell, C.; Gregis, V.; Grandi, V.; Coupland, G.; Colombo, L.; Kater, M.M. The Arabidopsis SOC1-like genes AGL42, AGL71 and AGL72 promote flowering in the shoot apical and axillary meristems. Plant J. 2011, 67, 1006–1017. [Google Scholar] [CrossRef]
- Van Mourik, H.; Van Dijk, A.D.J.; Stortenbeker, N.; Angenent, G.C.; Bemer, M. Divergent regulation of Arabidopsis SAUR genes: A focus on the SAUR10-clade. BMC Plant Biol. 2017, 17, 245. [Google Scholar] [CrossRef] [Green Version]
- Becker, A. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Adamczyk, B.J.; Lehti-Shiu, M.D.; Fernandez, D.E. The MADS domain factors AGL15 and AGL18 act redundantly as repressors of the floral transition in Arabidopsis. Plant J. 2007, 50, 1007–1019. [Google Scholar] [CrossRef]
- Nakaminami, K.; Hill, K.; Perry, S.E.; Sentoku, N.; Long, J.A.; Karlson, D.T. Arabidopsis cold shock domain proteins: Relationships to floral and silique development. J. Exp. Bot. 2009, 60, 1047–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.-K.; Hsu, W.-H.; Lee, P.-F.; Thiruvengadam, M.; Chen, H.-I.; Yang, C.-H. The MADS box gene, Forever Young Flower, acts as a repressor controlling floral organ senescence and abscission in Arabidopsis. Plant J. 2011, 68, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Peng, X.-Y.; Yu, Y.-C.; Sun, Z.-Y.; Han, L. The Effects of DNA Methylation Inhibition on Flower Development in the Dioecious Plant Salix Viminalis. Forests 2019, 10, 173. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Chang, F.; You, C.; Cui, J.; Zhu, G.; Wang, L.; Zheng, Y.; Qi, J.; Ma, H. Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J. 2014, 81, 268–281. [Google Scholar] [CrossRef]
- Soppe, W.J.; Jacobsen, E.S.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J. The Late Flowering Phenotype of fwa Mutants is Caused by Gain-of-Function Epigenetic Alleles of a Homeodomain Gene. Mol. Cell 2000, 6, 791–802. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Zhang, Y.; He, X.; Zhao, H.; Xu, W.; Deng, H.; Wang, H.; Wang, S.; Su, D.; Zheng, Z.; Yang, B.; et al. Genome-Wide Identification of DNA Methylases and Demethylases in Kiwifruit (Actinidia chinensis). Front. Plant Sci. 2020, 11, 1380. [Google Scholar] [CrossRef]
- Kim, S.-C.; Uhm, Y.K.; Ko, S.; Oh, C.J.; Kwack, Y.-B.; Kim, H.L.; Lee, Y.; An, C.S.; Park, P.B.; Kim, H.B. KiwiPME1 encoding pectin methylesterase is specifically expressed in the pollen of a dioecious plant species, kiwifruit (Actinidia chinensis). Hortic. Environ. Biotechnol. 2015, 56, 402–410. [Google Scholar] [CrossRef]
- Wormit, A.; Usadel, B. The Multifaceted Role of Pectin Methylesterase Inhibitors (PMEIs). Int. J. Mol. Sci. 2018, 19, 2878. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.Y.; Feng, J.; Wu, J.; Wang, X.W. BoPMEI1, a pollen-specific pectin methylesterase inhibitor, has an essential role in pollen tube growth. Planta 2010, 231, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.S.; Song, J.T.; Cheong, J.-J.; Lee, Y.-H.; Lee, Y.W.; Hwang, I.; Lee, J.S.; Choi, Y.D. Jasmonic acid carboxyl methyltransferase: A key enzyme for jasmonate-regulated plant responses. Proc. Natl. Acad. Sci. USA 2001, 98, 4788–4793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasternack, C. Jasmonates: An Update on Biosynthesis, Signal Transduction and Action in Plant Stress Response, Growth and Development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, J.W. The Hormonal Regulation of Flower Development. J. Plant Growth Regul. 2011, 30, 242–254. [Google Scholar] [CrossRef]
- Manzano, S.; Martínez, C.; García, J.M.; Megías, Z.; Jamilena, M. Involvement of ethylene in sex expression and female flower development in watermelon (Citrullus lanatus). Plant Physiol. Biochem. 2014, 85, 96–104. [Google Scholar] [CrossRef]
- Sun, J.-J.; Li, F.; Li, X.; Liu, X.-C.; Rao, G.-Y.; Luo, J.-C.; Wang, D.-H.; Xu, Z.-H.; Bai, S.-N. Why is ethylene involved in selective promotion of female flower development in cucumber? Plant Signal. Behav. 2010, 5, 1052–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.-H.; Li, P.-F.; Chen, M.-K.; Lee, Y.-I.; Yang, C.-H. Forever young Flower Negatively Regulates Ethylene Response DNA-Binding Factors by Activating an Ethylene-Responsive Factor to Control Arabidopsis Floral Organ Senescence and Abscission. Plant Physiol. 2015, 168, 1666–1683. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Wang, H.; Fu, Z.; Liu, J.; Yu, Y. Identification and expression analysis of ERF transcription factor genes in petunia during flower senescence and in response to hormone treatments. J. Exp. Bot. 2010, 62, 825–840. [Google Scholar] [CrossRef]
- Nakano, T.; Fujisawa, M.; Shima, Y.; Ito, Y. The AP2/ERF transcription factor SlERF52 functions in flower pedicel abscission in tomato. J. Exp. Bot. 2014, 65, 3111–3119. [Google Scholar] [CrossRef] [Green Version]
- Salman-Minkov, A.; Levi, A.; Wolf, S.; Trebitsh, T. ACC Synthase Genes are Polymorphic in Watermelon (Citrullus spp.) and Differentially Expressed in Flowers and in Response to Auxin and Gibberellin. Plant Cell Physiol. 2008, 49, 740–750. [Google Scholar] [CrossRef]
- Lin, Z.; Zhong, S.; Grierson, D. Recent advances in ethylene research. J. Exp. Bot. 2009, 60, 3311–3336. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zhou, Y.; Liu, X.; Yu, P.; Cohen, J.D.; Meyerowitz, E.M. LEAFY Controls Auxin Response Pathways in Floral Primordium Formation. Sci. Signal. 2013, 6, ra23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-E.; Kim, Y.-S.; Yoon, H.-K.; Park, C.-M. Functional characterization of a small auxin-up RNA gene in apical hook development in Arabidopsis. Plant Sci. 2007, 172, 150–157. [Google Scholar] [CrossRef]
- Fu, X.; Sudhakar, D.; Peng, J.; Richards, D.E.; Christou, P.; Harberd, N.P. Expression of Arabidopsis GAI in Transgenic Rice Represses Multiple Gibberellin Responses. Plant Cell 2001, 13, 1791–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Kang, S.G.; Hah, C.; Jang, J.-C. Molecular and cellular characterization of GA-Stimulated Transcripts GASA4 and GASA6 in Arabidopsis thaliana. Plant Sci. 2016, 246, 1–10. [Google Scholar] [CrossRef]
- Gomez, M.D.; Barro-Trastoy, D.; Escoms, E.; Saura-Sanchez, M.; Sánchez, I.; Briones-Moreno, A.; Vera-Sirera, F.; Carrera, E.; Ripoll, J.J.; Yanofsky, M.F.; et al. Gibberellins negatively modulate ovule number in plants. Development 2018, 145, 163865. [Google Scholar] [CrossRef] [Green Version]
- Zentella, R.; Yamauchi, D.; Ho, T.-H.D. Molecular Dissection of the Gibberellin/Abscisic Acid Signaling Pathways by Transiently Expressed RNA Interference in Barley Aleurone Cells. Plant Cell 2002, 14, 2289–2301. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ren, L.; Yue, J.-H.; Wang, L.; Zhuo, L.-H.; Shen, X.-H. A comprehensive analysis of flowering transition in Agapanthus praecox ssp. orientalis (Leighton) Leighton by using transcriptomic and proteomic techniques. J. Proteom. 2013, 80, 1–25. [Google Scholar] [CrossRef]
- Dill, A.; Thomas, S.G.; Hu, J.; Steber, C.M.; Sun, T.-P. The Arabidopsis F-Box Protein SLEEPY1 Targets Gibberellin Signaling Repressors for Gibberellin-Induced Degradation. Plant Cell 2004, 16, 1392–1405. [Google Scholar] [CrossRef] [Green Version]
- Matías-Hernández, L.; Jiang, W.; Yang, K.; Tang, K.; Brodelius, P.E.; Pelaz, S. AaMYB1 and its orthologue AtMYB61 affect terpene metabolism and trichome development inArtemisia annua and Arabidopsis thaliana. Plant J. 2017, 90, 520–534. [Google Scholar] [CrossRef] [Green Version]
- Kurakawa, T.; Ueda, N.; Maekawa, M.; Kobayashi, K.; Kojima, M.; Nagato, Y.; Sakakibara, H.; Kyozuka, J. Direct control of shoot meristem activity by a cytokinin-activating enzyme. Nature 2007, 445, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Kuroha, T.; Tokunaga, H.; Kojima, M.; Ueda, N.; Ishida, T.; Nagawa, S.; Fukuda, H.; Sugimoto, K.; Sakakibara, H. Functional Analyses of Lonely Guy Cytokinin-Activating Enzymes Reveal the Importance of the Direct Activation Pathway in Arabidopsis. Plant Cell 2009, 21, 3152–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]






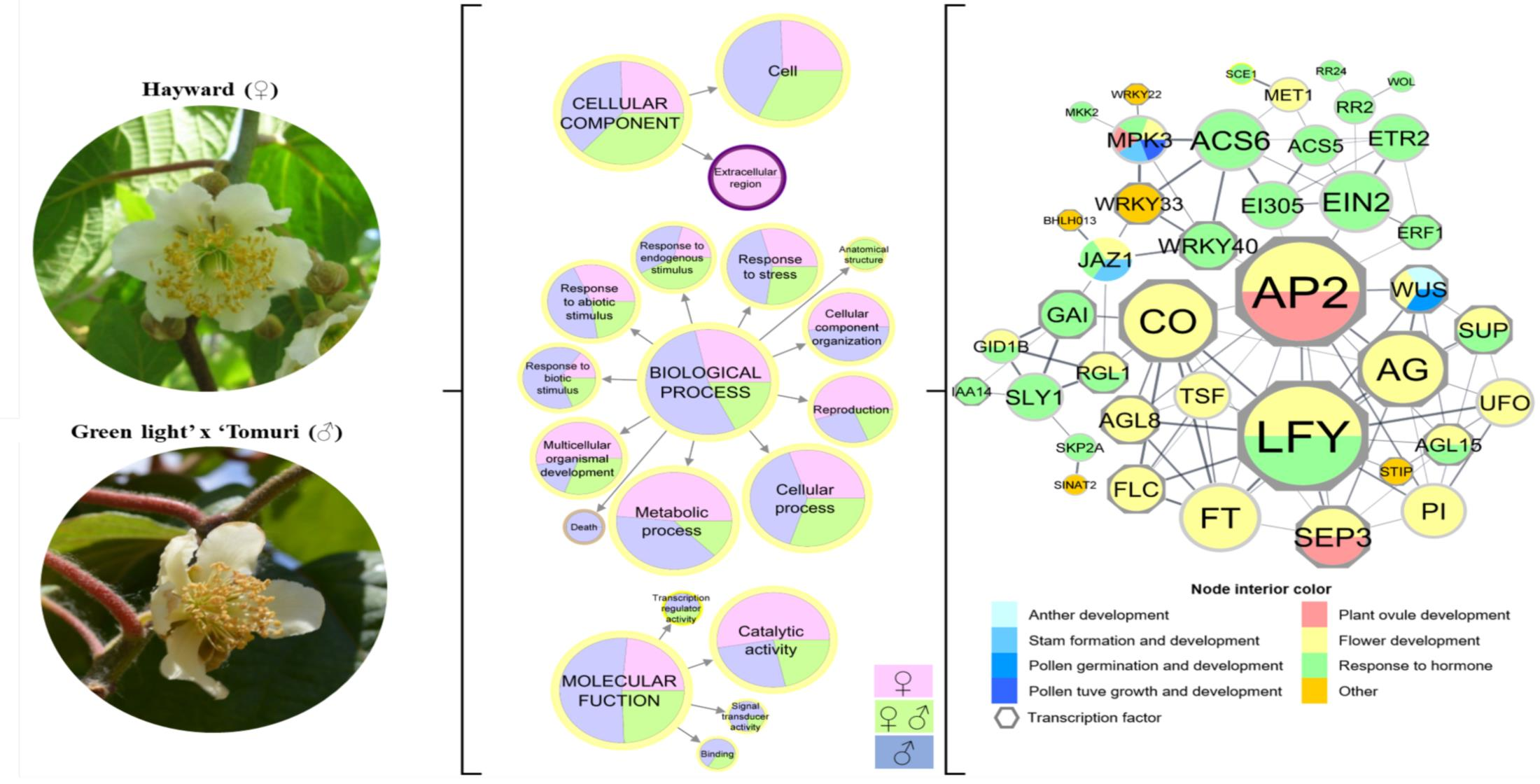{kind=link}
{kind=link}
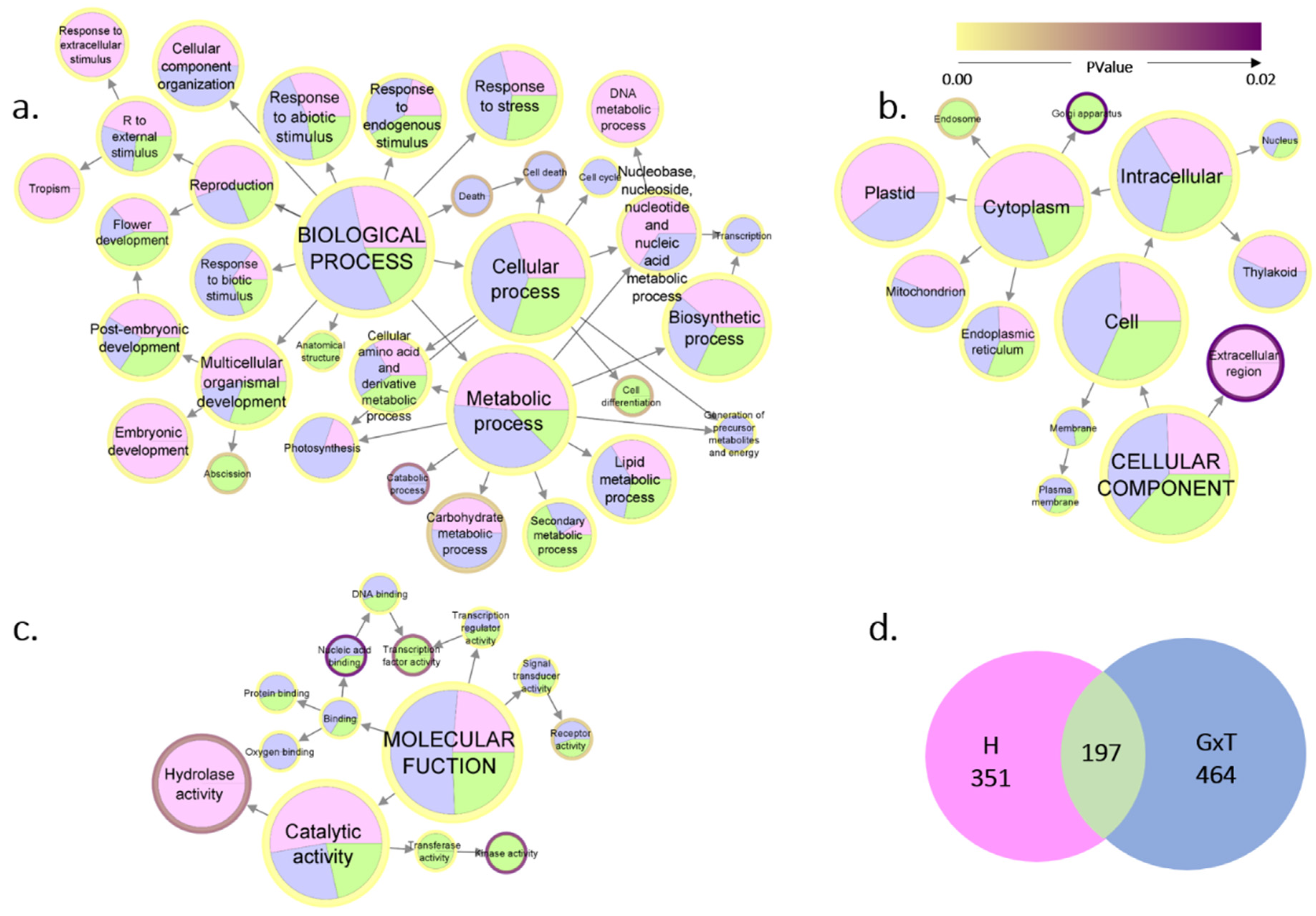{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G × T | H | |||
|---|---|---|---|---|
| Alignment Statistics | Counts | % of Total Reads | Counts | % of Total Reads |
| Reads Input | 80,209,378 | - | 73,001,231 | - |
| Reads mapped left | 46,514,175 | 57.99% | 36,135,609 | 49.50% |
| Multiple alignments left | 3,114,693 | 6.70% | 4,088,069 | 5.60% |
| Reads mapped right | 45,190,039 | 56.34% | 34,602,583 | 47.40% |
| Multiple alignments right | 3,009,555 | 6.66% | 4,088,069 | 5.60% |
| Overall read mapping rate | 45,852,107 | 57.20% | 35,405,597 | 48.50% |
| Aligned pairs | 36,321,861 | 45.28% | 29,930,505 | 41.00% |
| Multiple alignments | 2,438,520 | 6.71% | 1,646,178 | 5.50% |
| Discordant alignments | 206,108 | 0.57% | 119,722 | 0.40% |
| Q20(%) | 97.87% | 97.32% | ||
| Q30(%) | 94.59% | 95.30% | ||
| GC(%) | 46.68% | 45.74% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapata, P.; González, M.; Pacheco, I.; Jorquera, C.; Silva-Andrade, C.; Garrido, M.I.; Infante, R.; Salazar, J.A. Transcriptomic Analysis of Sex-Associated DEGs in Female and Male Flowers of Kiwifruit (Actinidia deliciosa [A. Chev] C. F. Liang & A. R. Ferguson). Horticulturae 2022, 8, 38. https://doi.org/10.3390/horticulturae8010038
Zapata P, González M, Pacheco I, Jorquera C, Silva-Andrade C, Garrido MI, Infante R, Salazar JA. Transcriptomic Analysis of Sex-Associated DEGs in Female and Male Flowers of Kiwifruit (Actinidia deliciosa [A. Chev] C. F. Liang & A. R. Ferguson). Horticulturae. 2022; 8(1):38. https://doi.org/10.3390/horticulturae8010038
Chicago/Turabian StyleZapata, Patricio, Makarena González, Igor Pacheco, Claudia Jorquera, Claudia Silva-Andrade, Marco Isaac Garrido, Rodrigo Infante, and Juan Alfonso Salazar. 2022. "Transcriptomic Analysis of Sex-Associated DEGs in Female and Male Flowers of Kiwifruit (Actinidia deliciosa [A. Chev] C. F. Liang & A. R. Ferguson)" Horticulturae 8, no. 1: 38. https://doi.org/10.3390/horticulturae8010038