
Characterization of Genomic Variation from Lotus (Nelumbo Adans.) Mutants with Wide and Narrow Tepals


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Grouping of Lotus Accessions
2.3. DNA Extraction
2.4. Library Construction and Whole Genome Resequencing
2.5. Data Filtering
2.6. Read Mapping, Variant Detection, and Annotation
2.7. Screening and Enrichment Analysis of Variant Genes
2.8. The Assessment of the Reliability of Candidate Genes
3. Results
3.1. Whole-Genome Resequencing Data of the Six Genotypes
3.2. SNPs and Indels in the Six Genotypes
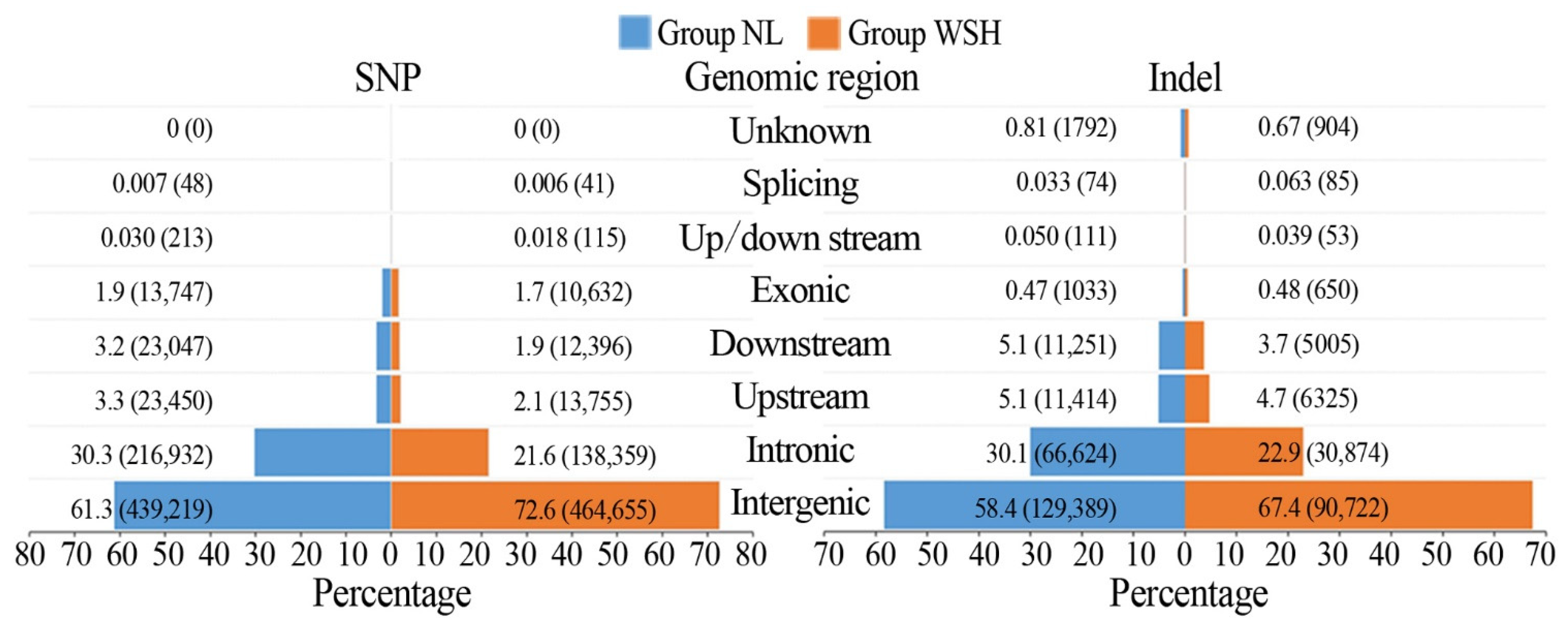
3.3. Analysis of SNPs and Indels between Mutant Accessions and Their Controls
3.4. Annotation and Analysis of Genes Harboring SNPs and Indels in the Exonic Regions
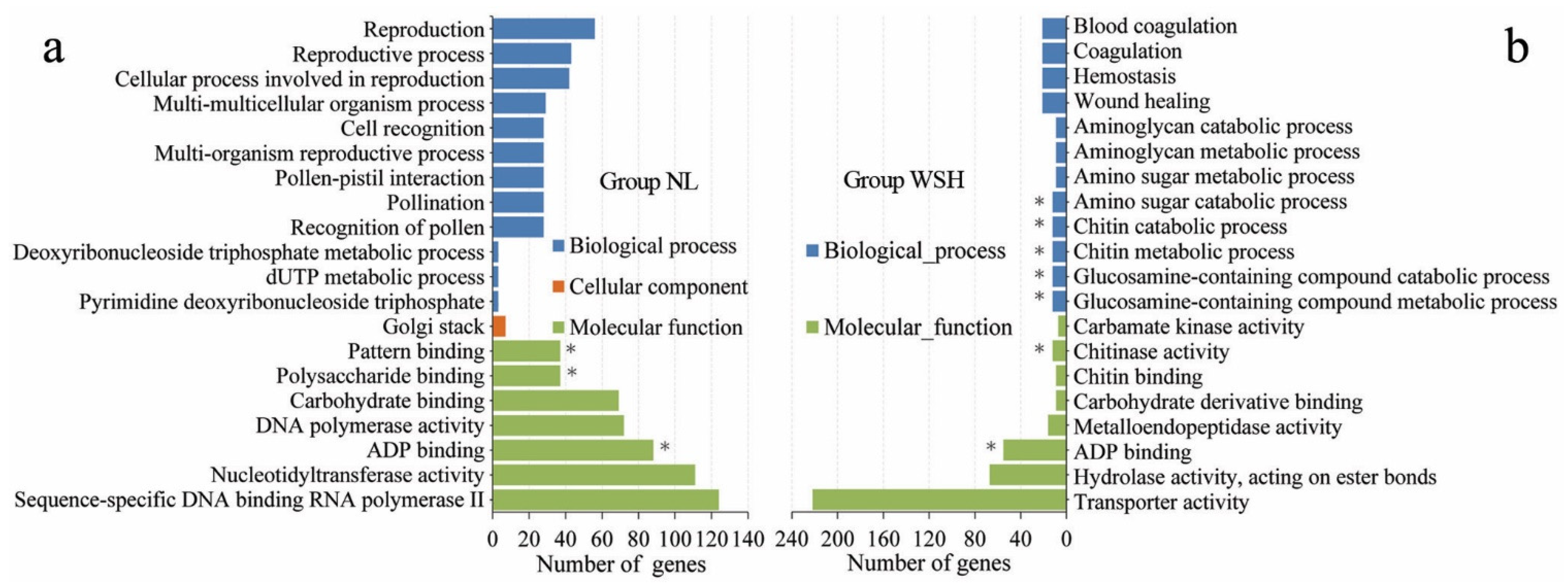
3.5. Sketchy Sampling of Biological Information of Candidate Genes
4. Discussion
4.1. Genetic Relationship of the Individuals in Each Group
4.2. Differences in Genome Mapping between the NL and WSH Groups
4.3. The Reliability of the Candidate Genes That May Associate with Tepal Shape in Lotus
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, H.B. Cultivation of lotus (Nelumbo nucifera Gaertn. ssp. nucifera) and its utilization in China. Genet. Resour. Crop Evol. 2009, 56, 323–330. [Google Scholar] [CrossRef]
- Li, H.L. Classification and phylogeny of Nymphaeaceae and allied families. Am. Midl. Nat. 1955, 54, 33–41. [Google Scholar] [CrossRef]
- Li, Y.; Svetlana, P.; Yao, J.X.; Li, C.S. A review on the taxonomic, evolutionary and phytogeographic studies of the lotus plant (Nelumbonaceae: Nelumbo). Acta Geol. Sin. 2014, 88, 1252–1261. [Google Scholar] [CrossRef]
- Gowthami, R.; Sharma, N.; Pandey, R.; Agrawal, A. A model for integrated approach to germplasm conservation of Asian lotus (Nelumbo nucifera Gaertn.). Genet. Resour. Crop Evol. 2021, 68, 1269–1282. [Google Scholar] [CrossRef]
- Wang, Q.C.; Zhang, X.Y. Lotus Flower Cultivars in China; Bao, M.Z., Translator; Forestry Publishing House Press: Beijing, China, 2004; p. 59. [Google Scholar]
- Liu, L.; Li, Y.C.; Min, J.; Tian, D.K. Analysis of the cultivar names and characteristics of global lotus (Nelumbo). Agric. Sci. 2019, 9, 163–181, (In Chinese with English Abstract). [Google Scholar] [CrossRef]
- Lin, Z.Y.; Zhang, C.H.; Cao, D.D.; Damaris, R.N.; Yang, P.F. The latest studies on lotus (Nelumbo nucifera)-an emerging horticultural model plant. Int. J. Mol. Sci. 2019, 20, 3680. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Xiang, Y.C.; Tian, D.K. Consumption demand and market potential of cut lotus flower in China. J. Anhui Agric. Sci. 2018, 46, 212–217, (In Chinese with English Abstract). [Google Scholar]
- Huang, L.Y.; Li, M.; Cao, D.D.; Yang, P.F. Genetic dissection of rhizome yield-related traits in Nelumbo nucifera through genetic linkage map construction and QTL mapping. Plant Physiol. Bioch. 2021, 160, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Chen, G.L.; Jin, J.; Wang, Y.; Xu, M.L.; Peng, J.; Ding, Y. Small RNA and transcriptome sequencing reveals miRNA regulation of floral thermogenesis in Nelumbo nucifera. Int. J. Mol. Sci. 2020, 21, 3324. [Google Scholar] [CrossRef]
- Gui, S.T.; Peng, J.; Wang, X.L.; Wu, Z.; Cao, R.; Salse, J.; Zhang, H.Y.; Zhu, Z.X.; Xia, Q.J.; Quan, Z.W.; et al. Improving Nelumbo nucifera genome assemblies using high-resolution genetic maps and BioNano genome mapping reveals ancient chromosome rearrangements. Plant J. 2018, 94, 721–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, R.; VanBuren, R.; Liu, Y.; Yang, M.; Han, Y.; Li, L.T.; Zhang, Q.; Kim, M.J.; Schatz, M.C.; Campbell, M.; et al. Genome of the long-living sacred lotus (Nelumbo nucifera Gaertn.). Genome Biol. 2013, 14, R41. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef]
- Say, Y.H. The association of insertions/deletions (INDELs) and variable number tandem repeats (VNTRs) with obesity and its related traits and complications. J. Physiol. Anthrop. 2017, 36, 25. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Pippucci, T.; Sidore, C. XCAVATOR: Accurate detection and genotyping of copy number variants from second and third generation whole-genome sequencing experiments. BMC Genom. 2017, 18, 747. [Google Scholar] [CrossRef] [Green Version]
- Neerman, N.; Faust, G.; Meeks, N.; Modai, S.; Kalfon, L.; Falik-Zaccai, T.; Kaplun, A. A clinically validated whole genome pipeline for structural variant detection and analysis. BMC Genom. 2019, 20, 545. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Gui, S.T.; Zhu, Z.X.; Wang, X.L.; Ke, W.D.; Ding, Y. Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred lotus (Nelumbo nucifera). PLoS ONE 2015, 11, e0143765. [Google Scholar] [CrossRef]
- Huang, L.Y.; Yang, M.; Li, L.; Li, H.; Yang, D.; Shi, T.; Yang, P.F. Whole genome re-sequencing reveals evolutionary patterns of sacred lotus (Nelumbo nucifera). J. Integr. Plant Biol. 2018, 60, 2–15. [Google Scholar] [CrossRef]
- Hase, Y.; Fujioka, S.; Yoshida, S.; Sun, G.Q.; Umeda, M.; Tanaka, A. Ectopic endoreduplication caused by sterol alteration results in serrated petals in Arabidopsis. J. Exp. Bot. 2005, 56, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.P.; Lord, E.M. Floral development in Arabidopsis thaliana: A comparison of the wild type and the homeotic pistillata mutant. Can. J. Bot. 1989, 67, 2922–2936. [Google Scholar] [CrossRef]
- Peaucelle, A.; Wightman, R.; Höfte, H. The control of growth symmetry breaking in the Arabidopsis hypocotyl. Curr. Biol. 2015, 25, 1746–1752. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, S.; Nii, K.; Yokoo, M. Three-dimensional formation of corolla shapes in relation to the developmental distortion of petals in Eustoma grandiflorum. Sci. Hortic. 2011, 132, 66–70. [Google Scholar] [CrossRef]
- Conner, J.; Liu, Z.C. LEUNIG, a putative transcriptional corepressor that regulates AGAMOUS expression during flower development. Proc. Natl. Acad. Sci. USA 2000, 97, 12902–12907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.C.; Meyerowitz, E.M. LEUNIG regulates AGAMOUS expression in Arabidopsis flowers. Development 1995, 121, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Franks, R.G.; Wang, C.X.; Levin, J.Z.; Liu, Z.C. SEUSS, a member of a novel family of plant regulatory proteins, represses floral homeotic gene expression with LEUNIG. Development 2002, 129, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Franks, R.G.; Liu, Z.C.; Fischer, R.L. SEUSS and LEUNIG regulate cell proliferation, vascular development and organ polarity in Arabidopsis petals. Planta 2006, 224, 801–811. [Google Scholar] [CrossRef]
- Dinneny, J.R.; Yadegari, R.; Fischer, R.L.; Yanofsky, M.F.; Weigel, D. The role of JAGGED in shaping lateral organs. Development 2004, 131, 1101–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauret-Güeto, S.; Schiessl, K.; Bangham, A.; Sablowski, R.; Coen, E. JAGGED controls Arabidopsis petal growth and shape by interacting with a divergent polarity field. PLoS Biol. 2013, 11, e1001550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiessl, K.; Muiño, J.M.; Sablowski, R. Arabidopsis JAGGED links floral organ patterning to tissue growth by repressing Kiprelated cell cycle inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 2830–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juntheikki-Palovaara, I.; Tähtiharju, S.; Lan, T.Y.; Broholm, S.K.; Rijpkema, A.S.; Ruonala, R.; Kale, L.; Albert, V.A.; Teeri, T.H.; Elomaa, P. Functional diversification of duplicated CYC2 clade genes in regulation of inflorescence development in Gerbera hybrida (Asteraceae). Plant J. 2014, 79, 783–796. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.L.; Ramm, S.; Kappel, C.; Ward, S.; Leyser, O.; Sakamoto, T.; Kurata, T.; Bevan, M.W.; Lenhard, M. The Tinkerbell (Tink) Mutation Identifies the dual-specificity MAPK phosphatase INDOLE-3-BUTYRIC ACID-RESPONSE5 (IBR5) as a novel regulator of organ size in Arabidopsis. PLoS ONE 2015, 10, e0131103. [Google Scholar]
- Ren, H.B.; Dang, X.; Yang, Y.Q.; Huang, D.Q.; Liu, M.T.; Gao, X.W.; Lin, D.S. SPIKE1 activates ROP GTPase to modulate petal growth and shape. Plant Physiol. 2016, 172, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.R.; Zhang, Y.; Li, Z.Z.; Liu, H.; Chen, J.M.; Yang, X.Y. Genetic diversity, population structure, and historical gene flow of Nelumbo lutea in USA using microsatellite markers. Aquat. Bot. 2020, 160, 103162. [Google Scholar] [CrossRef]
- Li, C.; Mo, H.B.; Tian, D.K.; Xu, Y.X.; Meng, J.; Tilt, K. Genetic diversity and structure of American lotus (Nelumbo lutea Willd.) in North America revealed from microsatellite markers. Sci. Hortic. 2015, 189, 17–21. [Google Scholar] [CrossRef]
- Guo, H.B.; Li, S.M.; Peng, J.; Ke, W.M. Genetic diversity of Nelumbo accessions revealed by RAPD. Genet. Resour. Crop Evol. 2007, 54, 741–748. [Google Scholar] [CrossRef]
- Liu, Z.W.; Zhu, H.L.; Zhou, J.H.; Jiang, S.J.; Wang, Y.; Kuang, J.; Ji, Q.; Peng, J.; Wang, J.; Gao, L.; et al. Resequencing of 296 cultivated and wild lotus accessions unravels its evolution and breeding history. Plant J. 2020, 104, 1673–1684. [Google Scholar] [CrossRef]
- Cao, Y.N.; Comes, H.P.; Sakaguchi, S.; Chen, L.Y.; Qiu, Y.X. Evolution of East Asia’s Arcto-Tertiary relict Euptelea (Eupteleaceae) shaped by Late Neogene vicariance and Quaternary climate change. BMC Evol. Biol. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.L.; Qin, M.; Liu, Q.Q.; Zhang, D.S.; Tian, D.K. Detection of genetic variation between broad and narrow-tepalled American lotus (Nelumbo lutea Willd.) by EST-SSR markers. Acta Agric. Boreali-Occident. Sin. 2020, 29, 306–314, (In Chinese with English Abstract). [Google Scholar]
- Li, Z.; Liu, X.Q.; Gituru, R.W.; Juntawong, N.; Zhou, M.Q.; Chen, L.Q. Genetic diversity and classification of Nelumbo germplasm of different origins by RAPD and ISSR analysis. Sci. Hortic. 2010, 125, 724–732. [Google Scholar] [CrossRef]
- Brock, M.T.; Weinig, C. Plasticity and environment-specific covariances: An investigation of floral-vegetative and within flower correlations. Evolution 2007, 61, 2913–2924. [Google Scholar] [CrossRef] [PubMed]
- Pyke, K.A.; Page, A.M. Plastid ontogeny during petal development in Arabidopsis. Plant Physiol. 1998, 116, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacón, B.; Ballester, R.; Birlanga, V.; Rolland-Lagan, A.G.; Pérez-Pérez, J.M. A quantitative framework for flower phenotyping in cultivated carnation (Dianthus caryophyllus L.). PLoS ONE 2013, 8, e82165. [Google Scholar] [CrossRef] [PubMed]
- Marsden-Jones, E.M.; Turrill, W.B. A quantitative study of petal size and shape in Saxifraga granulata L. J. Genet. 1947, 48, 206–218. [Google Scholar] [CrossRef] [PubMed]
- He, Z.H.; Cheeseman, I.; He, D.Z.; Kohorn, B.D. A cluster of five cell wall-associated receptor kinase genes, Wak1–5, are expressed in specific organs of Arabidopsis. Plant Mol. Biol. 1999, 39, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Kohorn, B.D.; Kohorn, S.L. The cell wall associated kinases, WAKs, as pectin receptors. Front. Plant Sci. 2012, 3, 88. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.A.; Kohorn, B.D. Wall-associated kinases are expressed throughout plant development and are required for cell expansion. Plant Cell 2001, 13, 303–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.C.; Metheetrairut, C.; Irish, V.F. Natural variation identifies multiple loci controlling petal shape and size in Arabidopsis thaliana. PLoS ONE 2013, 8, e56743. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Database | Sample Group | No. of Variant Genes | No. of Annotated Genes | Percentage of Annotated Genes | Distributed Times of Annotated Genes | |||
|---|---|---|---|---|---|---|---|---|
| Total Times | Biological Process | Cellular Component | Molecular Function | |||||
| GO | NL | 6715 | 4890 | 72.8 | 197,885 | 99,311 | 72,072 | 26,502 |
| WSH | 5353 | 1272 | 23.8 | 52,425 | 25,824 | 20,751 | 5850 | |
| Database | Sample group | No. of variant genes | No. of genes annotated | Percentage of annotated genes | No. of pathways | No. of annotated genes in the top three pathways | ||
| Metabolic pathways | Biosynthesis of secondary metabolites | Starch and sucrose metabolism | ||||||
| KEGG | NL | 6715 | 2286 | 34.0 | 122 | 430 | 256 | 67 |
| WSH | 5353 | 645 | 12.0 | 68 | 128 | 84 | 39 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Qin, M.; Li, S.; Zhang, D.; Liu, Q.; Yan, M.; Tian, D. Characterization of Genomic Variation from Lotus (Nelumbo Adans.) Mutants with Wide and Narrow Tepals. Horticulturae 2021, 7, 593. https://doi.org/10.3390/horticulturae7120593
Liu F, Qin M, Li S, Zhang D, Liu Q, Yan M, Tian D. Characterization of Genomic Variation from Lotus (Nelumbo Adans.) Mutants with Wide and Narrow Tepals. Horticulturae. 2021; 7(12):593. https://doi.org/10.3390/horticulturae7120593
Chicago/Turabian StyleLiu, Fengluan, Mi Qin, Shuo Li, Dasheng Zhang, Qingqing Liu, Mengxiao Yan, and Daike Tian. 2021. "Characterization of Genomic Variation from Lotus (Nelumbo Adans.) Mutants with Wide and Narrow Tepals" Horticulturae 7, no. 12: 593. https://doi.org/10.3390/horticulturae7120593