
Identification of a Novel Dehydrogenase from Gluconobacter oxydans for Degradation of Inhibitors Derived from Lignocellulosic Biomass
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains and Materials
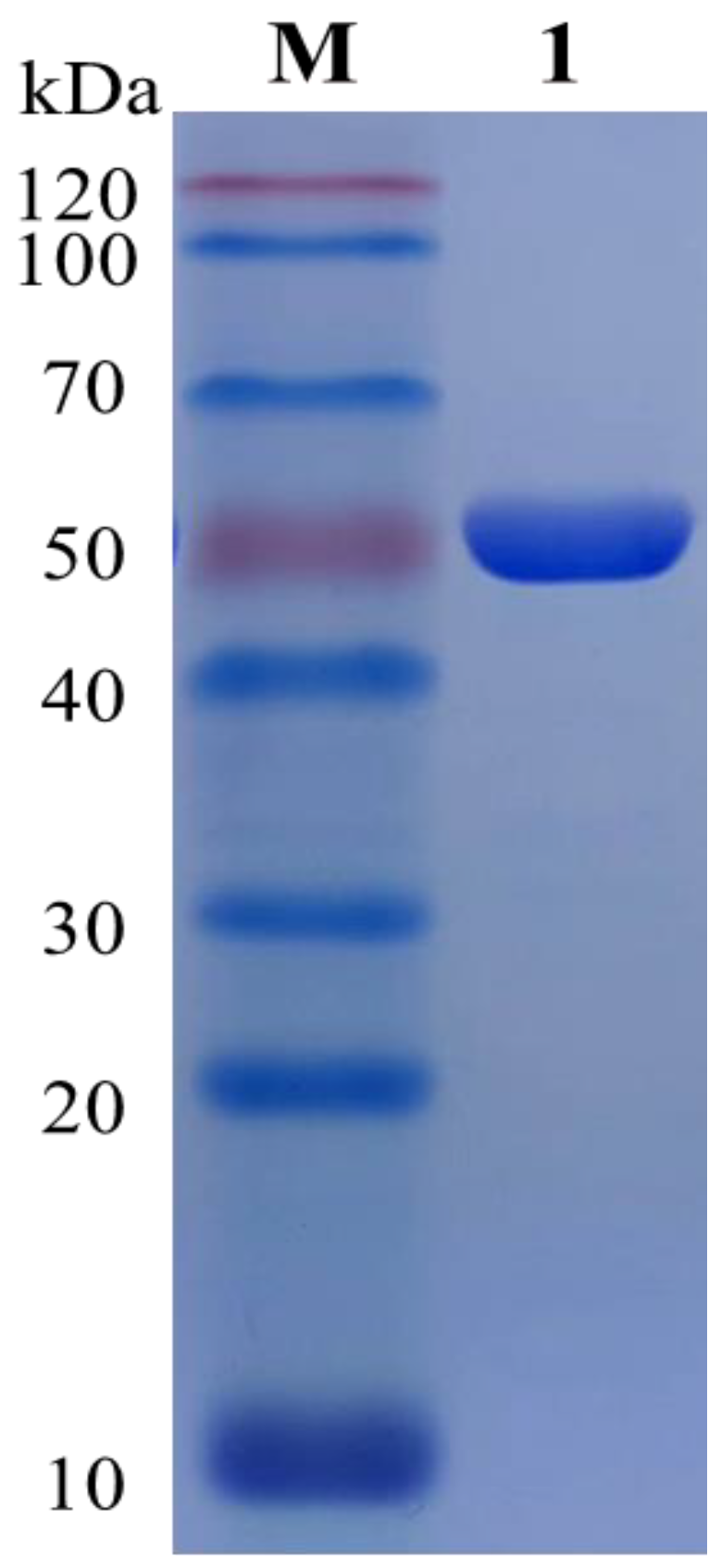
2.2. Cloning, Expression, and Purification
2.3. Enzyme Activity Assay
2.4. Effects of Temperature and pH on the Activity and Stability
2.5. Inhibitor Analytical Methods
2.6. Protein Mass Spectrometry
3. Results and Discussion
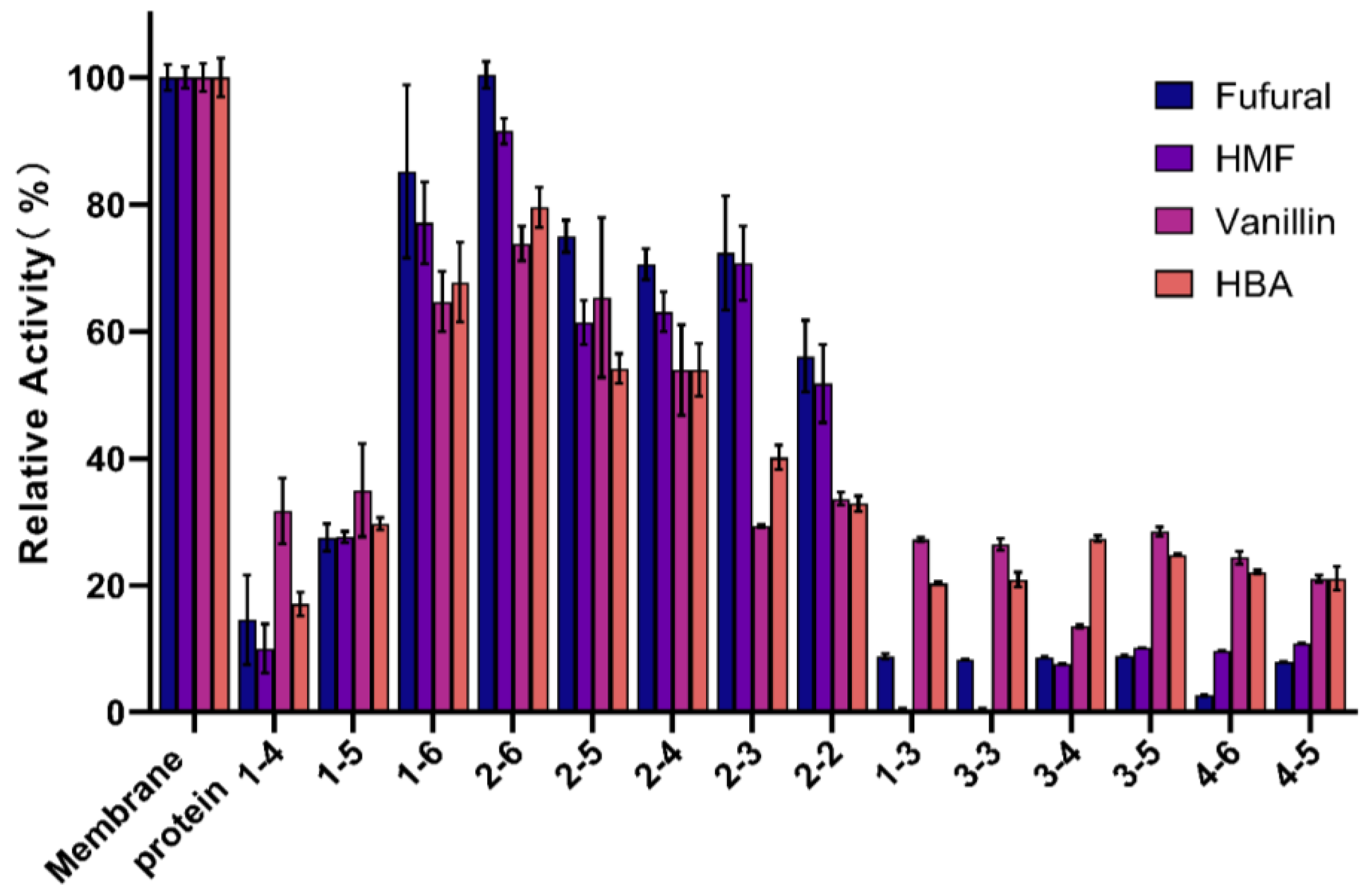
3.1. Screening of Aldehyde Inhibitor Degradation Genes Derived from G. oxydans DSM 2003
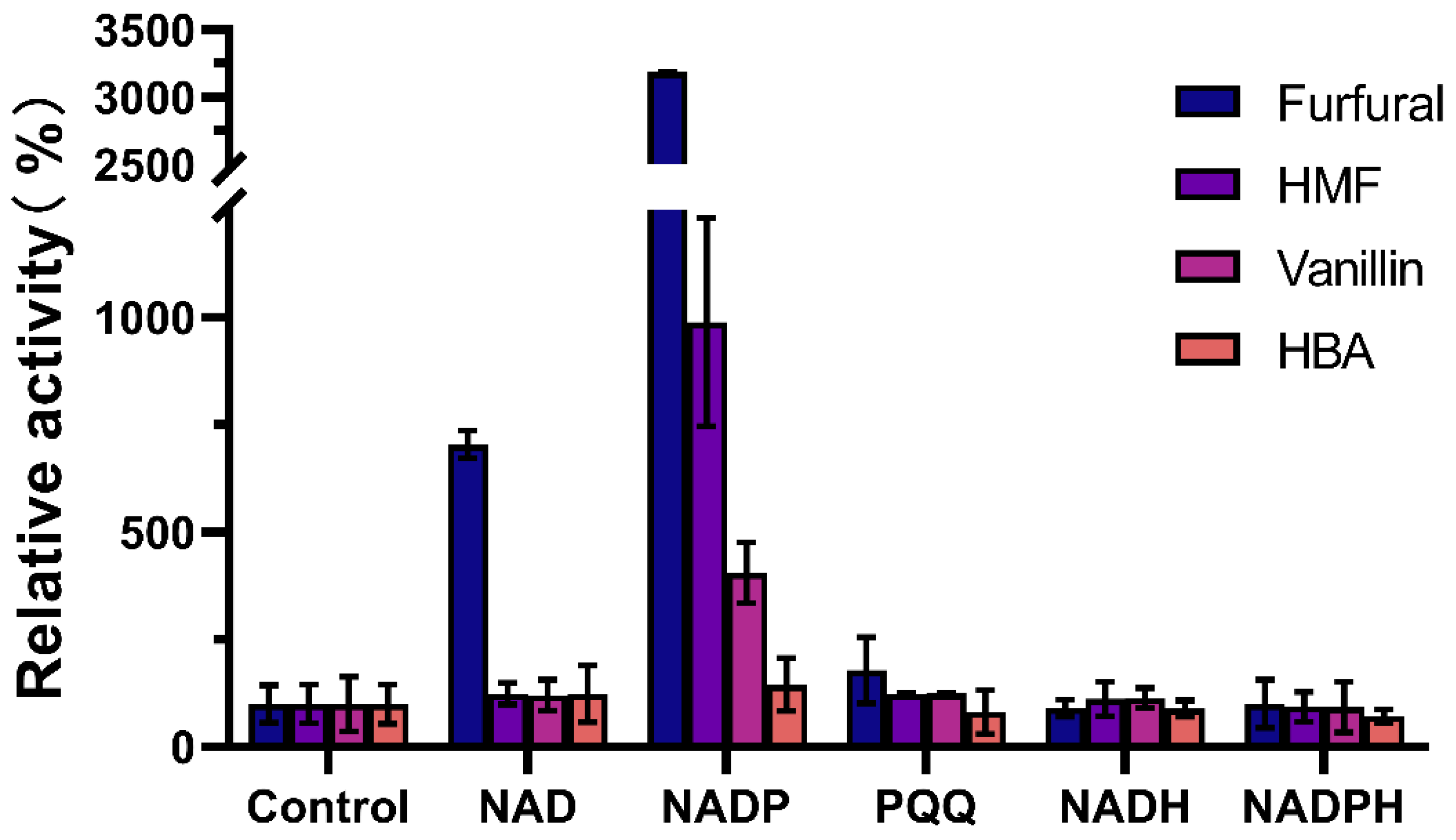
3.2. Effect of Cofactors on Enzymatic Activity of W826-RS0111485
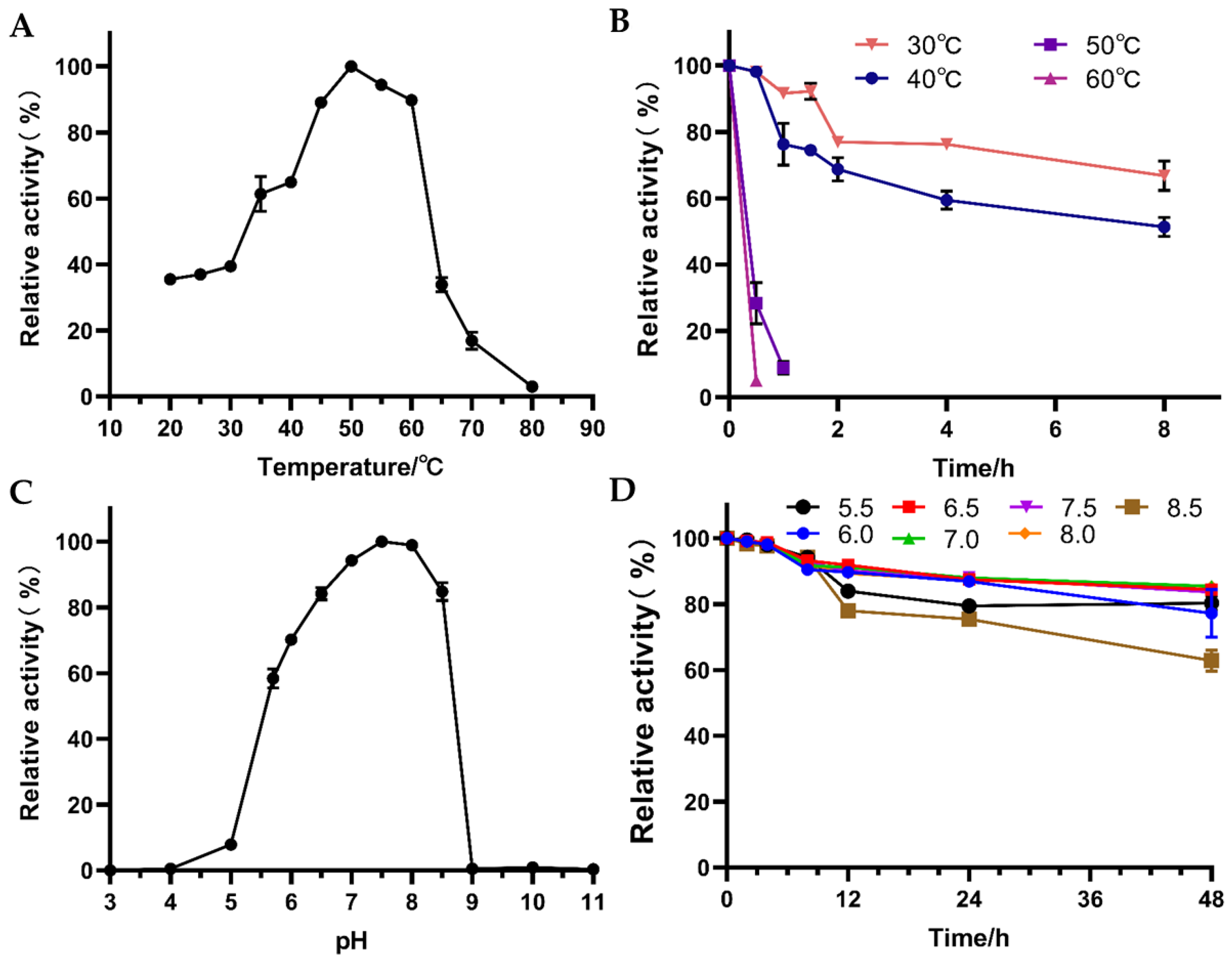
3.3. Effects of Temperature and pH on the Activity and Stability of W826-RS0111485
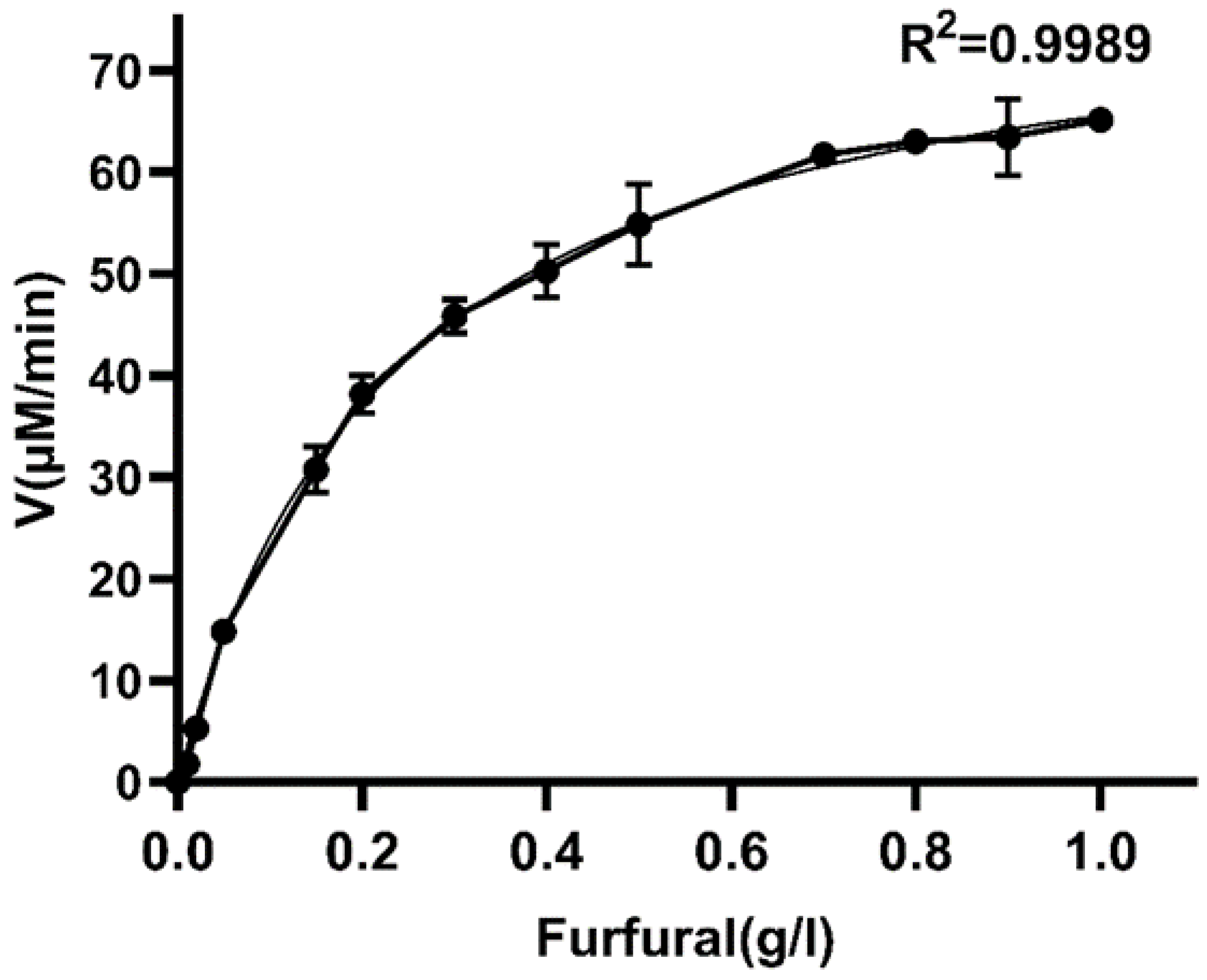
3.4. Enzyme Kinetic Parameters of W826-RS0111485 for Furfural
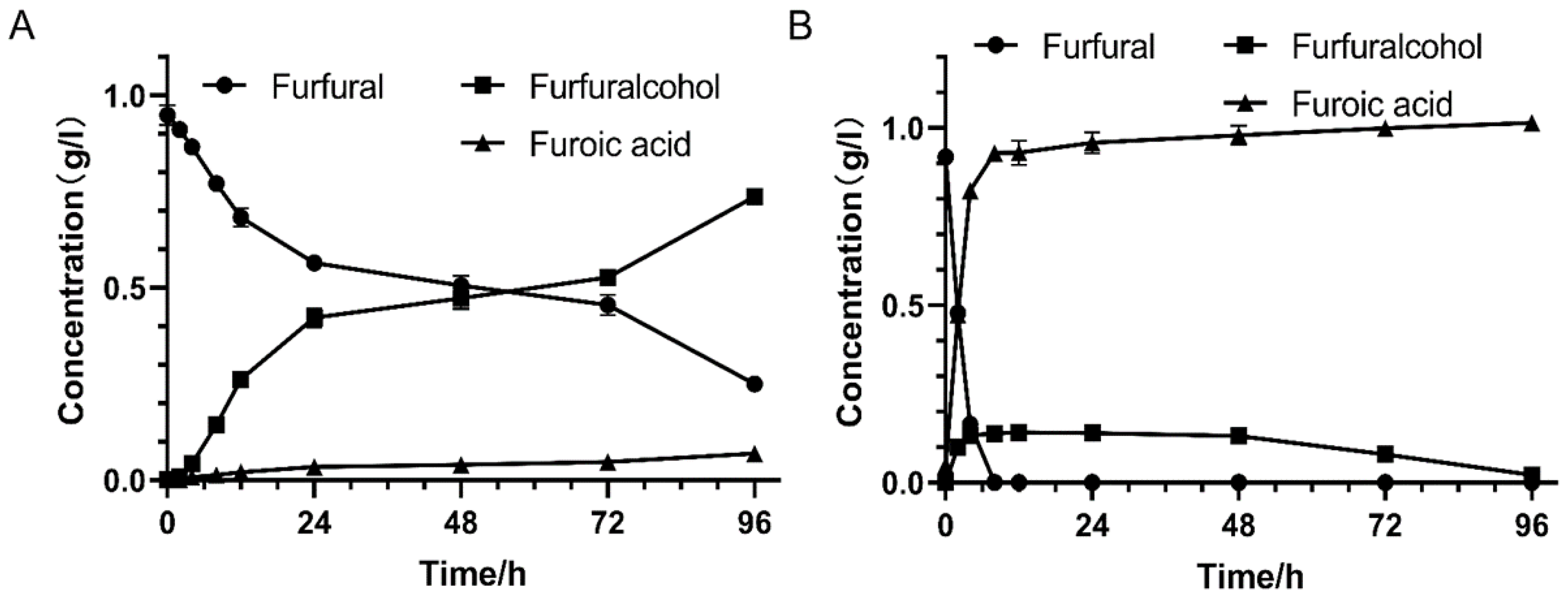
3.5. Biodetoxification Performance of Recombinant E. coli with W826-RS0111485
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chandel, A.K.; Garlapati, V.K.; Singh, A.K.; Antunes, F.A.F.; da Silva, S.S. The path forward for lignocellulose biorefineries: Bottlenecks, solutions, and perspective on commercialization. Bioresour. Technol. 2018, 264, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Yadav, S.K.; Kumar, J.; Ahluwalia, V. A critical review on current strategies and trends employed for removal of inhibitors and toxic materials generated during biomass pretreatment. Bioresour. Technol. 2020, 299, 122633. [Google Scholar] [CrossRef] [PubMed]
- Ragauskas, A.J.; Beckham, G.T.; Biddy, M.J.; Chandra, R.; Chen, F.; Davis, M.F.; Davison, B.H.; Dixon, R.A.; Gilna, P.; Keller, M.; et al. Lignin valorization: Improving lignin processing in the biorefinery. Science 2014, 344, 1246843. [Google Scholar] [CrossRef]
- Jönsson, L.J.; Alriksson, B.; Nilvebrant, N.-O. Bioconversion of lignocellulose: Inhibitors and detoxification. Biotechnol. Biofuels 2013, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Liu, H.; Shan, J.; Sun, B.; Chen, Y.; Ji, L.; Ji, X.; Wang, J.; Zhu, C.; Ying, H. Ammonia–Mechanical Pretreatment of Wheat Straw for the Production of Lactic Acid and High-Quality Lignin. Fermentation 2023, 9, 177. [Google Scholar] [CrossRef]
- Zabed, H.M.; Akter, S.; Yun, J.; Zhang, G.; Awad, F.N.; Qi, X.; Sahu, J.N. Recent advances in biological pretreatment of microalgae and lignocellulosic biomass for biofuel production. Renew. Sustain. Energy Rev. 2019, 105, 105–128. [Google Scholar] [CrossRef]
- Dong, H.-W.; Bao, J. Metabolism: Biofuel via biodetoxification. Nat. Chem. Biol. 2010, 6, 316–318. [Google Scholar] [CrossRef]
- Moreno, A.D.; Ibarra, D.; Alvira, P.; Tomás-Pejó, E.; Ballesteros, M. A review of biological delignification and detoxification methods for lignocellulosic bioethanol production. Crit. Rev. Biotechnol. 2015, 35, 342–354. [Google Scholar] [CrossRef]
- Parawira, W.; Tekere, M. Biotechnological strategies to overcome inhibitors in lignocellulose hydrolysates for ethanol production: Review. Crit. Rev. Biotechnol. 2011, 31, 20–31. [Google Scholar] [CrossRef]
- Candido, J.A.-O.; Claro, E.M.T.; de Paula, C.B.C.; Shimizu, F.L.; de Oliveria Leite, D.A.N.; Brienzo, M.; de Angelis, D.F. Detoxification of sugarcane bagasse hydrolysate with different adsorbents to improve the fermentative process. World J. Microbiol. Biotechnol. 2020, 36, 43. [Google Scholar] [CrossRef]
- Tomek, K.J.; Saldarriaga, C.R.C.; Velasquez, F.P.C.; Liu, T.; Hodge, D.B.; Whitehead, T.A. Removal and upgrading of lignocellulosic fermentation inhibitors by in situ biocatalysis and liquid-liquid extraction. Biotechnol. Bioeng. 2015, 112, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Geddes, R.; Shanmugam, K.T.; Ingram, L.O. Combining treatments to improve the fermentation of sugarcane bagasse hydrolysates by ethanologenic Escherichia coli LY180. Bioresour. Technol. 2015, 189, 15–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmqvist, E.; Hahn-Hägerdal, B.; Szengyel, Z.; Zacchi, G.; Rèczey, K. Simultaneous detoxification and enzyme production of hemicellulose hydrolysates obtained after steam pretreatment. Enzym. Microb. Technol. 1997, 20, 286–293. [Google Scholar] [CrossRef]
- Bowman, M.; Jordan, D.; Vermillion, K.; Braker, J.; Moon, J.; Liu, L. Stereochemistry of Furfural Reduction by a Saccharomyces cerevisiae Aldehyde Reductase That Contributes to In Situ Furfural Detoxification. Appl. Environ. Microbiol. 2010, 76, 4926–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.L. Molecular mechanisms of yeast tolerance and in situ detoxification of lignocellulose hydrolysates. Appl. Microbiol. Biotechnol. 2011, 90, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yao, R.; Zhang, H.; Bao, J.A.-O. Unique glucose oxidation catalysis of Gluconobacter oxydans constitutes an efficient cellulosic gluconic acid fermentation free of inhibitory compounds disturbance. Biotechnol. Bioeng. 2019, 116, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wan, H.; Li, J.; Zhou, J. Enhanced production of L-sorbose in an industrial Gluconobacter oxydans strain by identification of a strong promoter based on proteomics analysis. J. Ind. Microbiol. Biotechnol. 2015, 42, 1039–1047. [Google Scholar] [CrossRef]
- Hahn, T.; Torkler, S.; van der Bolt, R.; Gammel, N.; Hesse, M.; Möller, A.; Preylowski, B.; Hubracht, V.; Patzsch, K.; Zibek, S. Determining different impact factors on the xylonic acid production using Gluconobacter oxydans DSM 2343. Process. Biochem. 2020, 94, 172–179. [Google Scholar] [CrossRef]
- Mao, G.; Wang, K.; Wang, F.; Li, H.; Zhang, H.A.-O.; Xie, H.; Wang, Z.; Wang, F.; Song, A. An Engineered Thermostable Laccase with Great Ability to Decolorize and Detoxify Malachite Green. Int. J. Mol. Sci. 2021, 22, 11755. [Google Scholar] [CrossRef]
- Sainz, F.; Jesús Torija, M.; Matsutani, M.; Kataoka, N.; Yakushi, T.; Matsushita, K.; Mas, A. Determination of Dehydrogenase Activities Involved in D-Glucose Oxidation in Gluconobacter and Acetobacter Strains. Front. Microbiol. 2016, 7, 1358. [Google Scholar] [CrossRef] [Green Version]
- Pronk, J.T.; Levering, P.R.; Olijve, W.; van Dijken, J.P. Role of NADP-dependent and quinoprotein glucose dehydrogenases in gluconic acid production by Gluconobacter oxydans. Enzym. Microb. Technol. 1989, 11, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wu, X.; Zhu, C.; Jin, Z.; Wang, W.; Xia, X. Metabolic engineering to improve the biomanufacturing efficiency of acetic acid bacteria: Advances and prospects. Crit. Rev. Biotechnol. 2020, 40, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Hanke, T.; Nöh, K.; Noack, S.; Polen, T.; Bringer, S.; Sahm, H.; Wiechert, W.; Bott, M. Combined fluxomics and transcriptomics analysis of glucose catabolism via a partially cyclic pentose phosphate pathway in Gluconobacter oxydans 621H. Appl. Environ. Microbiol. 2013, 79, 2336–2348. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ji, L.; Xu, S.; Bilal, M.; Ehrenreich, A.; Deng, Z.; Cheng, H. Membrane-bound sorbitol dehydrogenase is responsible for the unique oxidation of D-galactitol to L-xylo-3-hexulose and D-tagatose in Gluconobacter oxydans. Biochim. Et Biophys. Acta (BBA)-Gen. Subj. 2023, 1867, 130289. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xie, J.; Wei, L.; Lin, J.; Zhao, L.; Wei, D. Identification of the enzymes responsible for 3-hydroxypropionic acid formation and their use in improving 3-hydroxypropionic acid production in Gluconobacter oxydans DSM 2003. Bioresour. Technol. 2018, 265, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Yang, X.; Lu, W. Research of 1,3-Dihydroxyacetone Production by Overexpressing Glycerol Transporter and Glycerol Dehydrogenase. Trans. Tianjin Univ. 2019, 25, 549–558. [Google Scholar] [CrossRef]
- Mientus, M.; Kostner, D.; Peters, B.; Liebl, W.; Ehrenreich, A.A.-O. Characterization of membrane-bound dehydrogenases of Gluconobacter oxydans 621H using a new system for their functional expression. Appl. Microbiol. Biotechnol. 2017, 101, 3189–3200. [Google Scholar] [CrossRef]
- Deppenmeier, U.; Schmidt, B.; Gottschalk, G.; Gottschalk, G. Purification and properties of a F420-nonreactive, membrane-bound hydrogenase from Methanosarcina strain Gö1. Arch. Microbiol. 1992, 157, 505–511. [Google Scholar] [CrossRef]
- Sakasegawa, S.I.; Yoshioka, I.; Koga, S.; Takahashi, M.; Matsumoto, K.; Misaki, H.; Ohshima, T. A novel glycerol kinase from Flavobacterium meningosepticum: Characterization, gene cloning and primary structure. Biosci. Biotechnol. Biochem. 1998, 62, 2388–2395. [Google Scholar] [CrossRef]
- Schweiger, P.; Volland, S.; Deppenmeier, U. Overproduction and characterization of two distinct aldehyde-oxidizing enzymes from Gluconobacter oxydans 621H. Microb. Physiol. 2007, 13, 147–155. [Google Scholar] [CrossRef]
- Li, X.; Yang, R.; Ma, M.; Wang, X.; Tang, J.; Zhao, X.; Zhang, X. A Novel Aldehyde Reductase Encoded by YML131W from Saccharomyces cerevisiae Confers Tolerance to Furfural Derived from Lignocellulosic Biomass Conversion. Bio Energy Res. 2015, 8, 119–129. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Km (mM) | Vmax (μM/min) | Kcat (min−1) | Kcat/Km (mM−1 min−1) | Cofactor | Product | Reference |
|---|---|---|---|---|---|---|---|
| ALDH | 2.451 | 80.97 | 232.33 | 94.80 | NADP | Furoic acid | This study |
| GRE2 | 2.18 | 5.16 | 189 | 86.70 | NADP | Furfuralcohol | [31] |
| YML131W | 6.96 | 11.76 | 470.09 | 67.59 | NAD | Furfuralcohol | [20] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Jiang, J.; Quan, C.; Zhao, G.; Mao, G.; Xie, H.; Wang, F.; Wang, Z.; Zhang, J.; Zhou, P.; et al. Identification of a Novel Dehydrogenase from Gluconobacter oxydans for Degradation of Inhibitors Derived from Lignocellulosic Biomass. Fermentation 2023, 9, 286. https://doi.org/10.3390/fermentation9030286
Zhang H, Jiang J, Quan C, Zhao G, Mao G, Xie H, Wang F, Wang Z, Zhang J, Zhou P, et al. Identification of a Novel Dehydrogenase from Gluconobacter oxydans for Degradation of Inhibitors Derived from Lignocellulosic Biomass. Fermentation. 2023; 9(3):286. https://doi.org/10.3390/fermentation9030286
Chicago/Turabian StyleZhang, Hongsen, Jiahui Jiang, Conghui Quan, Guizhong Zhao, Guotao Mao, Hui Xie, Fengqin Wang, Zhimin Wang, Jian Zhang, Pingping Zhou, and et al. 2023. "Identification of a Novel Dehydrogenase from Gluconobacter oxydans for Degradation of Inhibitors Derived from Lignocellulosic Biomass" Fermentation 9, no. 3: 286. https://doi.org/10.3390/fermentation9030286