

LncRNA PNKY Is Upregulated in Breast Cancer and Promotes Cell Proliferation and EMT in Breast Cancer Cells

Abstract
:1. Introduction
2. Results
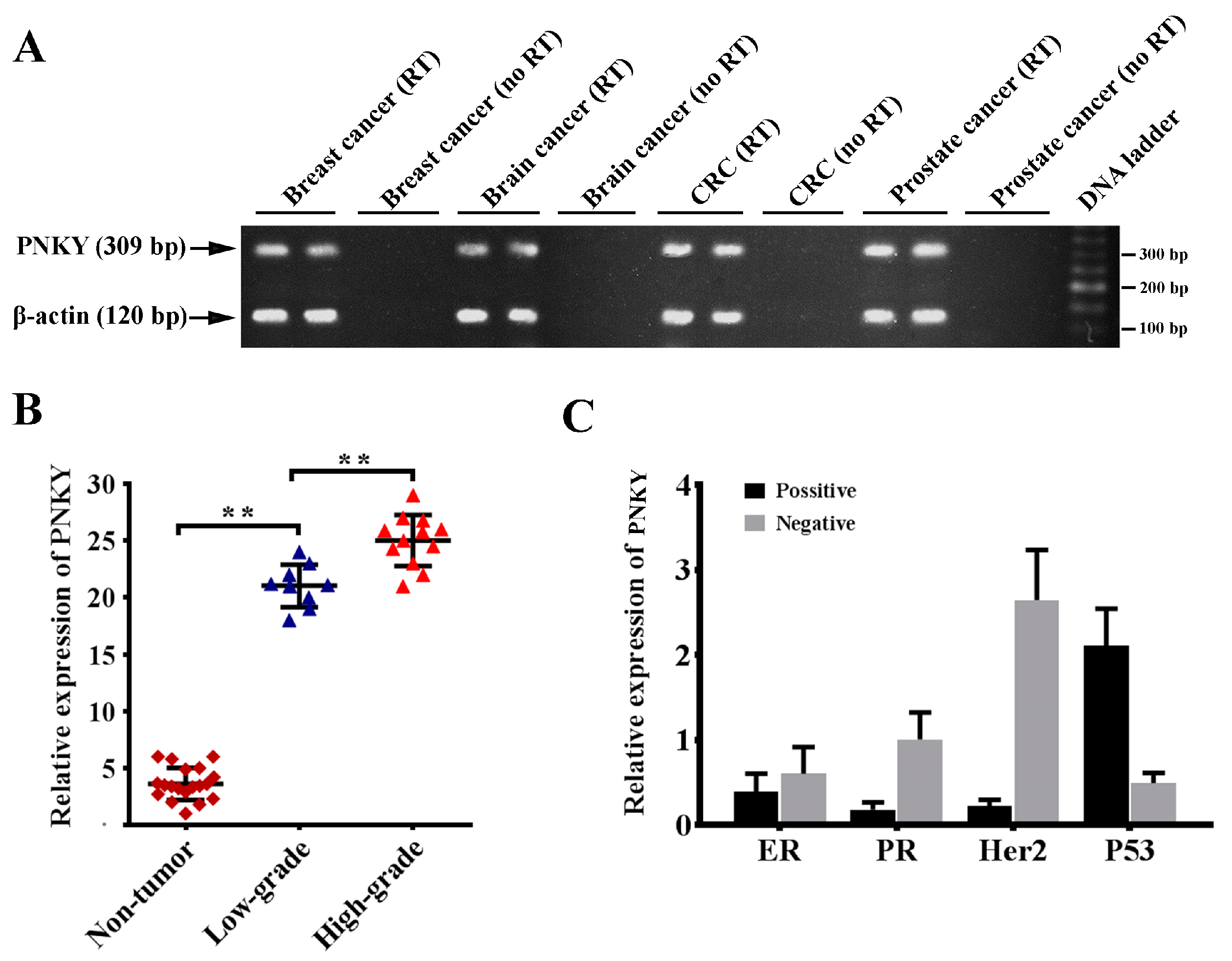
2.1. PNKY Is Expressed in Different Cancer Types and Its Expression Is Strongly Upregulated in High-Grade Breast Cancer
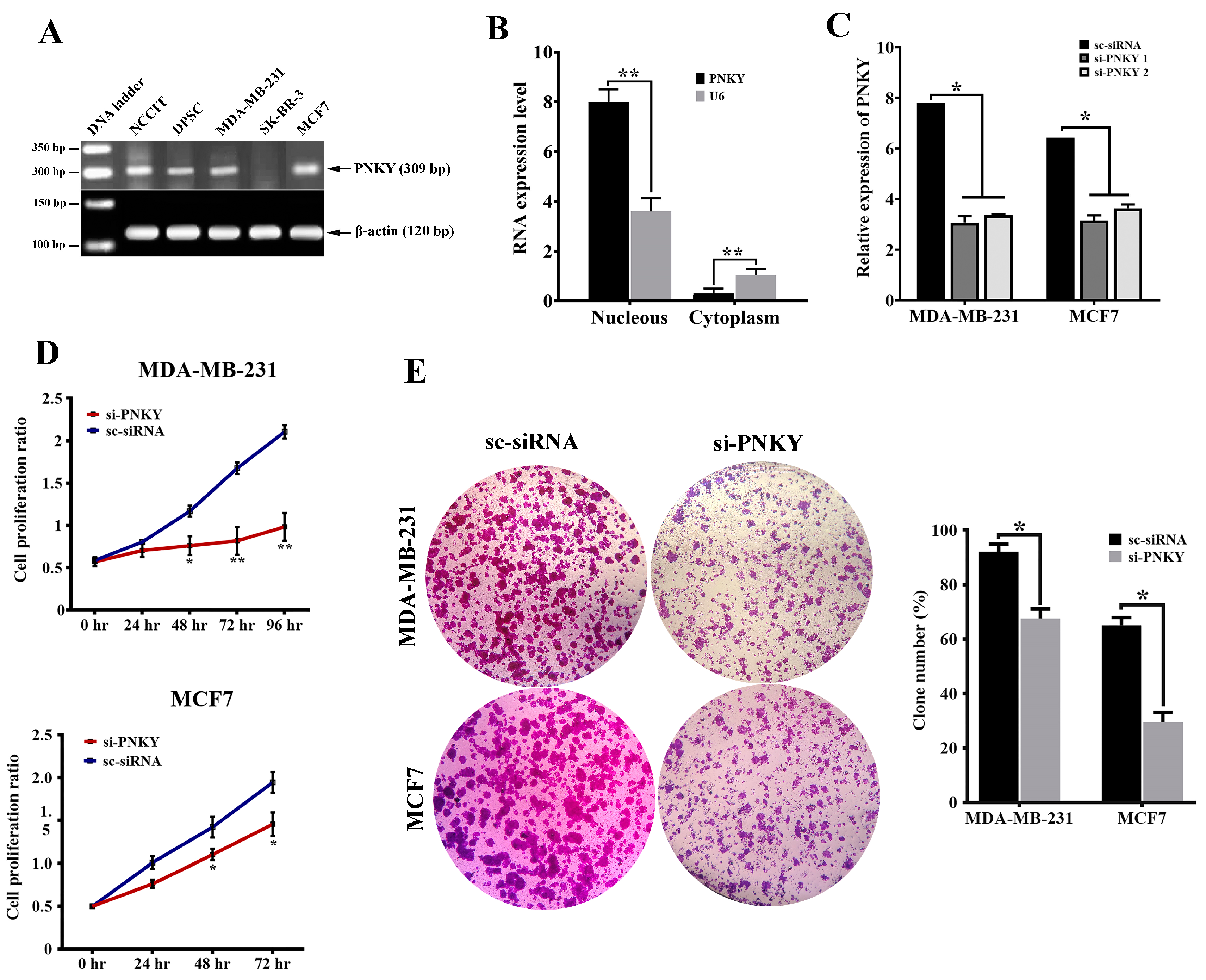
2.2. PNKY Transcript Was Localized in the Nucleus and Its Expression Was Significantly Downregulated during Neural Differentiation
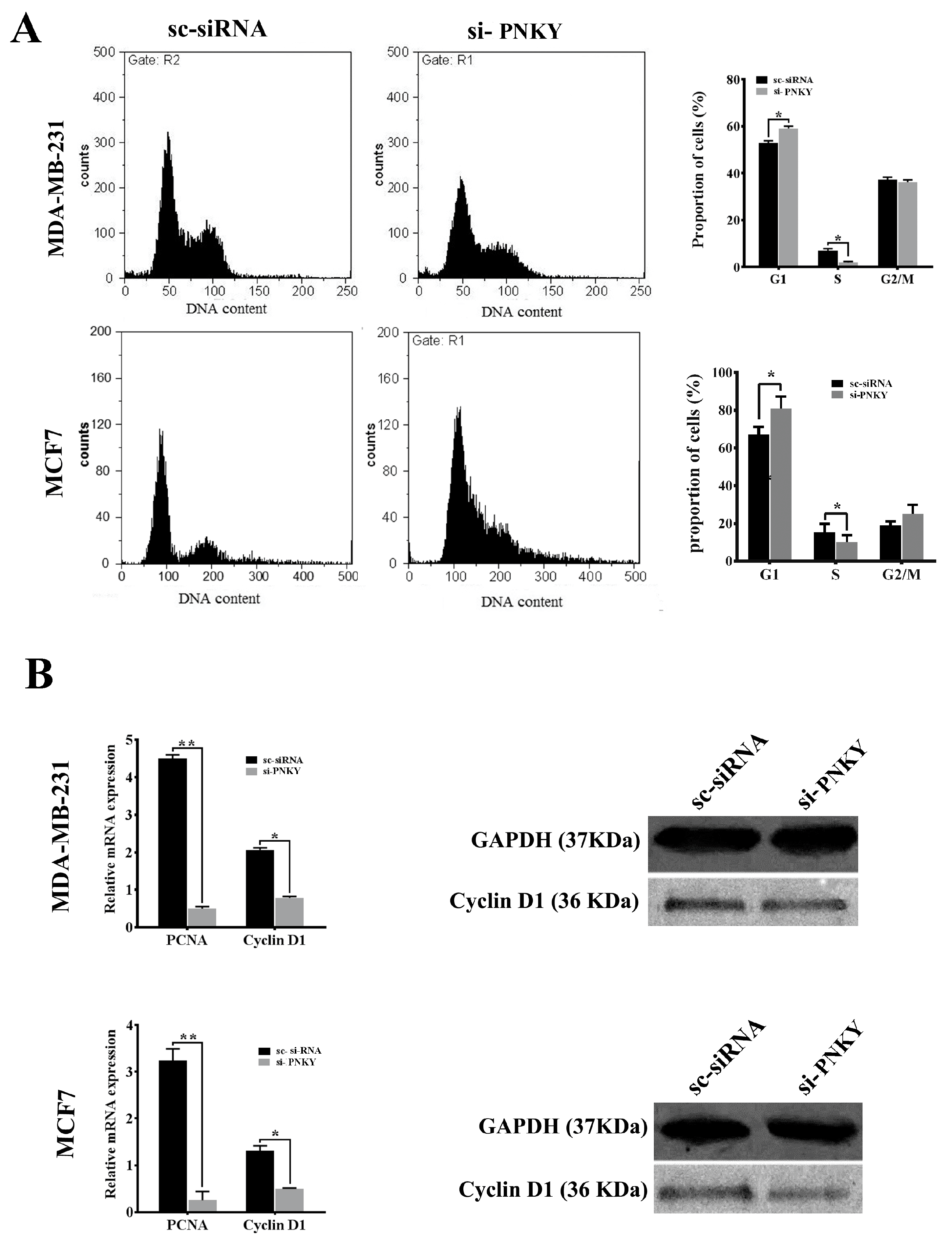
2.3. PNKY Promotes Cell Cycle Progression and Induces Proliferation of Breast Cancer Cells
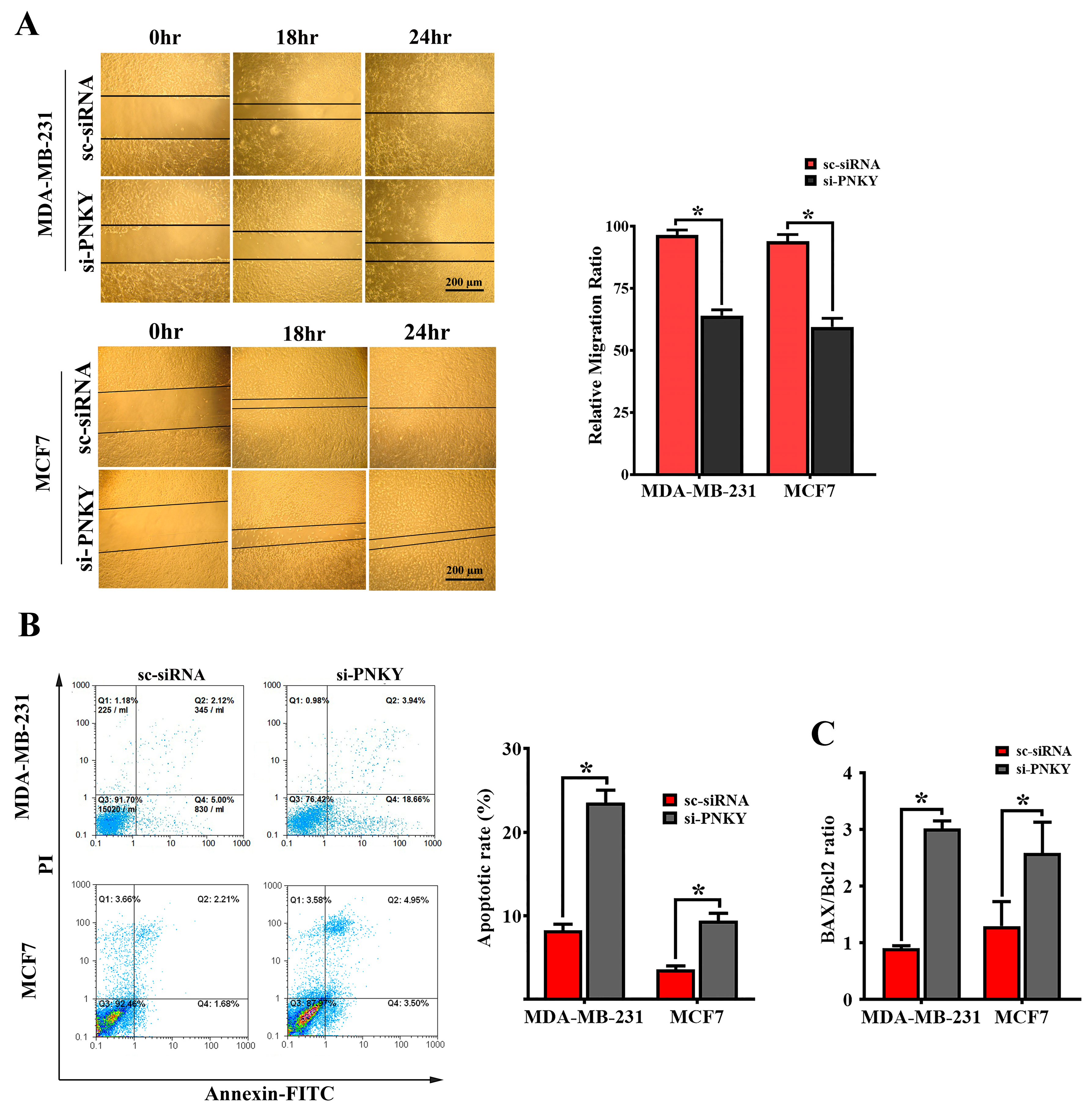
2.4. PNKY Induces Migration and Suppresses Apoptosis in MDA-MB-231 and MCF7 Cells
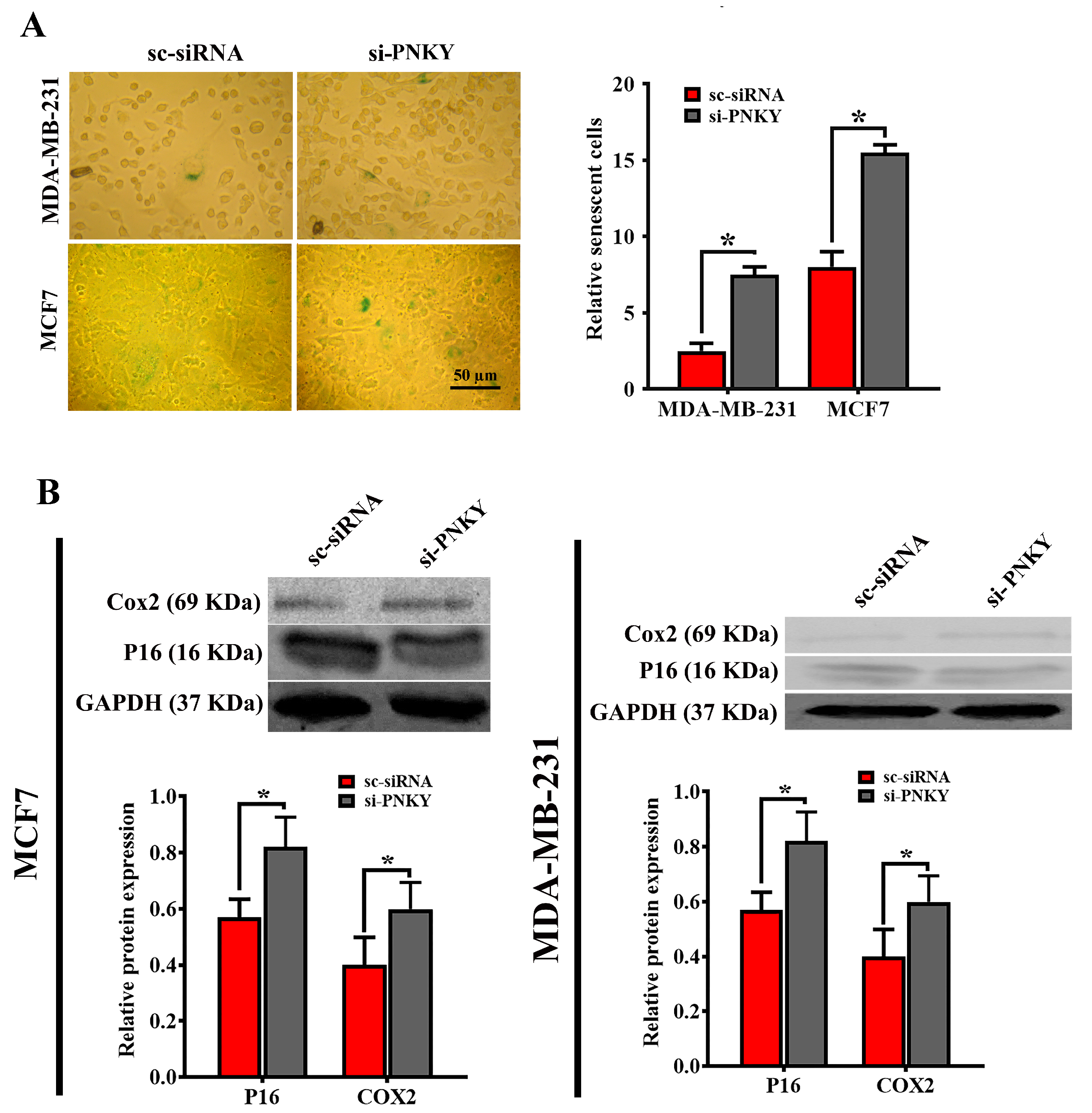
2.5. PNKY Restricts Cellular Senescence in MDA-MB-231 and MCF7 Cells
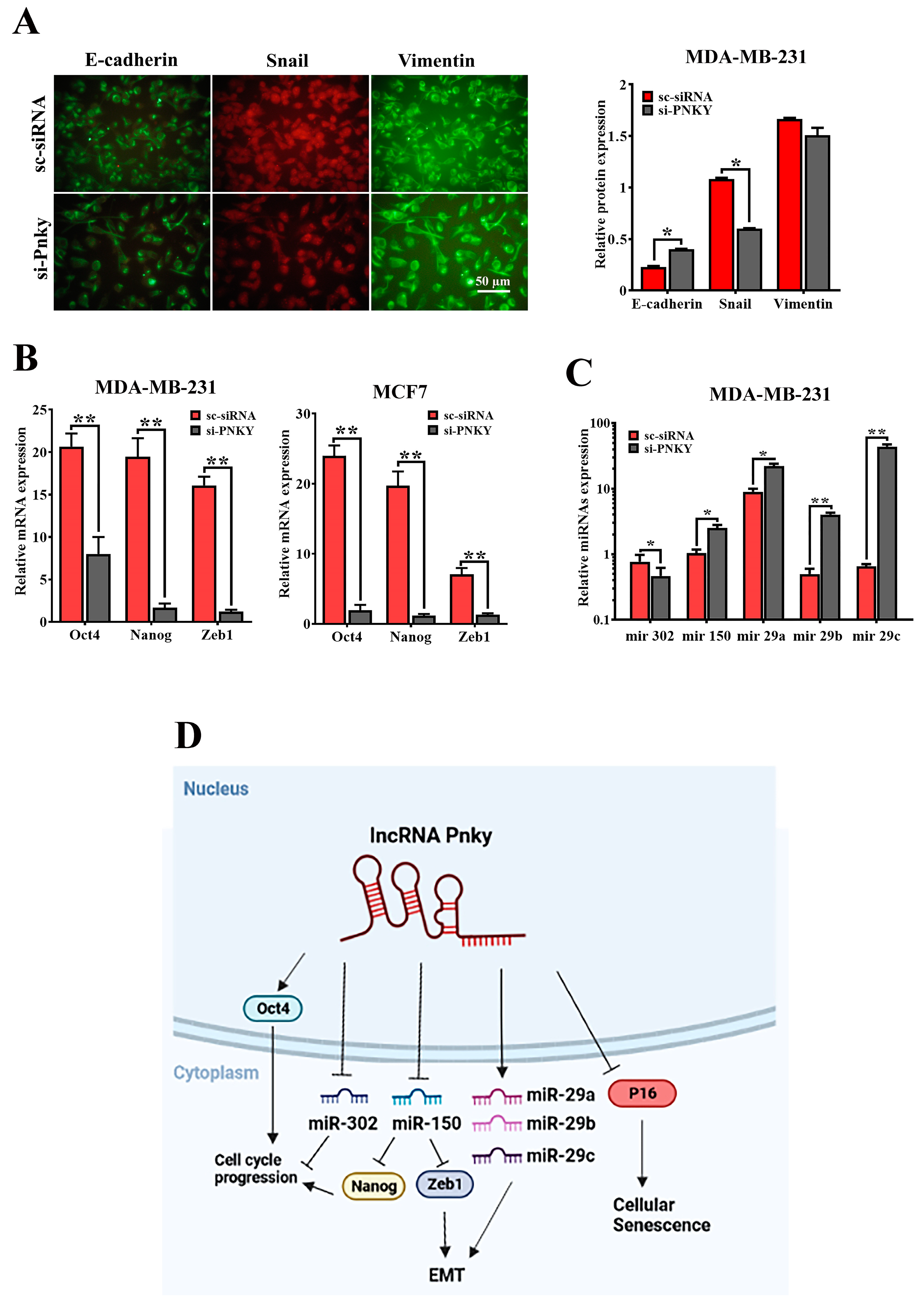
2.6. PNKY Induces EMT in Breast Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Clinical Sample Collection
4.2. Cell Culture
4.3. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
4.4. Isolation of Cytoplasmic and Nuclear Fractionation
4.5. Neural Differentiation of NCCIT Cells
4.6. PNKY Silencing by siRNA Transfection
4.7. Colony Formation Assay
4.8. Cell Senescence-Associated Beta-Galactosidase Assay
4.9. Flow Cytometric Analysis
4.10. Cell Proliferation Assay
4.11. Migration Assay
4.12. Western Blotting
4.13. MicroRNA Expression Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wapinski, O.; Chang, H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011, 21, 354–361. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013, 20, 300–307. [Google Scholar] [CrossRef]
- Eades, G.; Zhang, Y.-S.; Li, Q.-L.; Xia, J.-X.; Yao, Y.; Zhou, Q. Long non-coding RNAs in stem cells and cancer. World J. Clin. Oncol. 2014, 5, 134–141. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, J.S.; Gaughwin, P.M.; Lim, B.; Robson, P.; Lipovich, L. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. Rna 2010, 16, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Sattari, A.; Siddiqui, H.; Moshiri, F.; Ngankeu, A.; Nakamura, T.; Kipps, T.J.; Croce, C.M. Upregulation of long noncoding RNA MIAT in aggressive form of chronic lymphocytic leukemias. Oncotarget 2016, 7, 54174–54182. [Google Scholar] [CrossRef] [Green Version]
- Keshavarz, M.; Asadi, M.H. Long noncoding RNA ES1 controls the proliferation of breast cancer cells by regulating the Oct4/Sox2/miR-302 axis. FEBS J. 2019, 286, 2611–2623. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Asadi, M.H.; Riahi-Madvar, A. Upregulation of pluripotent long noncoding RNA ES3 in HER2-positive breast cancer. J. Cell. Biochem. 2019, 120, 18398–18405. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.D.; Andersen, R.E.; Liu, S.J.; Nowakowski, T.J.; Hong, S.J.; Gertz, C.C.; Salinas, R.D.; Zarabi, H.; Kriegstein, A.R.; Lim, D.A. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 2015, 16, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Zur Hausen, A. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Huang, X.; Xiao, R.; Pan, S.; Yang, X.; Yuan, W.; Tu, Z.; Xu, M.; Zhu, Y.; Yin, Q.; Wu, Y. Uncovering the roles of long non-coding RNAs in cancer stem cells. J. Hematol. Oncol. 2017, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.L.; Shepard, C.R.; Wells, A. Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 2010, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Larue, L.; Bellacosa, A. Epithelial–mesenchymal transition in development and cancer: Role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene 2005, 24, 7443–7454. [Google Scholar] [CrossRef] [Green Version]
- Yokobori, T.; Suzuki, S.; Tanaka, N.; Inose, T.; Sohda, M.; Sano, A.; Sakai, M.; Nakajima, M.; Miyazaki, T.; Kato, H. MiR-150 is associated with poor prognosis in esophageal squamous cell carcinoma via targeting the EMT inducer ZEB1. Cancer Sci. 2013, 104, 48–54. [Google Scholar] [CrossRef]
- Jin, M.; Yang, Z.; Ye, W.; Xu, H.; Hua, X. MicroRNA-150 predicts a favorable prognosis in patients with epithelial ovarian cancer, and inhibits cell invasion and metastasis by suppressing transcriptional repressor ZEB1. PLoS ONE 2014, 9, e103965. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Xie, J.; Shen, C.; Cheng, D.; Shi, Y.; Wu, Z.; Zhan, Q.; Deng, X.; Chen, H.; Shen, B. miR-150-5p inhibits hepatoma cell migration and invasion by targeting MMP14. PLoS ONE 2014, 9, e115577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Zhang, G.; Wu, J.-H.; Jiang, C.-P. Diverse roles of miR-29 in cancer. Oncol. Rep. 2014, 31, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Kalluri, R. miR-29b moulds the tumour microenvironment to repress metastasis. Nat. Cell Biol. 2013, 15, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Alipoor, F.J.; Asadi, M.H.; Torkzadeh-Mahani, M. MIAT lncRNA is overexpressed in breast cancer and its inhibition triggers senescence and G1 arrest in MCF7 cell line. J. Cell. Biochem. 2018, 119, 6470–6481. [Google Scholar] [CrossRef] [PubMed]
- Amirmahani, F.; Asadi, M.H.; Alipoor, F.J. LncRNA MIAT Promotes the Proliferation and Invasion of Colorectal Cancer via Suppressing Apoptosis and Senescence. Middle East J. Cancer 2022. [Google Scholar] [CrossRef]
- Rio, D.C.; Ares, M., Jr.; Hannon, G.J.; Nilsen, T.W. Preparation of cytoplasmic and nuclear RNA from tissue culture cells. Cold Spring Harb. Protoc. 2010, 2010, pdb.prot5441. [Google Scholar] [CrossRef] [PubMed]
- Gasimli, L.; Stansfield, H.E.; Nairn, A.V.; Liu, H.; Paluh, J.L.; Yang, B.; Dordick, J.S.; Moremen, K.W.; Linhardt, R.J. Structural remodeling of proteoglycans upon retinoic acid-induced differentiation of NCCIT cells. Glycoconj. J. 2013, 30, 497–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinicopathologic Parameters | Number of Cases | p Value |
|---|---|---|
| Tumor samples Normal samples | 47 47 | 0.004 * |
| Age (years) | 0.76 | |
| ≤65 | 35 | |
| >65 | 12 | |
| Tumor size | 0.63 | |
| ≤5 | 35 | |
| >5 | 12 | |
| Histologic grade | 0.008 ** | |
| Low grade | 26 | |
| High grade | 21 | |
| ER status | 0.1 | |
| Negative | 13 | |
| Positive | 34 | |
| PR status | 0.7 | |
| Negative | 19 | |
| Positive | 28 | |
| HER2 status | 0.08 | |
| Negative | 32 | |
| Positive | 15 | |
| TNM stage | 0.6 | |
| I, II | 19 | |
| III, IV | 28 | |
| P53 status | 0.17 | |
| Negative | 27 | |
| Positive | 12 | |
| Lymphatic node invasion | ||
| Negative | 18 | 0.011 * |
| Positive | 29 | |
| Histology | 0.07 | |
| Lobular | 7 | |
| Ductal | 40 |
| Gene | Primer (Forward) 5′–3′ | Primer (Reverse) 5′–3′ | Amplicon Size |
|---|---|---|---|
| PNKY | AGCTCTCGCTGGTTTTAGG | GTGAGGGAGATATCAAGACACC | 309 bp |
| PNKY | AAGCACGTTGAAGGTGTCTC | CATTGTCCTAGCGAGTGATC | 214 bp |
| β-actin | ACCACCTTCAACTCCATCATG | CTCCTTCTGCATCCTGTCG | 120 bp |
| U6 | GCTTCGGCAGCACATATACTAAAAT | CGCTTCACGAATTTGCGTGTCAT | 89 bp |
| Nestin | GAGAAACAGGGCCTACAGAG | GCTGAGGGAAGTCTTGGAG | 168 bp |
| Nucleostemin | GGGAAGATAACCAAGCGTGTG | CCTCCAAGAAGTTTCCAAAGG | 98 bp |
| Oct4(POU5F1) | AGTGAGAGGCAACCTGGAGA | TTACAGAACCACACTCGGACC | 140 bp |
| MBP | CACATGTACAAGGACTCACAC | GAAGAAGTGGACTACTGGGT | 108 bp |
| Nanog | TAACCTTGGCTGCCGTCTCT | AAGCAAAGCCTCCCAATCC | 154 bp |
| Bax | GGACGAACTGGACAGTAACATGG | GCAAAGTAGAAAAGGGCGACAAC | 150 bp |
| Bcl2 | CTGCACCTGACGCCCTTCACC | CACATGACCCCACCGAACTCAAAGA | 119 bp |
| Cyclin D1 | ACAAACAGATCATCCGCAAACAC | TGTTGGGGCTCCTCAGGTTC | 144 bp |
| PCNA | AGGTGGAGAACTTGGAAATGG | CGTTGAAGAGAGTGGAGTGG | 160 bp |
| Vimentin | CTCTTCCAAACTTTTCCTCCC | AGTTTCGTTGATAACCTGTCC | 134 bp |
| Zeb1 | ACCCTTGAAAGTGATCCAGC | CATTCCATTTTCTGTCTTCCGC | 142 bp |
| Name | Sequence | |
|---|---|---|
| siRNA 1 | Sense | 5′ACACAAACCUCCCAAAUdtdt3′ |
| Antisence | 5′UGUGUUUGGAGGGUUUAdtdt3′ | |
| siRNA 2 | Sense | 5′CAAAUUUCCACCUGAGUAAdtdt3′ |
| Antisence | 5′UUACUCAGGUGGAAAUUUGdtdt3′ | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hakiminia, F.; Jannat Alipoor, F.; Keshavarz, M.; Asadi, M.H. LncRNA PNKY Is Upregulated in Breast Cancer and Promotes Cell Proliferation and EMT in Breast Cancer Cells. Non-Coding RNA 2023, 9, 25. https://doi.org/10.3390/ncrna9020025
Hakiminia F, Jannat Alipoor F, Keshavarz M, Asadi MH. LncRNA PNKY Is Upregulated in Breast Cancer and Promotes Cell Proliferation and EMT in Breast Cancer Cells. Non-Coding RNA. 2023; 9(2):25. https://doi.org/10.3390/ncrna9020025
Chicago/Turabian StyleHakiminia, Forough, Firooz Jannat Alipoor, Mostafa Keshavarz, and Malek Hossein Asadi. 2023. "LncRNA PNKY Is Upregulated in Breast Cancer and Promotes Cell Proliferation and EMT in Breast Cancer Cells" Non-Coding RNA 9, no. 2: 25. https://doi.org/10.3390/ncrna9020025