4. Materials and Methods
The synthetic procedures and compound characterization is included in the PhD thesis of Dr. Helena Vilaça, accessible in the Repositorium of the University of Minho (RepositoriUM;
http://hdl.handle.net/1822/40563; accessed on 10 March 2023) [
44]. Melting points (mp, °C) were determined in a Gallenkamp apparatus, Cambridge, UK and are uncorrected.
1H and
13C NMR spectra were recorded on a Bruker Avance III (Billerica, MA, USA) at 400 and 100.6 MHz, respectively, or in a Varian Unity Plus 300 (Hansen Way Palo Alto, CA, USA) at 300 and 75.4 MHz, respectively. 1H-1H spin-spin decoupling and DEPT θ 45° were used. HMQC and HMBC were used to attribute some signals. Chemical shifts (δ) are given in parts per million (ppm), downfield from tetramethylsilane (TMS), and coupling constants (J) in Hertz (Hz). High-resolution mass spectrometry (HRMS) data were recorded by the mass spectrometry service of the University of Vigo, Spain. Elemental analysis was performed on a LECO CHNS 932 (Lakeview, IL, USA) elemental analyzer. Column chromatography was performed on Macherey–Nagel silica gel 230–400 mesh. Petroleum ether refers to the boiling range 40–60 °C. Some reactions were monitored by thin layer chromatography (TLC), using pre-coated TLC-sheets Alugram Xtra SIL G/UV254. The sheets were revealed in UV light (254 nm). Acetonitrile (ACN) was dried over silica and calcium hydride (CaH
2) and then distilled and stored under molecular sieves.
Synthesis: Compounds
3a (CAS 7524-50-7, Fluorochem cat. nº 003826, UK) and
3b (CAS 2491-20-5, Fluorochem cat. nº M02946, UK) are commercially available. The synthesis of compounds
9a and
9b was described elsewhere [
28,
29].
Synthesis of amino acid methyl esters (3a,b): To methanol (1 M of amino acid) in an ice bath was slowly added thionyl chloride (3.4 equiv.). The amino acid was added slowly and the mixture was left stirring at 40 °C for 4 h. The solvent was removed under reduced pressure and ethyl ether was added. The mixture was stored in the freezer for 1 h and then the solid was filtered.
H-L-Phe-OMe, HCl (3a): H-L-Phe-OH (5.26 g, 31.8 mmol) gave compound 3a (6.72 g, 98%) as a white solid; mp: 153–155 °C (mplit.: 156.0–160.0 °C); 1H NMR (400 MHz, DMSO-d6, δ): 3.09 (dd, J = 7.4 and 14.0 Hz, 1H, βCH), 3.21 (dd, J = 5.6 and 14.0 Hz, 1H, βCH), 3.64 (s, 3H, OCH3), 4.21 (dd, J = 5.6 and 7.4 Hz, 1H, αCH), 7.22–7.34 (m, 5H, Ar H), 8.77 (s, 3H, NH3+).
H-L-Ala-OMe,HCl (3b): H-L-Ala-OH (5.18 g, 58.1 mmol) gave compound 3b (8.11 g, quant.) as a white solid; 1H NMR (400 MHz, DMSO-d6, δ): 1.41 (d, J = 6.8 Hz, 3H, CH3), 3.71 (s, 3H, OCH3), 4.11 (brs, 1H, αCH), 8.73 (brs, 3H, NH3+).
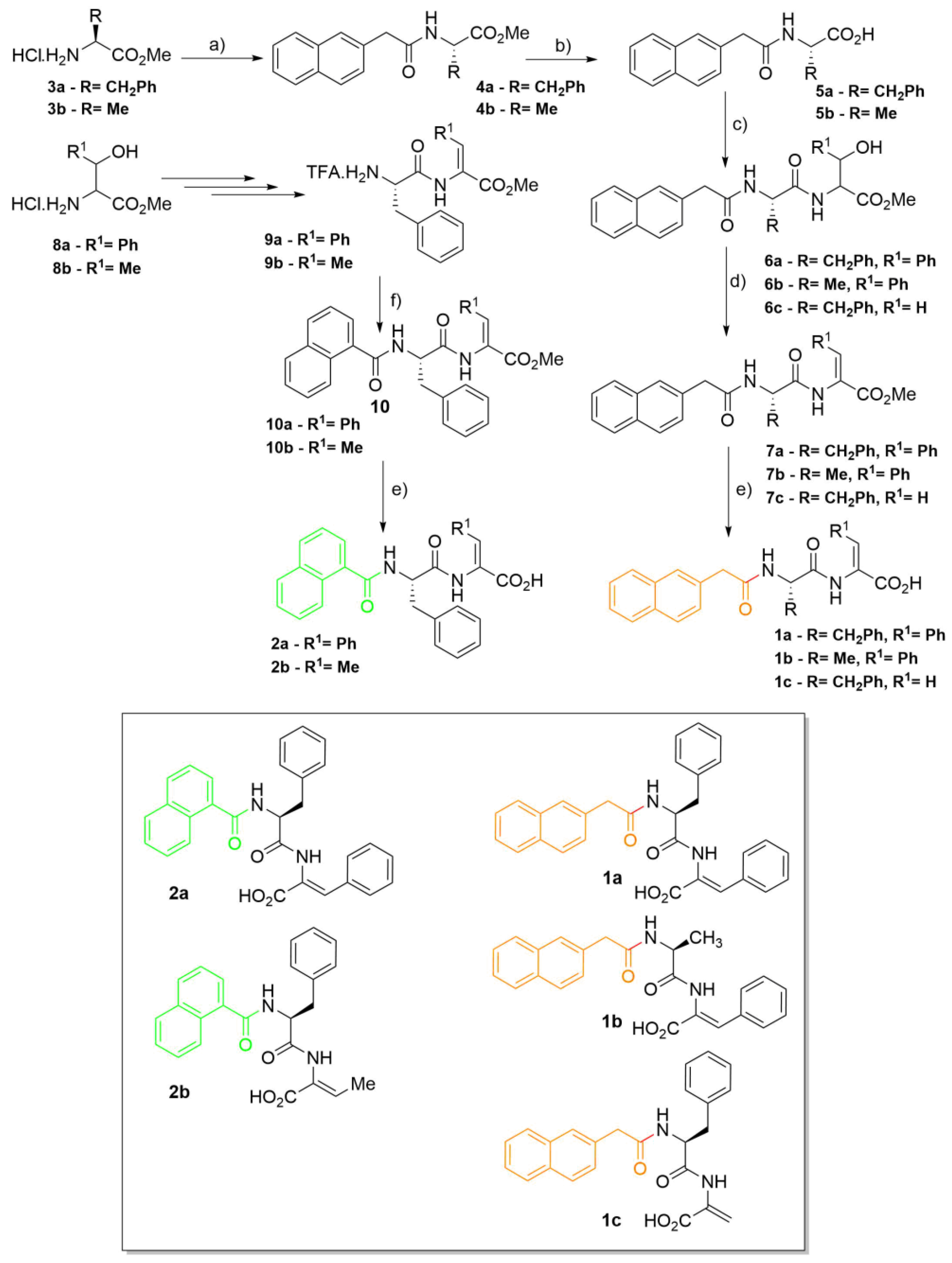
Synthesis of 2-naphthylacetylamino acids (4a,b): 2-Naphthylacetic acid was dissolved in acetonitrile (10 mL mmol-1) and put in an ice bath. HOBt (1.0 equiv.), DCC (1.0 equiv.), amino acid methyl ester (1.0 equiv.) and triethylamine (2.0 equiv.) were added, waiting about 2 min between each addition. The mixture was left stirring at rt overnight (~18 h). The urea was filtered and the solvent removed under reduced pressure. Acetone was added and the mixture was stored in the freezer for 2 h. The urea was filtered again. Evaporation at reduced pressure gave a residue that was partitioned between ethyl acetate (50 mL) and KHSO4 (30 mL, 1 M). The organic phase was thoroughly washed with KHSO4 (1 M), NaHCO3 (1 M) and brine (3 × 30 mL, each), and dried with MgSO4. Removal of the solvent afforded compounds 4a,b.
2-Naph-L-Phe-OMe (4a): 2-Naphthylacetic acid (0.37 g, 2.00 mmol) and H-L-Phe-OMe,HCl (3a) gave compound 4a as a white solid (0.64 g, 93%); mp: 85.0–87.0 °C; 1H NMR (400 MHz, CDCl3, δ): 3.01 (dq, J = 5.6 and 14.0 Hz, 2H, βCH2), 3.70 (s, 3H, OCH3), 3.72 (s, 2H, CH2), 4.86 (td, J = 5.6 and 7.6 Hz, 1H, αCH), 5.83 (d, J = 7.2 Hz, 1H, NH), 6.79 (d, J = 7.4 Hz, 2H, Ho Phe), 6.99 (t, J = 8.0 Hz, 2H, Hm Phe), 7.10 (tt, J = 2.0 and 7.4 Hz, 1H, Hp Phe), 7.30 (dd, J = 1.6 and 8.4 Hz, 1H, H-3 Naph), 7.49–7.54 (m, 2H, 2 × Ar H Naph), 7.66 (s, 1H, H-1 Naph), 7.79–7.87 (m, 3H, 3 × Ar H Naph); 13C NMR (100.6 MHz, CDCl3, δ): 37.52 (βCH2), 43.81 (CH2), 52.30 (OCH3), 52.89 (αCH), 126.05 (CH Naph), 126.37 (CH Naph), 126.97 (Cp Phe), 127.19 (CH-3 Naph), 127.70 (2 × CH Naph), 128.26 (CH-1 Naph), 128.40 (Cm Phe), 128.80 (CH Naph), 128.99 (Co Phe), 131.85 (C-2 Naph), 132.57 (C-4a Naph), 133.56 (C-8a Naph), 135.33 (Ci Phe), 170.42 (C=O Naph), 171.72 (C=O Phe); HRMS (ESI) m/z: [M+H]+ calcd for C22H22NO3+ 348.1594; found, 348.1592.
2-Naph-L-Ala-OMe (4b): 2-Naphthylacetic acid (0.55 g, 2.95 mmol) and H-L-Ala-OMe,HCl (3b) gave compound 4b as a white solid (0.62 g, 78%); mp: 93.5–96.0 °C; 1H NMR (400 MHz, CDCl3, δ): 1.33 (d, J = 7.2 Hz, 3H, βCH3), 3.71 (s, 3H, OCH3), 3.77 (s, 2H, CH2), 4.61 (quint, J = 7.2 Hz, 1H, αCH), 6.05 (d, J = 6.4 Hz, 1H, NH), 7.41 (dd, J = 2.0 and 8.4 Hz, 1H, H-3), 7.46–7.53 (m, 2H, Ar H), 7.75 (s, 1H, H-1), 7.82–7.87 (m, 3H, Ar H); 13C NMR (100.6 MHz, CDCl3, δ): 18.26 (βCH3), 43.72 (CH2), 48.10 (αCH), 52.40 (OCH3), 125.99 (CH), 126.35 (CH), 127.19 (CH-3), 127.66 (CH), 127.70 (CH), 128.23 (CH-1), 128.74 (CH), 131.96 (C-2), 132.54 (C-4a), 133.54 (C-8a), 170.41 (C=O Naph), 173.30 (C=O Ala); HRMS (ESI) m/z: [M+H]+ calcd for C16H18NO3+ 272.1281; found, 272.1280.
Synthesis of compounds 5a,b: The amino acid derivative was dissolved in methanol (until 10 mL) and NaOH (1 M) (1.5 equiv.). The reaction was followed by TLC (Thin Layer Chromatography) until no starting material was detected. The organic solvent was removed under reduced pressure, and the reaction mixture was acidified to pH 3 with KHSO4 (1 M). The solid formed was filtered, affording compounds 5a,b.
2-Naph-L-Phe-OH (5a): 2-Naph-L-Phe-OMe (4a) (0.55 g, 1.58 mmol) gave compound 5a as a white solid (0.48 g, 91%); mp: 145.0–147.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 2.88 (dd, J = 9.6 and 14.0 Hz, 1H, βCH), 3.07 (dd, J = 4.8 and 14.0 Hz, 1H, βCH), 3.58 (dd, J = 14.0 and 21.2 Hz, 2H, CH2), 4.41–4.48 (m, 1H, αCH), 7.19 (brs, 5H, Ar H Phe), 7.27 (dd, J = 1.6 and 8.4 Hz, 1H, Ar H), 7.43–7.50 (m, 2H, Ar H), 7.65 (s, 1H, Ar H), 7.76–7.80 (m, 2H, Ar H), 7.85 (d, J = 8.0 Hz, 1H, Ar H), 8.44 (d, J = 8.0 Hz, 1H, NH), 12.63 (brs, 1H, CO2H); 13C NMR (100.6 MHz, DMSO-d6, δ): 36.71 (βCH2), 42.09 (CH2), 53.49 (αCH), 125.45 (CH), 125.99 (CH), 126.36 (Cp Phe), 127.21 (CH), 127.32 (CH), 127.43 (CH), 127.45 (CH), 127.56 (CH), 128.10 (Cm Phe), 129.11 (Co Phe), 131.72 (C), 132.91 (C), 133.85 (C), 137.53 (Ci Phe), 169.96 (C=O), 172.99 (CO2H); HRMS (ESI) m/z: [M+H]+ calcd for C21H20NO3+ 334.1438; found, 334.1435.
2-Naph-L-Ala-OH (5b): 2-Naph-L-Ala-OMe (4b) (0.37 g, 1.36 mmol) gave compound 5b as a white solid (0.28 g, 80%); mp: decomposes above 157.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 1.27 (d, J = 7.2 Hz, 3H, βCH3), 3.62 (s, 2H, CH2), 4.21 (t, J = 7.2 Hz, 1H, αCH), 7.41–7.48 (m, 3H, Ar H), 7.76 (s, 1H, Ar H), 7.82–7.87 (m, 3H, Ar H), 8.45 (d, J = 7.2 Hz, 1H, NH), 12.49 (brs, 1H, CO2H); 13C NMR (100.6 MHz, DMSO-d6, δ): 17.21 (βCH3), 41.94 (CH2), 47.58 (αCH), 125.46 (CH), 126.02 (CH), 127.23 (CH), 127.34 (CH), 127.43 (CH), 127.48 (CH), 127.63 (CH), 131.73 (C), 132.94 (C), 133.94 (C), 169.83 (C=O), 174.13 (CO2H); HRMS (ESI) m/z: [M+H]+ calcd for C15H16NO3+ 258.1125; found, 258.1125.
Synthesis of dipeptide derivatives (6a–c): The procedure is similar to the one described for amino acid derivatives 4a,b, but 2-naphthylacetic acid is substituted by the N-protected amino acid derivative (5a,b).
2-Naph-L-Phe-D,L-Phe(β-OH)-OMe (6a): 2-Naph-L-Phe-OH (5a) (0.24 g, 0.72 mmol) and H-D,L-Phe(β-OH)-OMe,HCl gave compound 6a as a light solid (0.27 g, 73%); mp: 130.0–133.0 °C; 1H NMR (400 MHz, CDCl3, δ): 2.56–2.61 (m, 1H, βCH), 2.72–2.79 (m, 2H, 2 × βCH), 2.86 (dd, J = 6.4 and 14.0 Hz, 1H, βCH), 3.55 (s, 4H, 2 × CH2), 3.61 (s, 6H, 2 × OCH3), 4.60–4.76 (m, 4H, 4 × αCH), 5.14 (d, J = 3.2 Hz, 1H, βCH), 5.21 (d, J = 3.2 Hz, 1H, βCH), 5.84–5.89 (m, 1H, NH), 5.94–5.99 (m, 1H, NH), 6.51–6.56 (m, 2H, Ar H), 6.82–6.89 (m, 4H, Ar H), 6.95–7.27 (m, 19H, 17 × Ar H, 2 × NH), 7.40–7.48 (m, 6H, Ar H), 7.66–7.69 (m, 3H, Ar H), 7.73–7.75 (m, 2H, Ar H); 13C NMR (100.6 MHz, CDCl3, δ): 37.25 (βCH2), 37.47 (βCH2), 43.58 (CH2), 43.63 (CH2), 52.61 (OCH3), 52. 67 (OCH3), 53.68 (αCH Phe), 53.97 (αCH Phe), 58.32 (αCH), 58.38 (αCH), 73.38 (βCH), 73.49 (βCH), 125.78 (CH), 125.84 (CH), 126.01 (CH), 126.09 (CH), 126.30 (CH), 126.38 (CH), 126.70 (CH), 126.76 (CH), 127.08 (2 × CH), 127.65 (CH), 127.68 (CH), 127.70 (CH), 127.95 (CH), 128.03 (CH), 128.08 (CH), 128.22 (CH), 128.25 (CH), 128.27 (CH), 128.30 (CH), 128.42 (CH), 128.45 (CH), 128.75 (CH), 128.81 (CH), 129.08 (CH), 129.15 (CH), 131.48 (C), 131.59 (C), 133.49 (2 × C), 133.52 (2 × C), 135.64 (Ci Phe), 135.90 (Ci Phe), 139.54 [Ci Phe(β-OH)], 139.68 [Ci Phe(β-OH)], 170.44 (C=O), 170.63 (C=O), 170.84 (C=O), 171.03 (C=O), 171.14 (C=O), 171.16 (C=O); HRMS (ESI) m/z: [M+H]+ calcd for C31H31N2O5+ 511.2228; found, 511.2237; [M+Na]+ calcd for C31H30N2NaO5+ 533.2047; found, 533.2061.
2-Naph-L-Ala-D,L-Phe(β-OH)-OMe (6b): 2-Naph-L-Ala-OH (5b) (0.44 g, 1.71 mmol) and H-D,L-Phe(β-OH)-OMe,HCl gave compound 6b as a light oil, that spontaneously crystallized (0.33 g, 45%); mp: 112.0–115.0 ◦C; 1H NMR (400 MHz, CDCl3, δ): 1.01 (d, J = 7.2 Hz, 3H, βCH3), 1.24 (d, J = 6.8 Hz, 3H, βCH3), 3.68 (s, 2H, CH2), 3.69 (s, 2H, CH2), 3.70 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 4.45–4.56 (m, 2H, αCH), 4.80 (dd, J = 3.6 and 8.8. Hz, 1H, αCH), 4.85 (dd, J = 3.2 and 8.8 Hz, 1H, αCH), 5.25 (d, J = 3.6 Hz, 1H, βCH), 5.32 (d, J = 3.2 Hz, 1H, βCH), 6.20 (d, J = 7.6 Hz, 1H, NH), 6.35 (d, J = 7.6 Hz, 1H, NH), 7.20–7.35 (m, 14H, 12 × Ar H and 2 × NH), 7.42–7.50 (m, 4H, Ar H), 7.66 (d, J = 8.0 Hz, 2H, Ar H), 7.76–7.82 (m, 6H, Ar H); 13C NMR (100.6 MHz, CDCl3, δ): 18.12 (βCH3), 18.53 (βCH3), 43.52 (CH2), 43.56 (CH2), 48.78 (αCH), 48.88 (αCH), 52.56 (OCH3), 52.61 (OCH3), 58.10 (αCH), 58.37 (αCH), 73.43 (βCH), 73.48 (βCH), 125.66 (2 × CH), 125.87 (2 × CH), 125.97 (CH), 126.02 (CH), 126.30 (CH), 126.36 (CH), 127.05 (2 × CH), 127.66 (2 × CH), 127.67 (2 × CH), 127.85 (CH), 127.93 (CH), 128.20 (2 × CH), 128.27 (2 × CH), 128.30 (2 × CH), 128.74 (CH), 128.76 (CH), 131.62 (C), 131.64 (C),132.51 (C), 132.52 (C), 133.49 (2 × C), 139.65 (2 × C), 170.59 (C=O), 170.62 (C=O), 171.13 (C=O Naph), 171.14 (C=O Naph), 172.08 (C=O Ala), 172.12 (C=O Ala); HRMS (ESI) m/z: [M+H]+ calcd for C25H27N2O5+ 435.1915; found, 435.1912.
2-Naph-L-Phe-D,L-Ser-OMe (6c): 2-Naph-L-Phe-OH (5a) (0.36 g, 1.08 mmol) and H-D,L-Ser-OMe,HCl gave compound 6c as a white solid (0.24 g, 51%), after a column chromatography (petroleum ether, ethyl acetate, mixtures of crescent polarity); mp: 166.0–168.0 °C; 1H NMR (300 MHz, CDCl3, δ): 2.92 (dd, J = 8.0 and 14.0 Hz, 1H, βCH Phe), 3.06 (dd, J = 5.9 and 14.0 Hz, 1H, βCH Phe), 3.58 (t, J = 6.5 Hz, 1H, OH), 3.65 (s, 2H, CH2 Naph), 3.73 (s, 3H, OCH3), 3.82–3.88 (m, 2H, βCH2 Ser), 4.54–4.59 (m, 1H, αCH Ser), 4.71–4.78 (m, 1H, αCH Phe), 6.37 (d, J = 7.8 Hz, 1H, NH Phe), 6.92–6.95 (m, 2H, 2 × Ho Phe), 7.00–7.05 (m, 2H, 2 × Hm Phe), 7.08–7.13 (m, 1H, Hp Phe), 7.21 (dd, J = 1.7 and 8.3 Hz, 1H, Ar H Naph), 7.25 (s, 1H, NH Ser), 7.46–7.52 (m, 12H, 2 × Ar H Naph), 7.58 (s, 1H, Ar H Naph), 7.73–7.76 (m, 2H, 2 × Ar H Naph), 7.80–7.83 (m, 1H, Ar H Naph); 13C NMR (75.5 MHz, CDCl3, δ): 37.84 (βCH2 Phe), 43.47 (CH2 Naph), 52.61 (OCH3), 54.48 (αCH Phe), 54.85 (αCH Ser), 62.51 (βCH2 Ser), 126.02 (CH Naph), 126.32 (CH Naph), 126.87 (CHp Phe), 127.08 (CH Naph), 127.64 (CH Naph), 127.69 (CH Naph), 128.22 (CH Naph), 128.42 (CHm Phe), 128.72 (CH Naph), 129.09 (CHo Phe), 131.60 (C Naph), 132.50 (C Naph), 133.46 (C Naph), 135.79 (Ci Phe), 170.53 (C=O Ser), 171.17 (C=O Phe), 171.70 (C=O Naph); Anal. calcd for C25H26N2O5: C 69.11, H 6.03, N 6.45; found: C 68.52, H 6.485, N 7.151; HRMS (ESI) m/z: [M+H]+ calcd for C25H27N2O5+ 435.1915; found, 435.1914.
Synthesis of dehydrodipeptide derivatives (7a–c): DMAP (0.1 equiv.) was added to solutions of compounds 6a–c in dry acetonitrile (1 M) followed by Boc2O (1.0 equiv.) under rapid stirring at room temperature. The reaction was monitored by 1H NMR until all the reactant had been consumed. Then TMG (2% in volume) was added, stirring was con-tinued and the reaction followed by 1H NMR. When all the reactant had been consumed, evaporation at reduced pressure gave a residue that was partitioned between ethyl acetate (50 mL) and KHSO4 (30 mL, 1 M). The organic phase was thoroughly washed with KHSO4 (1 M), NaHCO3 (1 M) and brine (3 × 30 mL, each), and dried with MgSO4. Removal of the solvent afforded compounds 7a–c.
2-Naph-L-Phe-Z-ΔPhe-OMe (7a): 2-Naph-L-Phe-D,L-Phe(β-OH)-OMe (6a) (0.27 g, 0.53 mmol) gave compound 7a as a light solid (0.16 g, 62%); mp: 197.0–200.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 2.84 (dd, J = 10.4 and 13.6 Hz, 1H, βCH), 3.13 (dd, J = 4.0 and 13.6 Hz, 1H, βCH), 3.59 (q, J = 14.0 Hz, 2H, CH2), 3.69 (s, 3H, OCH3), 4.72 (ddd, J = 4.0, 8.4 and 10.4 Hz, 1H, αCH), 7.16–7.25 (m, 6H, 5 × Ar H and βCH), 7.29–7.31 (m, 4H, Ar H), 7.42–7.49 (m, 2H, Ar H), 7.60–7.62 (m, 3H, Ar H), 7.73–7.77 (m, 2H, Ar H), 7.83–7.85 (m, 1H, Ar H), 8.53 (d, J = 8.4 Hz, 1H, NH), 9.92 (s, 1H, NH); 13C NMR (100.6 MHz, DMSO-d6, δ): 37.11 (βCH2), 42.15 (CH2 Naph), 52.17 (OCH3), 54.09 (αCH Phe), 125.41 (CH), 125.86 (αC ΔPhe), 125.94 (CH), 126.30 (CH), 127.18 (CH), 127.31 (CH), 127.40 (CH), 127.56 (CH), 128.03 (CH), 128.93 (CH), 129.23 (CH), 128.40 (CH), 130.02 (CH), 131.69 (C), 132.01 (CH), 132.01 (βCH ΔPhe), 132.89 (C), 133.93 (C-2 Naph), 137.76 (Ci Phe), 138.18 (Ci ΔPhe), 165.33 (C=O ΔPhe), 170.05 (C=O Naph), 171.47 (C=O Phe); HRMS (ESI) m/z: [M+H]+ calcd for C31H29N2O4+ 493.2122; found, 493.2128; [M+Na]+ calcd for C31H28N2NaO4+ 515.1941; found, 515.1949.
2-Naph-L-Ala-Z-ΔPhe-OMe (7b): 2-Naph-L-Ala-D,L-Phe(β-OH)-OMe (6b) (0.26 g, 0.60 mmol) gave compound 7b as a light yellow solid (0.21 g, 84%); mp: 159.5–167.0 ◦C; 1H NMR (400 MHz, CDCl3, δ): 1.36 (d, J = 6.8 Hz, 3H, βCH3), 3.69 (s, 2H, CH2), 3.78 (s, 3H, OCH3), 4.70–4.77 (m, 1H, αCH), 6.24 (d, J = 7.2 Hz, 1H, NH), 7.28–7.33 (m, 4H, Ar H), 7.37 (s, 1H, βCH), 7.43–7.50 (m, 4H, Ar H), 7.67 (s, 1H, Ar H), 7.75–7.82 (m, 3H, Ar H), 8.04 (s, 1H, NH); 13C NMR (100.6 MHz, CDCl3, δ): 17.53 (βCH3), 43.51 (CH2), 49.10 (αCH), 52.59 (OCH3), 123.88 (αC), 126.01 (CH), 126.35 (CH), 127.03 (CH), 127.65 (CH), 127.67 (CH), 128.20 (CH), 128.53 (CH), 128.78 (CH), 129.54 (CH), 129.67 (CH), 131.64 (C), 132.52 (C), 133.03 (βCH), 133.41 (C), 133.49 (C), 165.25 (C=O ΔPhe), 170.83 (C=O Ala), 171.31 (C=O Naph); HRMS (ESI) m/z: [M+H]+ calcd for C25H25N2O4+ 417.1809; found, 417.1806; [M+Na]+ calcd for C25H24N2NaO4+ 439.1628; found, 439.1626.
2-Naph-L-Phe-ΔAla-OMe (7c): 2-Naph-L-Phe-D,L-Ser-OMe (6c) (0.30 g, 0.69 mmol) gave compound 7c as a white solid (0.21 g, 72%); mp: 65.0–67.0 °C; 1H NMR (400 MHz, CDCl3, δ): 2.99–3.01 (m, 2H, βCH2), 3.73 (s, 2H, CH2), 3.81 (s, 3H, OCH3), 4.71–4.76 (m, 1H, αCH), 5.89 (s, 1H, βCH), 5.99 (d, J = 7.6 Hz, 1H, NH), 6.55 (s, 1H, βCH), 6.89 (d, J = 7.2 Hz, 2H, Ho Phe), 7.02 (t, J = 7.4 Hz, 2H, Hm Phe), 7.11 (t, J = 7.4 Hz, 1H, Hp Phe), 7.23–7.29 (m, 1H, H-3 Naph), 7.50–7.54 (m, 2H, Ar H Naph), 7.63 (s, 1H, H-1 Naph), 7.78–7.81 (m, 2H, Ar H Naph), 7.84.7.87 (m, 1H, Ar H Naph), 8.24 (s, 1H, NH); 13C NMR (100.6 MHz, CDCl3, δ): 37.29 (βCH2), 43.62 (CH2 Naph), 52.91 (OCH3), 54.94 (αCH Phe), 109.37 (βCH2), 126.11 (CH Naph), 126.38 (CH Naph), 126.95 (Cp Phe), 127.09 (CH-3 Naph), 127.67 (CH Naph), 127.71 (CH Naph), 128.34 (CH-1 Naph), 128.56 (Cm Phe), 128.91 (CH Naph and Co Phe), 130.58 (αC), 131.45 (C-2 Naph), 132.57 (C-4a Naph), 133.53 (C-8a Naph), 135.55 (Ci Phe), 163.95 (C=O ΔAla), 169.41 (C=O Phe), 171.19 (C=O Naph); HRMS (ESI) m/z: [M+H]+ calcd for C25H25N2O4+ 417.18088; found, 417.18060; [M+Na]+ calcd for C25H24N2NaO4+ 439.16283; found, 439.16256.
Synthesis of 1-naphthoyl dehydrodipeptides (10a,b): The methyl ester of the peptide (1.1 equiv.) was dissolved in DCM (5 mL) and put in an ice bath. Triethylamine (2.2 equiv.) was added and, slowly, 1-naphthoyl chloride. The mixture was left stirring at rt overnight (~18 h). The mixture was filtered. Evaporation at reduced pressure gave a residue that was partitioned between ethyl acetate (50 mL) and KHSO4 (30 mL, 1 M). The organic phase was thoroughly washed with KHSO4 (1 M), NaHCO3 (1 M) and brine (3 × 30 mL, each), and dried with MgSO4. Removal of the solvent afforded compounds 10a,b.
1-Nap-L-Phe-Z-ΔPhe-OMe (10a): H-L-Phe-Z-ΔPhe-OMe.TFA (9a) (0.39 g, 0.90 mmol) and 1-naphthoyl chloride gave compound 10a as a white solid (0.38 g, 88%); mp: 197.0–198.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 2.95–3.02 (dd, J = 11.2 and 2.4 Hz, 1H, βCH2), 3.23–3.28 (dd, J = 3.6 and 10.4 Hz, 1H, βCH2 Phe), 3.74 (s, 3H, OCH3), 4.99–5.06 (m, 1H, αCH Phe), 7.25–7.29 (m, 1H, Ar H), 7.32–7.41 (m, 7H, Ar H and βCH ΔPhe), 7.42–7.46 (m, 3H, Ar H), 7.48–7.53 (m, 2H, Ar H), 7.74–7.77 (m, 2H, Ar H), 7.87 (d, J = 8.4 Hz, 1H, Ar H), 7.93 (d, J = 8.0 Hz, 1H, Ar H), 7.98 (d, J = 8.4 Hz, 1H, Ar H), 8.86 (d, J = 8.4 Hz, 1H, NH Phe), 10.02 (s, 1H, NH ΔPhe); 13C NMR (100.6 MHz, DMSO-d6, δ): 36.63 (βCH2 Phe), 52.25 (OCH3), 54.85 (αCH Phe), 124.82 (CH), 125.21 (CH), 125.54 (CH), 125.96 (αC), 126.12 (CH), 126.40 (CH), 126.42 (CH), 127.99 (CH), 128.15 (CH), 128.60 (CH), 129.31 (CH), 129.53 (CH), 129.68 (C), 129.77 (CH), 130.14 (CH), 132.10 (βCH ΔPhe), 132.99 (C), 133.31 (C), 134.38 (C),138.20 (C), 165.41 (C=O), 168.70 (C=O), 171.57 (C=O); Anal. calcd for C30H26N2O4: C 75.30, H 5.48, N 5.85; found: C 74.84, H 5.57, N 5.92.
1-Nap-L-Phe-Z-ΔAbu-OMe (10b): H-L-Phe-Z-ΔAbu-OMe.TFA (9b) (0.53 g, 1.4 mmol) and 1-naphthoyl chloride gave compound 10b as a white solid (0.47 g, 80%); mp: 164.0–165.0 °C; 1H NMR (400 MHz, CDCl3, δ): 1.70 (d, J = 7.2 Hz, 3H, γCH3 ΔAbu), 3.22–3.25 (m, 1H, βCH2), 3.36–3.40 (m, 1H, βCH2 Phe), 3.73 (s, 3H, OCH3), 5.22 (q, J = 7.6 Hz, 1H, αCH Phe), 6.74–6.83 (m, 2H, NH Phe and βCH ΔAbu), 7.28–7.41 (m, 6H, Ar H), 7.43–7.52 (m, 3H, Ar H), 7.84 (d, J = 8.0 Hz, 2H, Ar H), 7.89 (d, J = 8.0 Hz, 1H, Ar H), 8.07 (d, J = 8.4 Hz, 1H, NH ΔPhe); 13C NMR (100.6 MHz, CDCl3, δ): 14.42 (γCH3 ΔAbu), 37.78 (βCH2 Phe), 52.30 (OCH3), 54.87 (αCH Phe), 124.62 (CH), 125.18 (CH), 125.39 (CH), 125.92 (C), 126.41 (CH), 127.11 (CH), 127.19 (CH), 128.25 (CH), 128.77 (CH), 129.41 (CH), 129.99 (C), 131.04 (CH), 133.21 (C), 133.58 (C), 134.69 (βCH ΔAbu), 136.38 (C), 164.53 (C=O), 169.38 (C=O), 169.74 (C=O); HRMS (micrO-TOF) m/z: [M+H]+ calcd. for C25H25N2O4+ 417.1814; found, 417.1809.
Synthesis of the N-capped C-deprotected dehydrodipeptides (1a–c, 2a,b): The N-protected dehydrodipeptide (7a–c and 10a,b) was dissolved in 1,4-dioxane (10 mL) and NaOH (1 M) (1.5 equiv.). The reaction was followed by TLC until no starting material was detected. The organic solvent was removed under reduced pressure, and the reaction mixture was acidified to pH 3 with KHSO4 (1 M). The solid formed was filtered, affording compounds 1a–c, 2a,b.
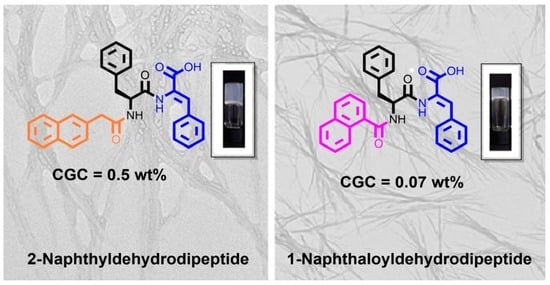
2-Naph-L-Phe-Z-ΔPhe-OH (1a): 2-Naph-L-Phe-Z-ΔPhe-OMe (7a) (0.34 g, 0.69 mmol) gave compound 1a as a white solid (0.31 g, 94%); mp: 165.0–167.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 2.78–2.84 (m, 1H, βCH), 3.11–3.16 (m, 1H, βCH), 3.51–3.59 (m, 2H, CH2), 4.68–4.74 (m, 1H, αCH), 7.15–7.24 (m, 5H, Ar H), 7.27–7.30 (m, 5H, Ar H and βCH), 7.42–7.48 (m, 2H, Ar H), 7.58–7.60 (m, 3H, Ar H), 7.72–7.76 (m, 2H, Ar H), 7.82–7.85 (m, 1H, Ar H), 8.47 (d, J = 8.4 Hz, 1H, NH), 9.73 (s, 1H, NH); 13C NMR (100.6 MHz, DMSO-d6, δ): 37.16 (βCH2), 42.18 (CH2), 54.14 (αCH), 125.43 (CH), 125.95 (CH), 126.27 (CH), 126.54 (αC), 127.19 (CH), 127.33 (CH), 127.41 (CH), 127.57 (CH), 128.02 (CH), 128.46 (CH), 129.18 (CH), 129.25 (CH), 129.93 (CH), 131.70 (C), 131.93 (βCH), 132.89 (C), 133.54 (C), 133.94 (C-2), 137.85 (Ci Phe), 166.19 (C=O), 170.06 (C=O), 171.16 (C=O); HRMS (ESI) m/z: [M+Na]+ calcd for C30H26N2NaO4+ 501.17848; found, 501.17831; [M+H]+ calcd for C30H27N2O4+ 479.1965; found, 479.1963.
2-Naph-L-Ala-Z-ΔPhe-OH (1b): 2-Naph-L-Ala-Z-ΔPhe-OMe (7b) (0.18 g, 0.42 mmol) gave compound 1b as a light solid (0.13 g, 76%); mp: 161.0–163.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 1.30 (d, J = 7.2 Hz, 3H, βCH3), 3.66 (s, 2H, CH2), 4.46 (quint, J = 7.2 Hz, 1H, αCH), 7.24–7.31 (m, 4H, 3 × Ar H and βCH), 7.43–7.49 (m, 3H, Ar H), 7.59–7.62 (m, 2H, Ar H), 7.76 (s, 1H, Ar H), 7.81–7.87 (m, 3H, Ar H), 8.40 (d, J = 7.2 Hz, 1H, NH Ala), 9.50 (s, 1H, NH ΔPhe), 12.68 (brs, 1H, CO2H); 13C NMR (100.6 MHz, DMSO-d6, δ): 17.84 (βCH3), 42.01 (CH2), 48.32 (αCH), 125.44 (CH), 125.98 (CH), 126.60 (C), 127.30 (CH), 127.35 (CH), 127.43 (CH), 127.49 (CH), 127.71 (CH), 128.37 (CH), 129.07 (CH), 129.93 (CH), 131.73 (βCH), 131.74 (C), 132.95 (C), 133.62 (C), 134.05 (C), 166.23 (C=O ΔPhe), 169.88 (C=O Naph), 171.93 (C=O Ala); HRMS (ESI) m/z: [M+H]+ calcd for C24H23N2O4+ 403.1652; found, 403.1659.
2-Naph-L-Phe-ΔAla-OH (1c): 2-Naph-L-Phe-ΔAla-OMe (7c) (0.21 g, 0.50 mmol) gave compound 1c as a white solid (0.13 g, 65%); mp: 159.0–160.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 2.82 (dd, J = 10.4 and 13.6 Hz, 1H, βCH), 3.08 (dd, J = 4.0 and 13.6 Hz, 1H, βCH), 3.52–3.61 (m, 2H, CH2), 4.72–4.76 (m, 1H, αCH), 5.73 (s, 1H, βCH), 6.31 (s, 1H, βCH), 7.16–7.23 (m, 4H, 2 × Hm Phe, Hp Phe, Ar H Naph), 7.27–7.29 (m, 2H, 2 × Ho Phe), 7.44–7.48 (m, 2H, 2 × Ar H Naph), 7.59 (s, 1H, H-1 Naph), 7.73 (d, J = 8.4 Hz, 1H, Ar H Naph), 7.77 (dd, J = 2.0 and 7.8 Hz, 1H, Ar H Naph), 8.56 (dd, J = 1.6 and 7.6 Hz, 1H, Ar H Naph), 8.56 (d, J = 8.0 Hz, 1H, NH Phe), 9.28 (s, 1H, NH ΔAla), 12.80 (brs, 1H, CO2H); 13C NMR (100.6 MHz, DMSO-d6, δ): 36.85 (βCH2), 42.07 (CH2 Naph), 54.67 (αCH Phe), 108.10 (βCH2), 125.46 (CH Naph), 125.98 (CH Naph), 126.27 (Cp Phe), 127.17 (CH-1 Naph), 127.32 (CH), 127.41 (CH), 127.48 (2 × CH), 127.99 (CH), 129.24 (Co Phe), 131.70 (C Naph), 132.80 (αC), 132.89 (C Naph), 133.69 (C Naph), 137.64 (Ci Phe), 164.79 (C=O ΔAla), 170.27 (C=O Naph), 170.92 (C=O Phe); HRMS (ESI) m/z: [M+H]+ calcd for C24H23N2O4+ 403.16578; found, 403.16522; [M+Na]+ calcd for C24H22N2NaO4+ 425.1477; found, 425.14716; [M+K]+ calcd for C24H22N2KO4+ 441.1217; found, 441.1210.
1-Nap-L-Phe-Z-ΔPhe-OH (2a): 1-Nap-L-Phe-Z-ΔPhe-OMe (10a) (0.32 g, 0.67 mmol) gave compound 2a as a white solid (0.29 g, 94%); mp: 181.0–182.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 2.92–2.98 (dd, J = 11.6 and 2.4 Hz, 1H, βCH2 Phe), 3.24–3.29 (dd, J = 3.2 and 10.4 Hz, 1H, βCH2 Phe), 4.97–5.03 (m, 1H, αCH Phe), 7.25–7.36 (m, 7H, Ar H and βCH ΔPhe), 7.38–7.51 (m, 6H, Ar H), 7.61 (d, J = 6.8 Hz, 2H, Ar H), 7.80 (d, J = 8.4 Hz, 1H, Ar H), 7.91 (d, J = 8.0 Hz, 1H, Ar H), 7.96 (d, J = 7.6 Hz, 1H, Ar H), 8.87 (d, J = 8.4 Hz, 1H, NH Phe), 9.74 (s, 1H, NH ΔPhe); 13C NMR (100.6 MHz, DMSO-d6, δ): 36.65 (βCH2 Phe), 55.09 (αCH Phe), 124.85 (CH), 125.17 (CH), 125.60 (CH), 126.12 (CH), 126.34 (CH), 126.42 (CH), 127.95 (CH), 128.14 (CH), 128.24 (CH), 128.44 (CH), 128.64 (C), 129.34 (CH), 129.65 (C), 129.71 (CH), 129.76 (CH), 132.98 (C), 134.53 (C), 134.67 (C), 138.37 (C), 166.14 (C=O), 170.93 (C=O), 173.33 (C=O); HRMS (micrOTOF) m/z: [M+Na]+ calcd. for C29H24N2NaO4+ 487.1634; found, 487.1629.
1-Nap-L-Phe-Z-ΔAbu-OH (2b): 1-Nap-L-Phe-Z-ΔAbu-OMe (10b) (0.33 g, 0.79 mmol) gave compound 2b as a white solid (0.31 g, 97%); mp: 200.0–201.0 °C; 1H NMR (400 MHz, DMSO-d6, δ): 1.69 (d, J = 7.2 Hz, 3H, γCH3 ΔAbu), 2.92–2.99 (dd, J = 11.2 and 2.4 Hz, 1H, βCH2), 3.20–3.25 (dd, J = 4.0 and 10.0 Hz, 1H, βCH2 Phe), 4.97–5.03 (m, 1H, αCH Phe), 6.62 (q, J = 7.2 Hz, 1H, γCH ΔAbu), 7.11 (dd, J = 2.8 and 6.0 Hz, 1H, Ar H), 7.18–7.31 (m, 6H, Ar H), 7.56 (s, 1H, Ar H), 7.23–7.27 (m, 1H, Ar H), 7.30–7.33 (m, 2H, Ar H), 7.39–7.43 (m, 4H, Ar H), 7.46–7.52 (m, 2H, Ar H), 7.78 (d, J = 8.4 Hz, 1H, Ar H), 7.91 (d, J = 8.0 Hz, 1H, Ar H), 7.96 (d, J = 8.0 Hz, 1H, Ar H), 8.76 (d, J = 8.4 Hz, 1H, NH Phe), 9.39 (s, 1H, NH ΔAbu), 12.55 (brs, 1H, CO2H); 13C NMR (100.6 MHz, DMSO-d6, δ): 13.79 (γCH3 ΔAbu), 37.32 (βCH2 Phe), 54.64 (αCH Phe), 124.83 (CH), 125.07 (CH), 125.54 (CH), 126.11 (CH), 126.32 (CH), 126.42 (CH), 127.95 (CH), 128.10 (CH), 128.20 (αC ΔAbu), 129.33 (CH), 129.64 (C), 129.67 (CH), 132.18 (βCH ΔAbu), 132.97 (C), 134.57 (C), 138.21 (C), 165.55 (C=O), 168.50 (C=O), 170.10 (C=O); HRMS (micrOTOF) m/z: [M+Na]+ calcd for C24H22N2NaO4+ 425.1477; found, 425.1488.
Self-assembly: All solutions were made up with ultra-filtered (18 MΩ) water from a Barnstead Nanopure system (Thermofisher, Waltham, MA, USA). Phosphate buffer was prepared from sodium dihydrogen phosphate monohydrate (NaH2PO4·H2O, Fluka BioChemika, Honeywell, Charlotte, NC, USA) and sodium phosphate dibasic dodecahydrate (Na2HPO4·12H2O, Fluka, Honeywell, Charlotte, NC, USA) with a final concentration of 0.1 M and pH 6.00, 7.19 or 8.06 (Mettler Toledo FiveEasy pH Meter, Merck, Darmstadt, Germany). D-glucono-δ-lactone (GdL, Sigma, St. Louis, MO, USA), aqueous NaOH 1 M and HCl 0.1 M were used.
Self-assembly with buffer: Compounds were weighed into sample vials. Buffer was added and the mixture was sonicated at room temperature, followed by heating to 80 °C under magnetic stirring. Solutions were left to cool to room temperature for 24 h.
Self-assembly with GdL: Compounds were weighed into sample vials. Water was added and the suspension was adjusted to pH circa 10 with aqueous NaOH 1 M. The mixture was briefly sonicated at room temperature and added into a vial with the weighed amount of GDL. The mixture was stirred (1000 rpm during 10 s) and left standing at room temperature overnight. The pH of the hydrogel was measured with pH paper.
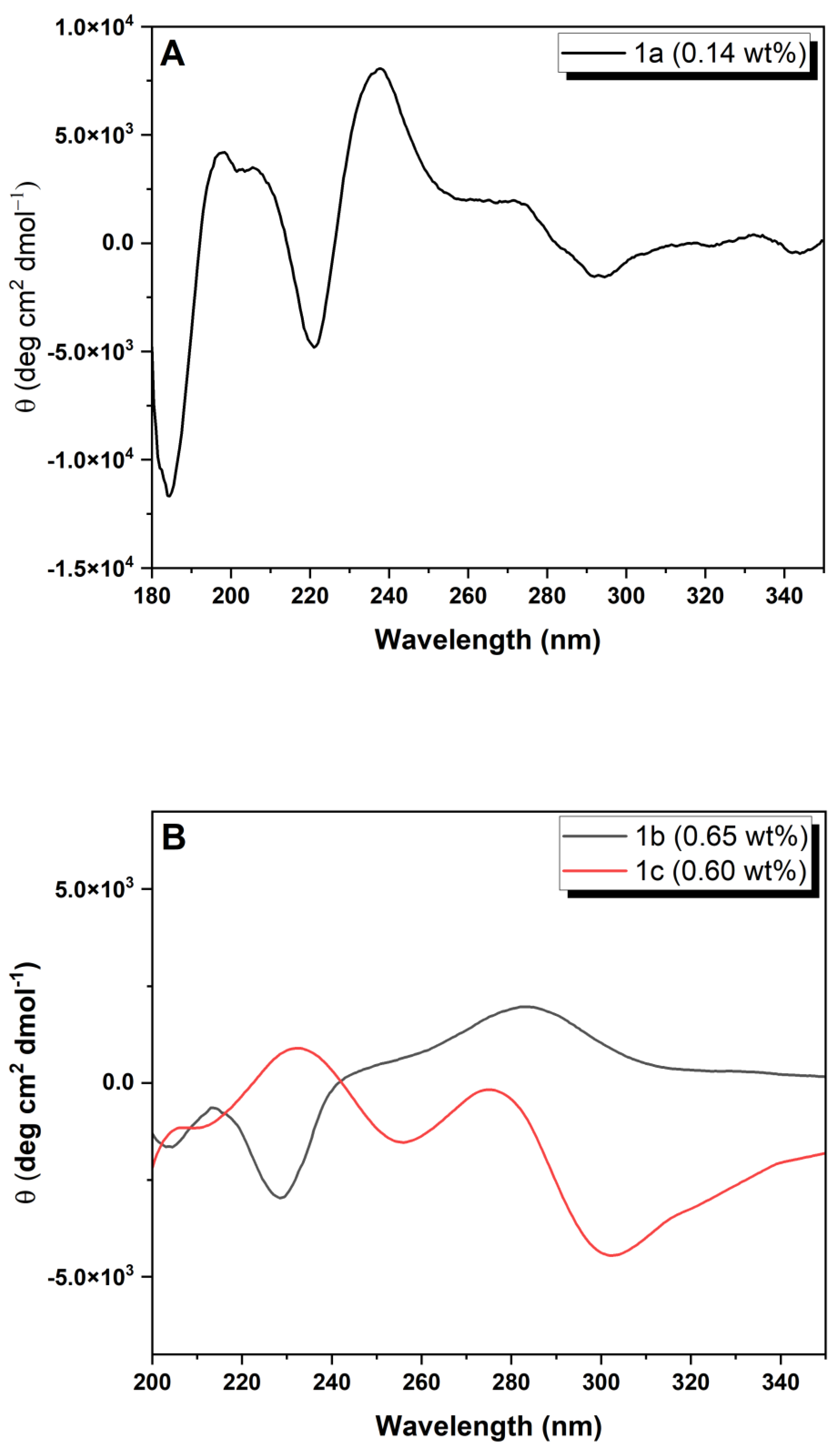
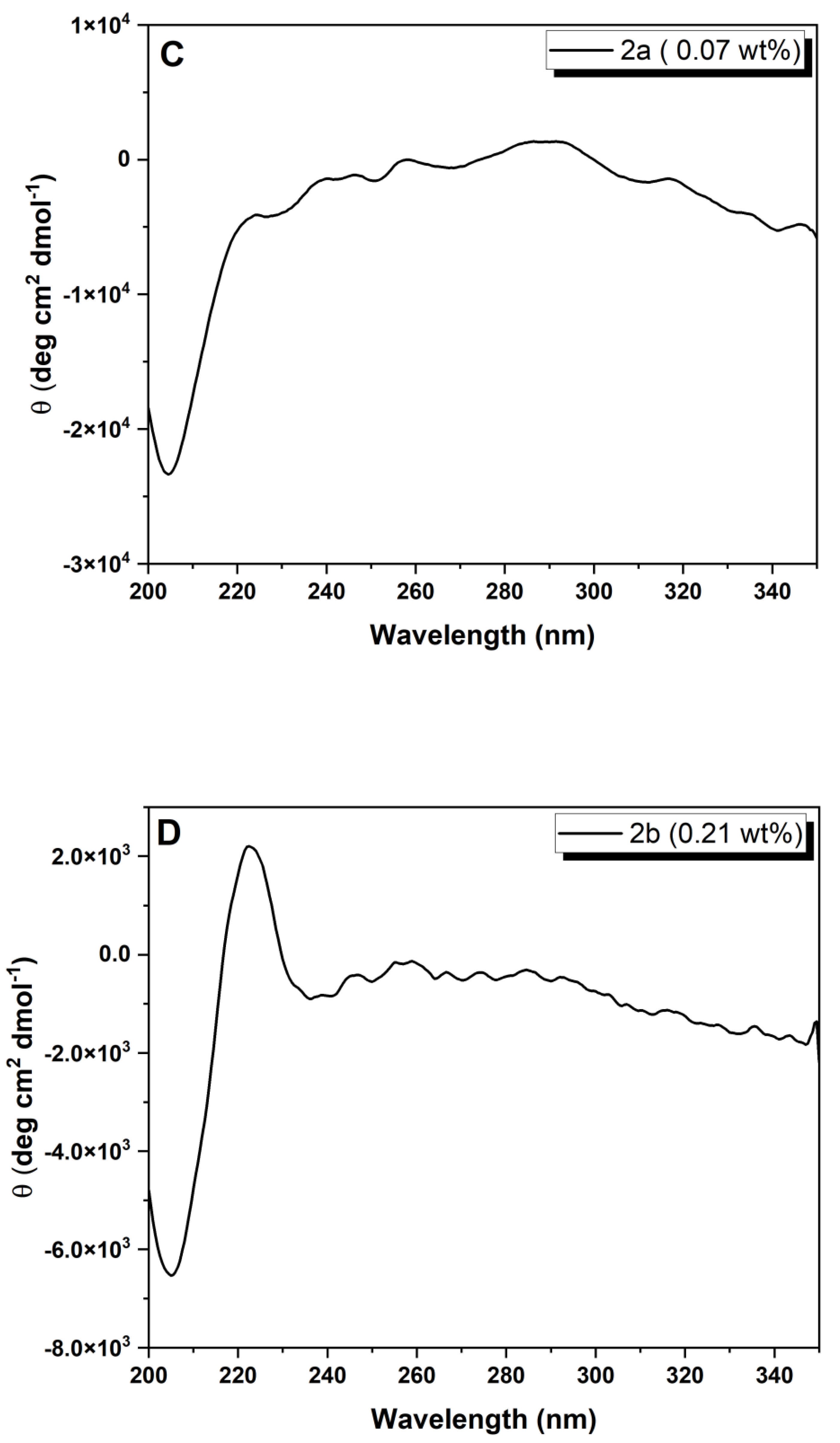
Circular dichroism: The CD spectra were recorded at 20 °C on a Chirascan spectropolarimeter (AppliedPhotophysics, Leatherhead, Surrey, UK). Peptide hydrogels were loaded into 0.1 mm quartz cells. Spectra display absorbance <2 at any measured point with 0.5 nm step, 1 nm bandwidth and 1 s collection time per step, taking three averages. The post-acquisition smoothing tool from Chirascan software (Version 4.13) was used to remove random noise elements from the averaged spectra. A residual plot was generated for each curve in order to check for spectral distortion effects during the smoothing process. CD data were normalized to molar mean residue ellipticity.
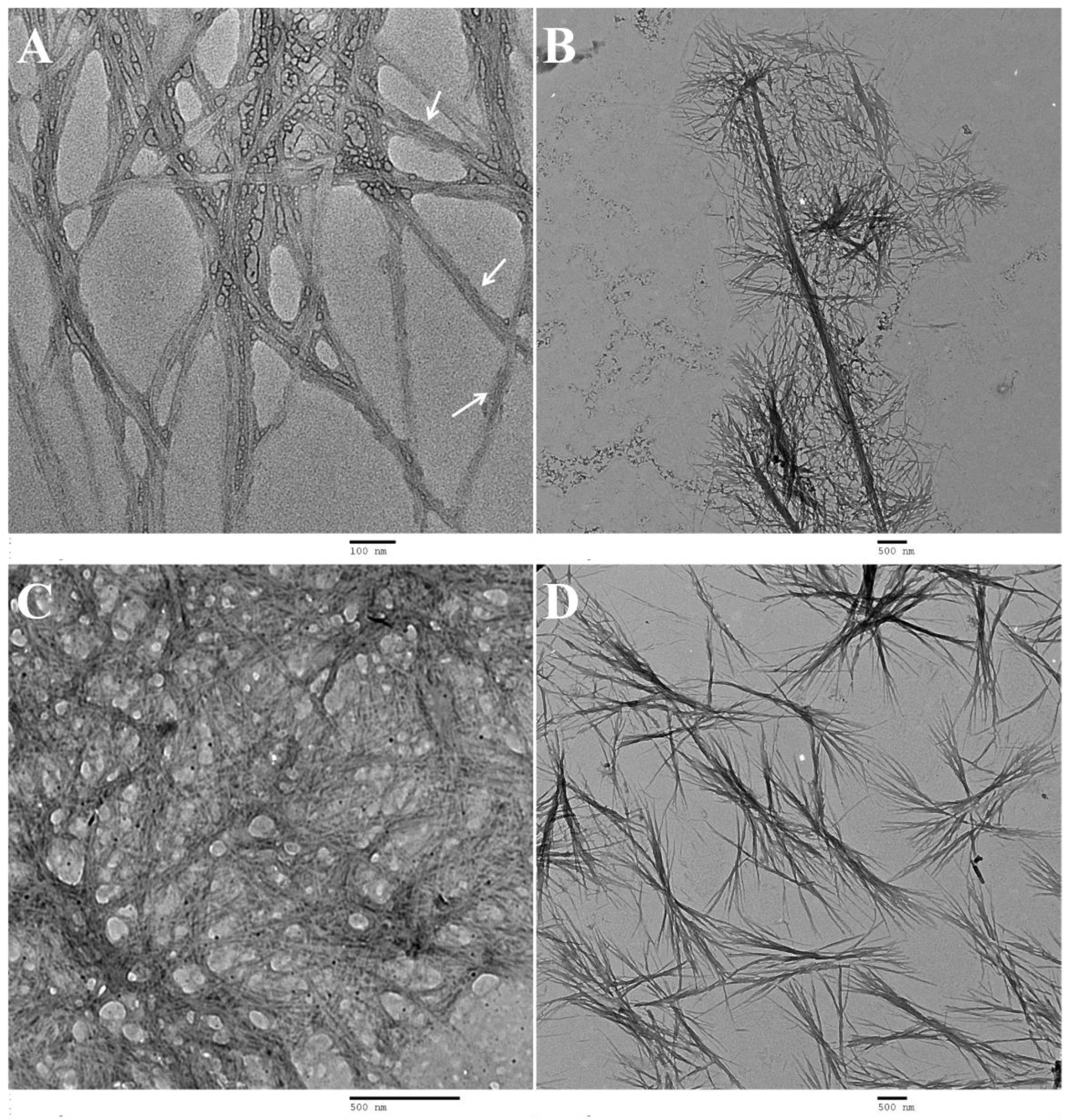
Scanning transmission electron microscopy: STEM experiments were performed using an ultra-high resolution field emission gun scanning electron microscopy (FEG-SEM), NOVA 200 Nano SEM, FEI Company, Hillsboro, Oregon, USA (SEMAT/UM), operated at 15 and 18.5 kV, using a STEM detector (PNDetector, Munich, Germany). Cu-C grids (S160-4 AGAR) were immersed in the peptide hydrogels. The grid was then allowed to dry at room temperature.
Transmission electron microscopy: TEM experiments were performed using a Philips CM20 (FELMI-ZFE, Graz, Austria) transmission electron microscope operating at 200 kV. Diluted solutions, 5 times more diluted than the measured cgc, were prepared in conditions similar to those used for preparation of the hydrogels. The shiny side of 300 mesh Cu grids coated with a carbon film (Agar Scientific, Stansted, Essex, UK) was placed over a drop of dehydrodipeptide solution (1a and 1c) during 1 min; the excess solution at the sides of the grid was carefully cleaned. One drop of the peptide solution (1b, 2a and 2b) was placed on the shiny side of 300 mesh Cu grids coated with a carbon film (Agar Scientific, Stansted, Essex, UK) for 1 min; the excess solution at the sides of the grid was carefully cleaned. In all cases, the shiny side of the grid was placed over a drop of aqueous uranyl acetate (1 wt%) (Agar Scientific, Stansted, Essex, UK) for 1 min. The excess at the sides of the grid was cleaned very carefully and the grid was then allowed to dry at room temperature.
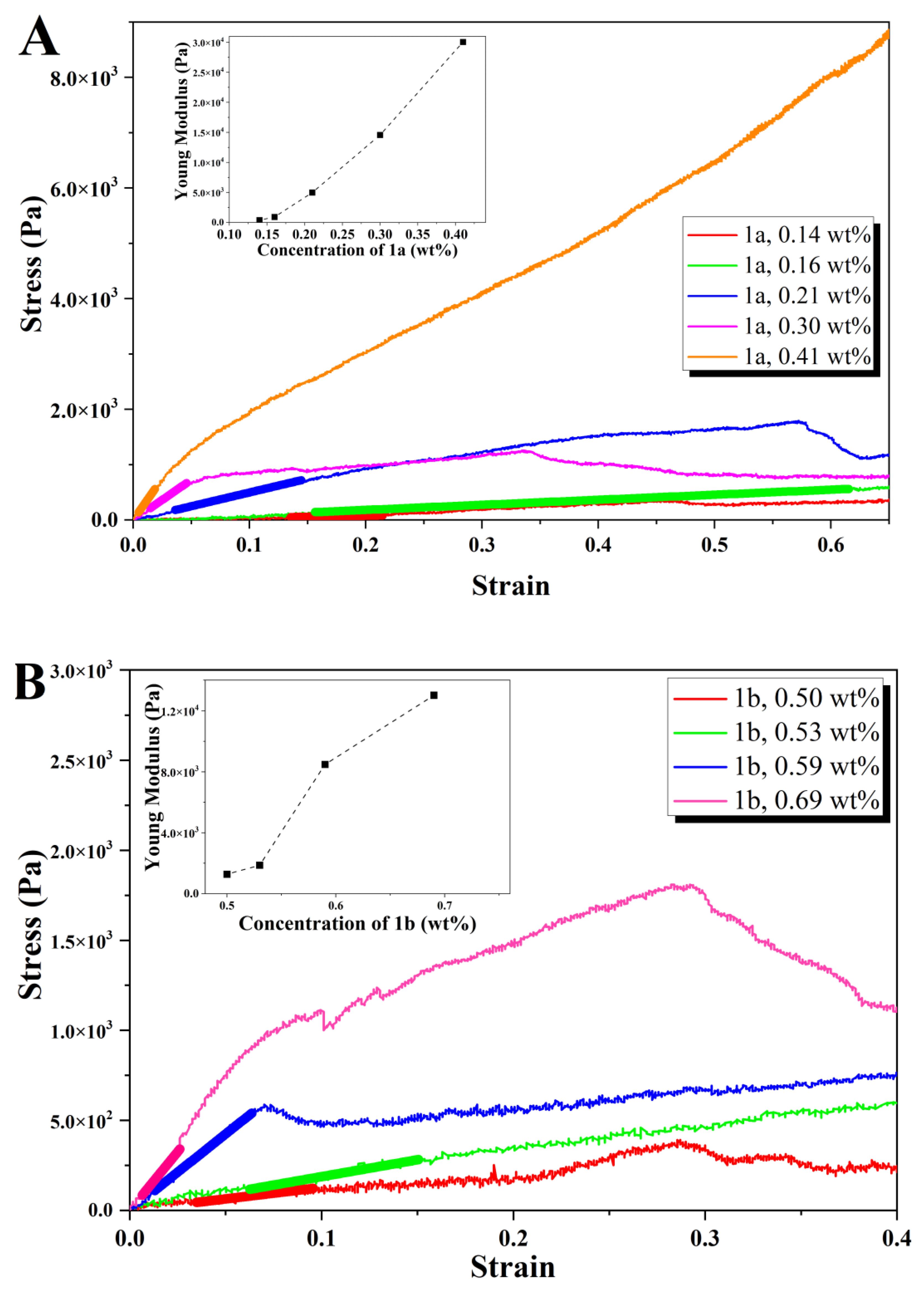
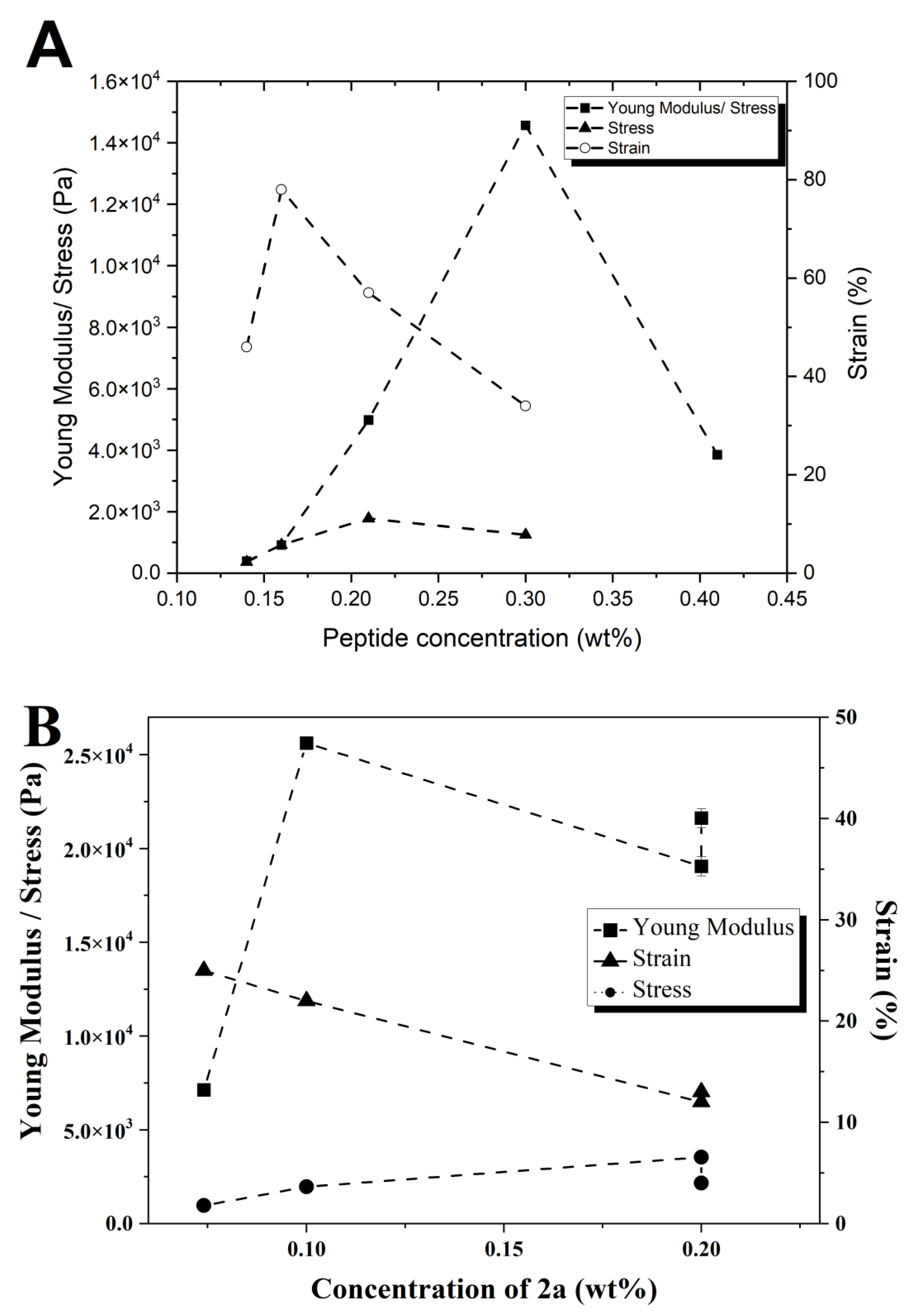
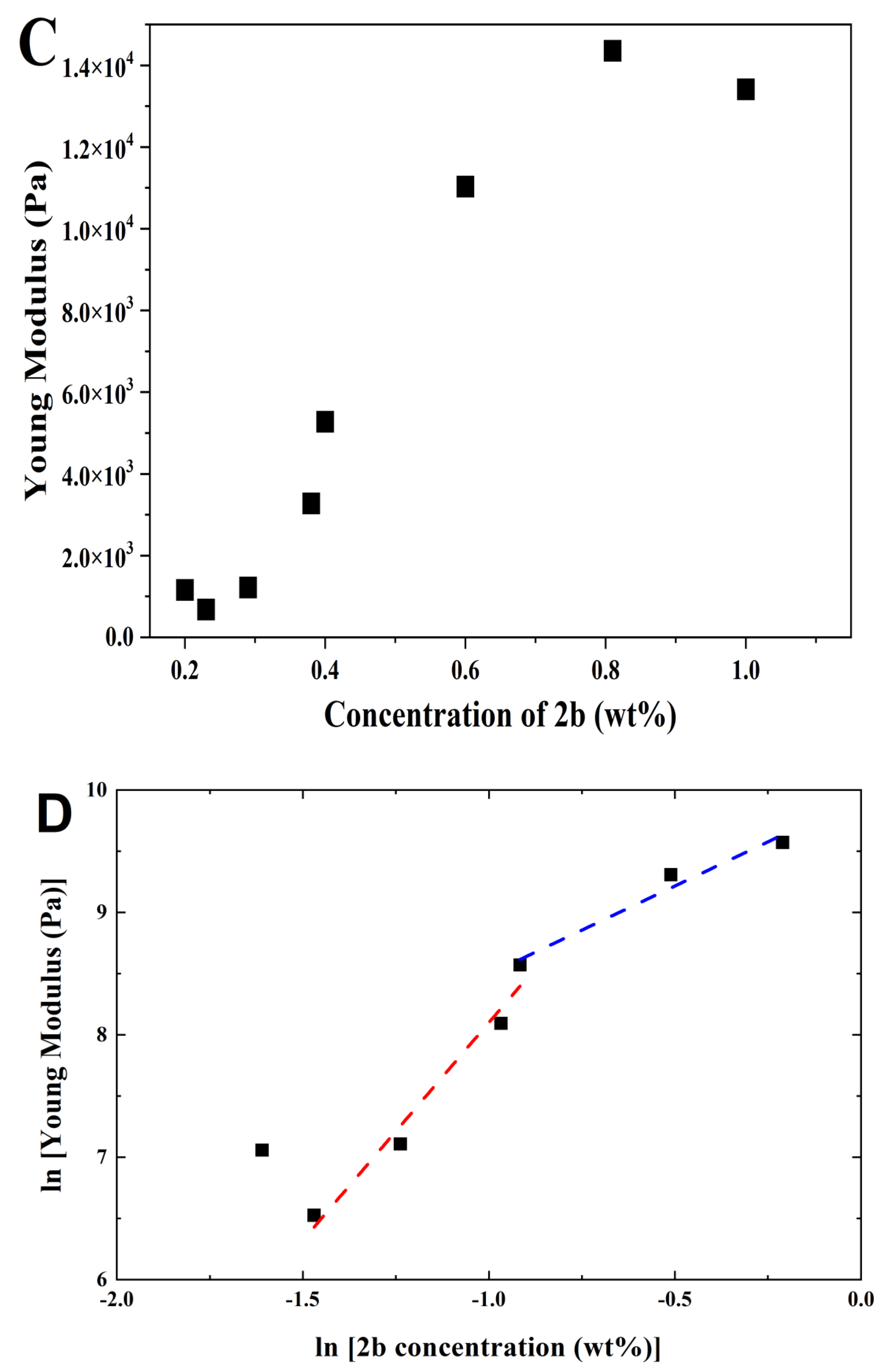
Rheology: Rheology experiments were carried out using a PaarPhysica MCR300 rheometer, Graz, Austria, equipped with a TEK 350-C plate with TC20/EDT/TEK temperature control and a cylindrical plunger with a diameter of 1 cm. Penetration tests on gelled samples were preferred to dynamic shear rheology performed on gelling samples in the rheometer. This is because very long (within 24 h) gel kinetics of samples would jeopardize the practicality of the study, which focuses on the viscoelastic comparison between gels and not on the gelation properties. The hydrogels were prepared in soda glass specimen tubes (Samco, Warwickshire, UK), which also served as the cups for the rheological measurements. The vials had diameters of 25 mm and the measurements were performed till a gap of 1.5 mm to the bottom of the vials was reached, avoiding significant end effects. The normal force was measured as a function of penetration distance in the gel. Young’s moduli were computed neglecting any plunger buoyancy effect, following the approach of Oakenfull et al. [
40]. The slope and associated standard error of the linear regime of the stress–strain responses (Young’s moduli) and the power law exponents for the concentration regimes in
2b were determined using OriginPro 8 software. Strain and stress at break correspond to the location of the first maximum in the stress–strain curves followed by a deep decrease in stress.
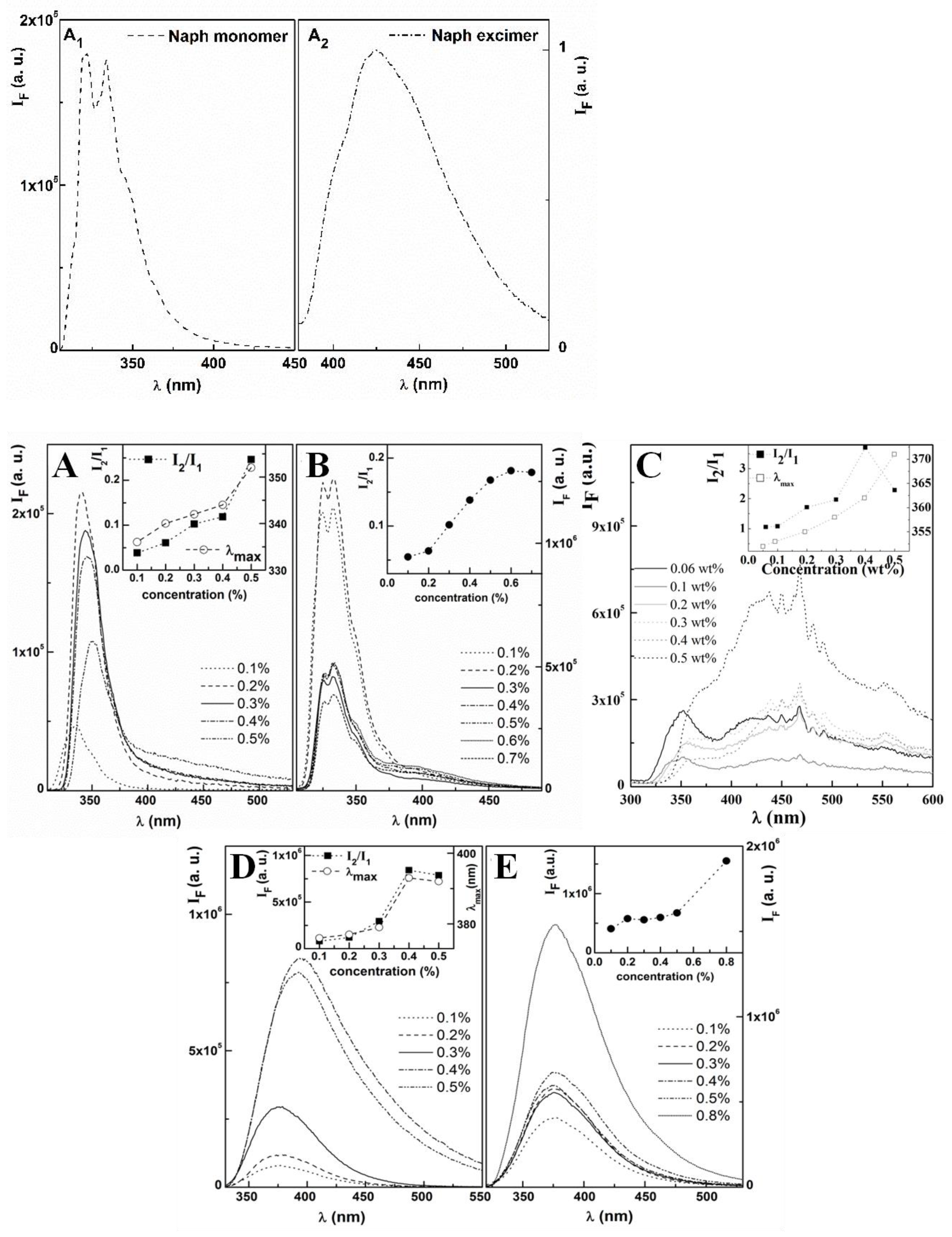
Spectroscopic measurements: Fluorescence measurements were performed using a Fluorolog 3 spectrofluorimeter (Kyoto, Japan), equipped with double monochromators in both excitation and emission, Glan-Thompson polarizers and a temperature-controlled cuvette holder. Fluorescence emission and excitation spectra were corrected for the instrumental response of the system.
Molecular dynamics (MD) simulations: The α,β-dehydroamino acids, ΔzPhe, ΔAla and ΔzAbu and Napthtalene were parameterized and validated in a previous work by some of the authors [
29], from bonded and non-bonded parameters based on equivalent encoded amino acids of the GROMOS 54a7 force field [
45]. The N-protected groups 2-naphthylacetyl (2-Naph) and 1-naphthoyl (1-Nyl) were parameterized based on the Naproxen group also used previously [
29]. Topology files for 2-Naph, 1-Nyl, Npx, ΔzPhe, ΔAla and ΔzAbu are available upon request.
The initial molecular structure of the dehydrodipeptides was built with the program PyMOL [
45]. MD simulations of dehydrodipeptides monomers were performed in octahedral boxes of SPC water with a hydration layer of at least 1.5 nm between the peptide and the periodic boundary conditions in all directions [
46,
47]. Boxes were made neutral with the addition of one Na
+ ion. Each system was subjected to 12,000 steps of energy minimization with the steepest descent algorithm. Then, 100 ns of MD simulations were run, 40 ns of equilibration and 60 ns of production. MD simulations were run with the GROMACS 4.5.4 software package [
48,
49,
50]. In all MD simulations, the system was maintained at constant temperature and pressure, 310 K and 1 atm, respectively, with the Berendsen thermostat and barostat methods, with ΔT = 0.10 ps and ΔP = 1.0 ps [
51]. The SETTLE algorithm was used to constrain bond lengths and angles of water molecules, while the bond lengths and angles of peptides were constrained with the LINCS algorithm, which allowed the use of a 2 fs time step [
52,
53]. For the treatment of long-range interactions, we used the reaction field method, with a cutoff of 1.4 nm and a dielectric constant of 54 (corresponding to SPC water). The van der Waals interactions were truncated with a twin-range cutoff of 0.8 and 1.4 nm. The conformation population was computed with a clustering algorithm through a single-linkage method by first fitting the heavy atoms on the dehydrodipeptides and then analyzing the RMSD matrix vs. time to determine the most likely conformations for each peptide. The MD procedure used to simulate the dimers was similar to that used for the monomers. In this case, the dimers were solvated with SPC water in cubic boxes of 5 × 5 × 5 nm neutralized with two Na
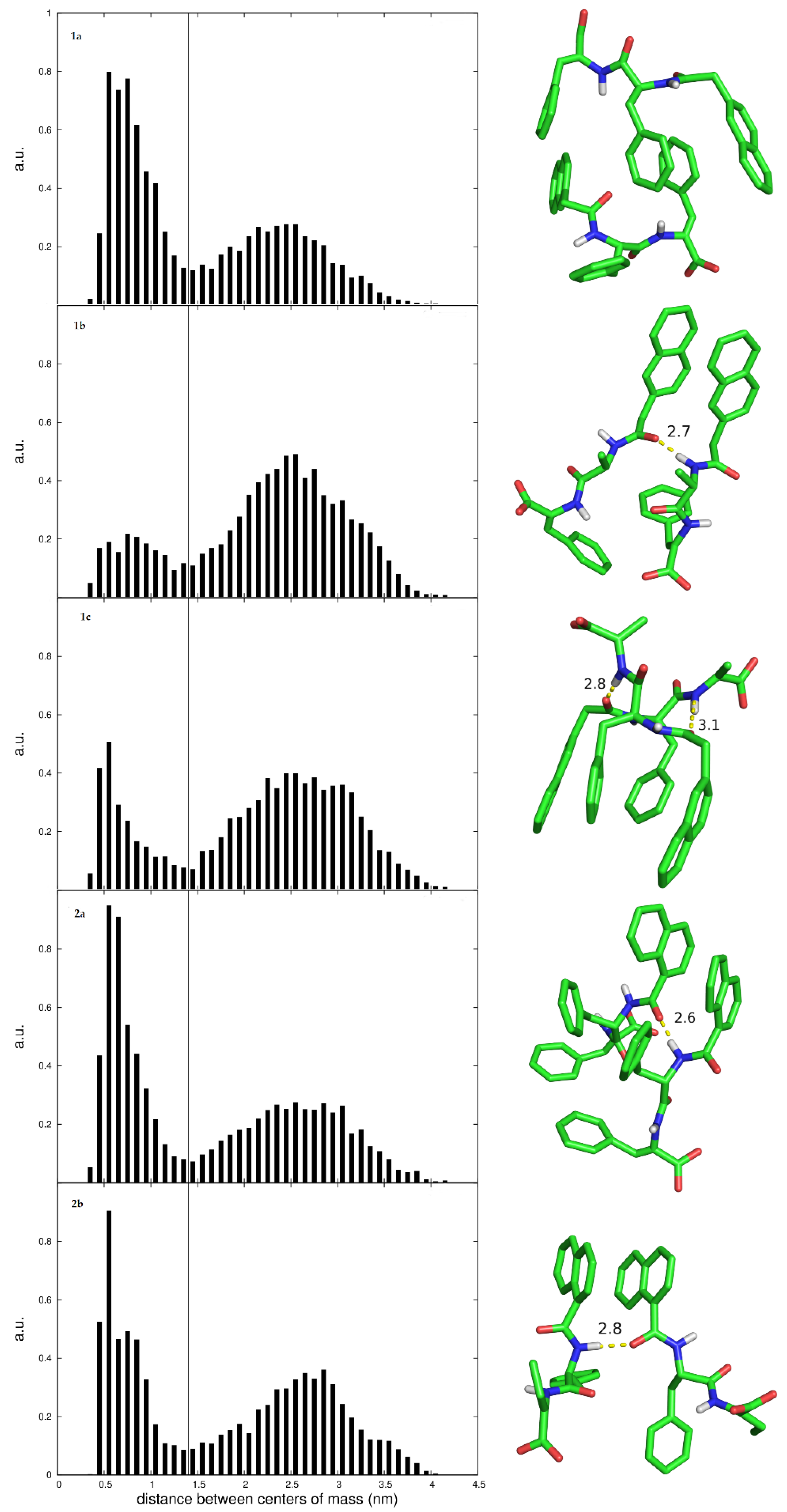
+ ions; the same energy minimization, equilibration and production run (40 + 60 ns) was also followed for the dimers. The aggregation analysis was done by monitoring the distance between the centers of mass of the peptides in the box vs. time for 60 ns and used to populate 0.1 nm bin histograms.
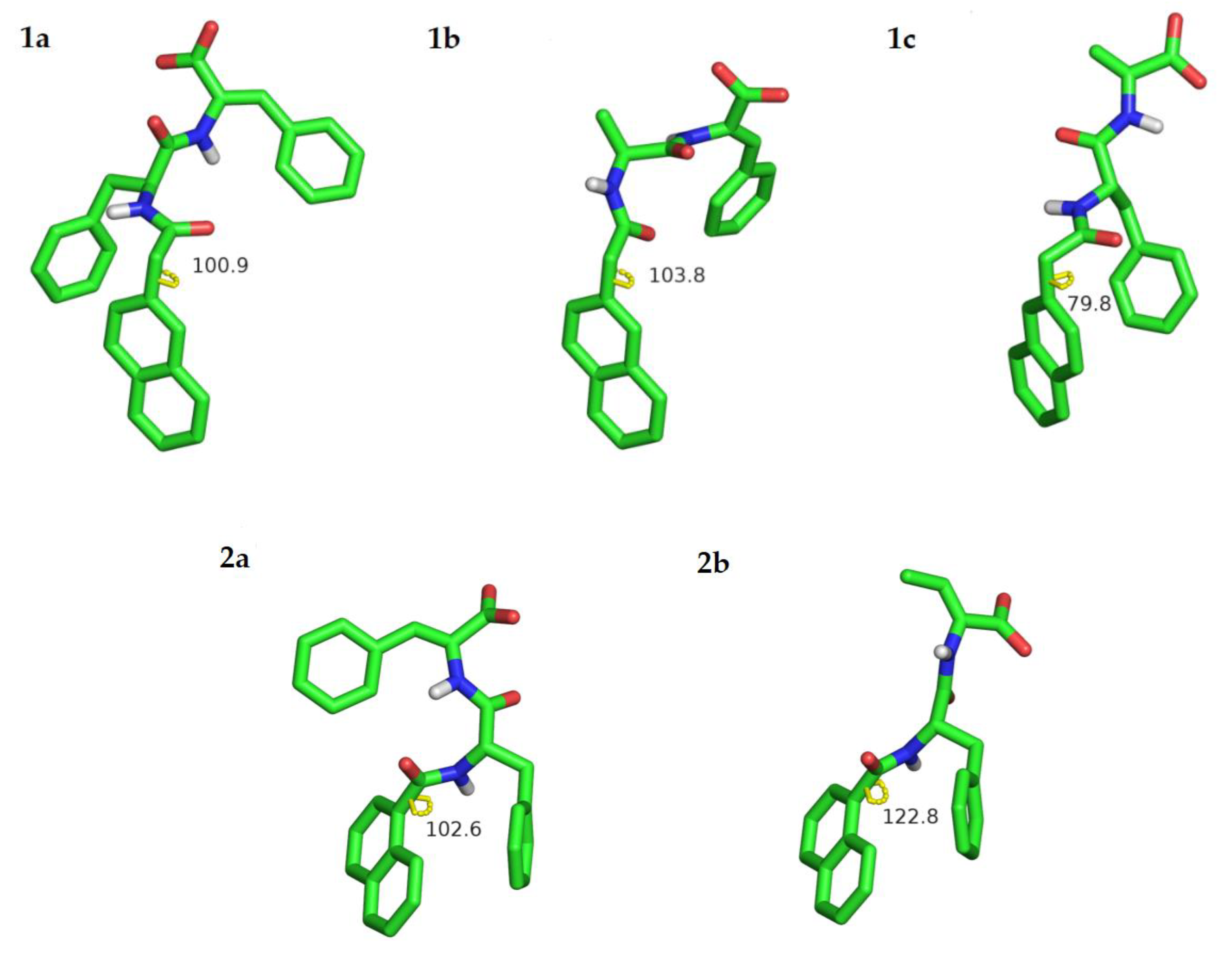
The most likely conformations for each molecule were also studied using quantum chemical DFT calculations. DFT calculations were performed with the program Gaussian 09 with the BLYP-D3 Hamiltonian that has been found to yield improved geometries in π–π stacked systems [
54,
55,
56]. Calculations were performed with the SMD implicit water solvent with the def2-TVZP basis set.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}