
Biomanufacturing Recombinantly Expressed Cripto-1 Protein in Anchorage-Dependent Mammalian Cells Growing in Suspension Bioreactors within a Three-Dimensional Hydrogel Microcarrier
 , and
, and 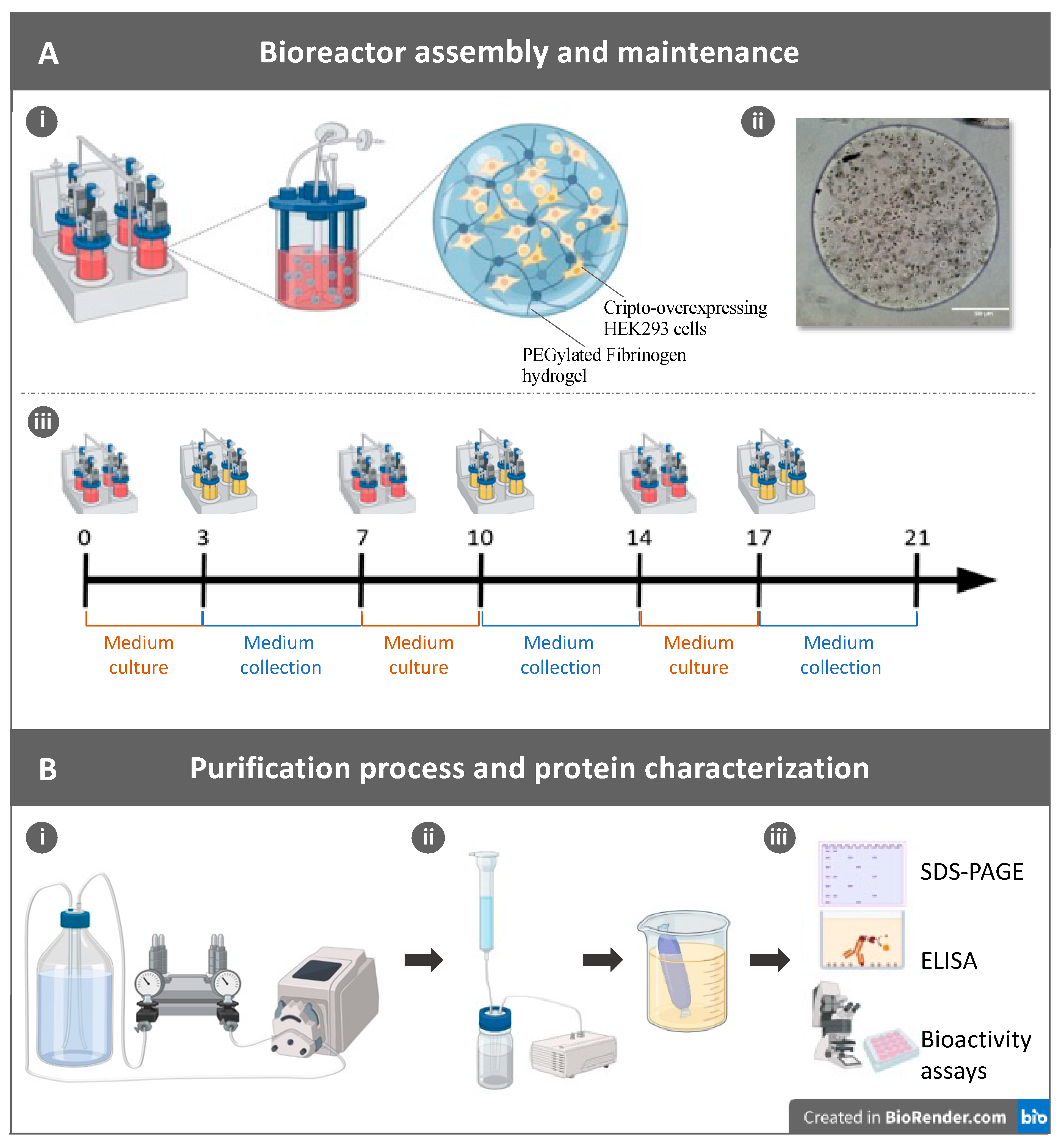{kind=link}
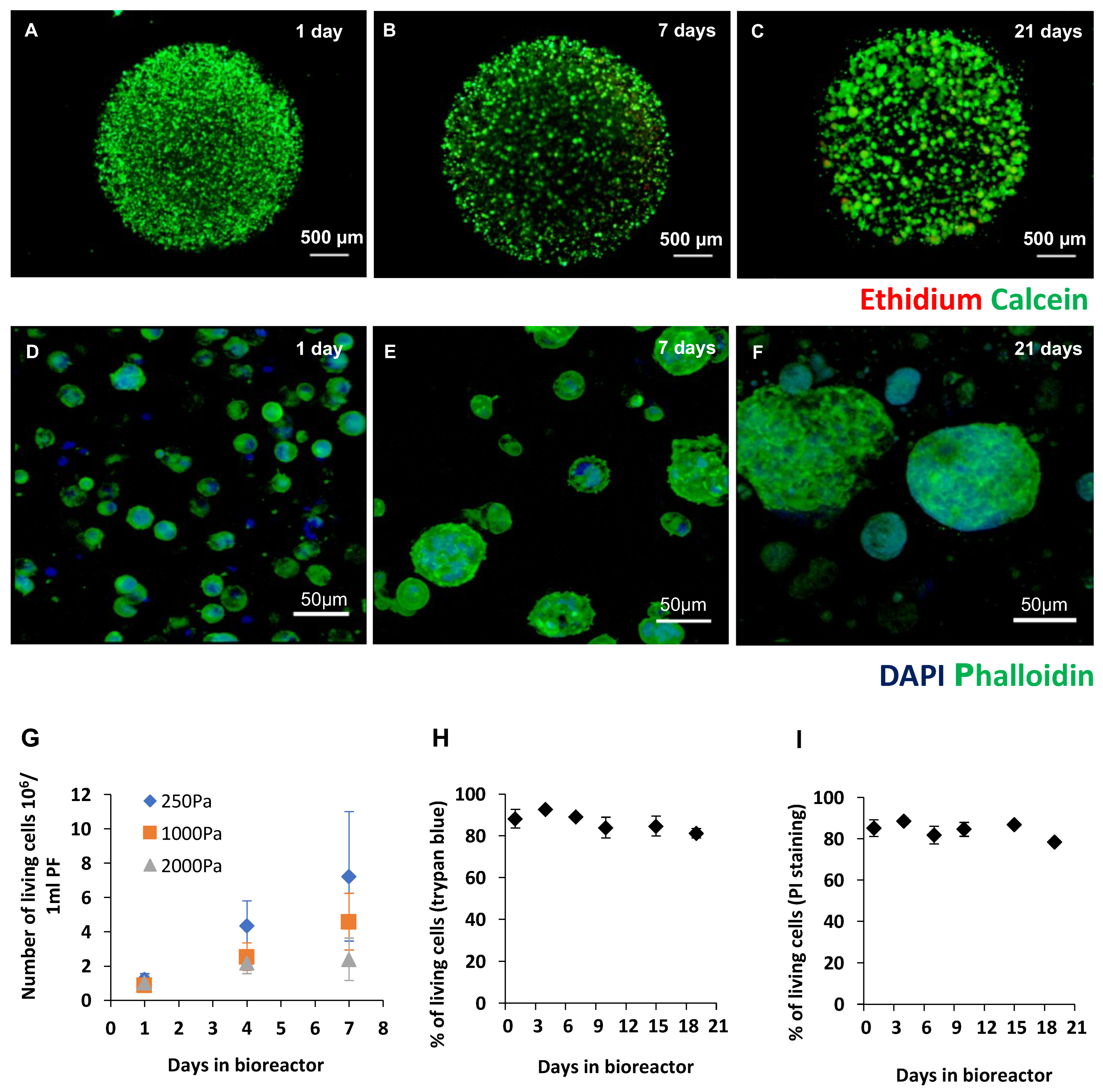{kind=link}
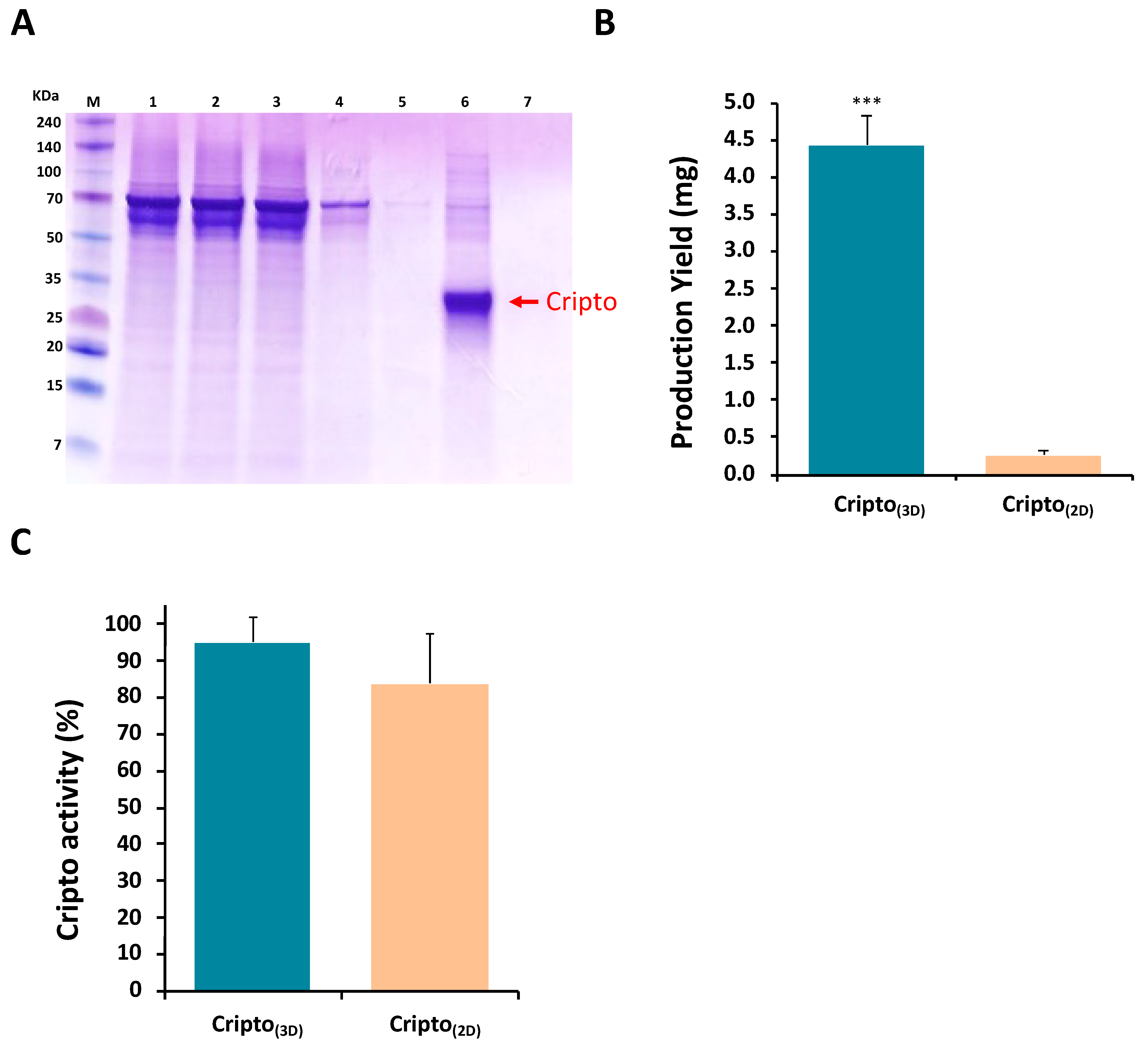{kind=link}
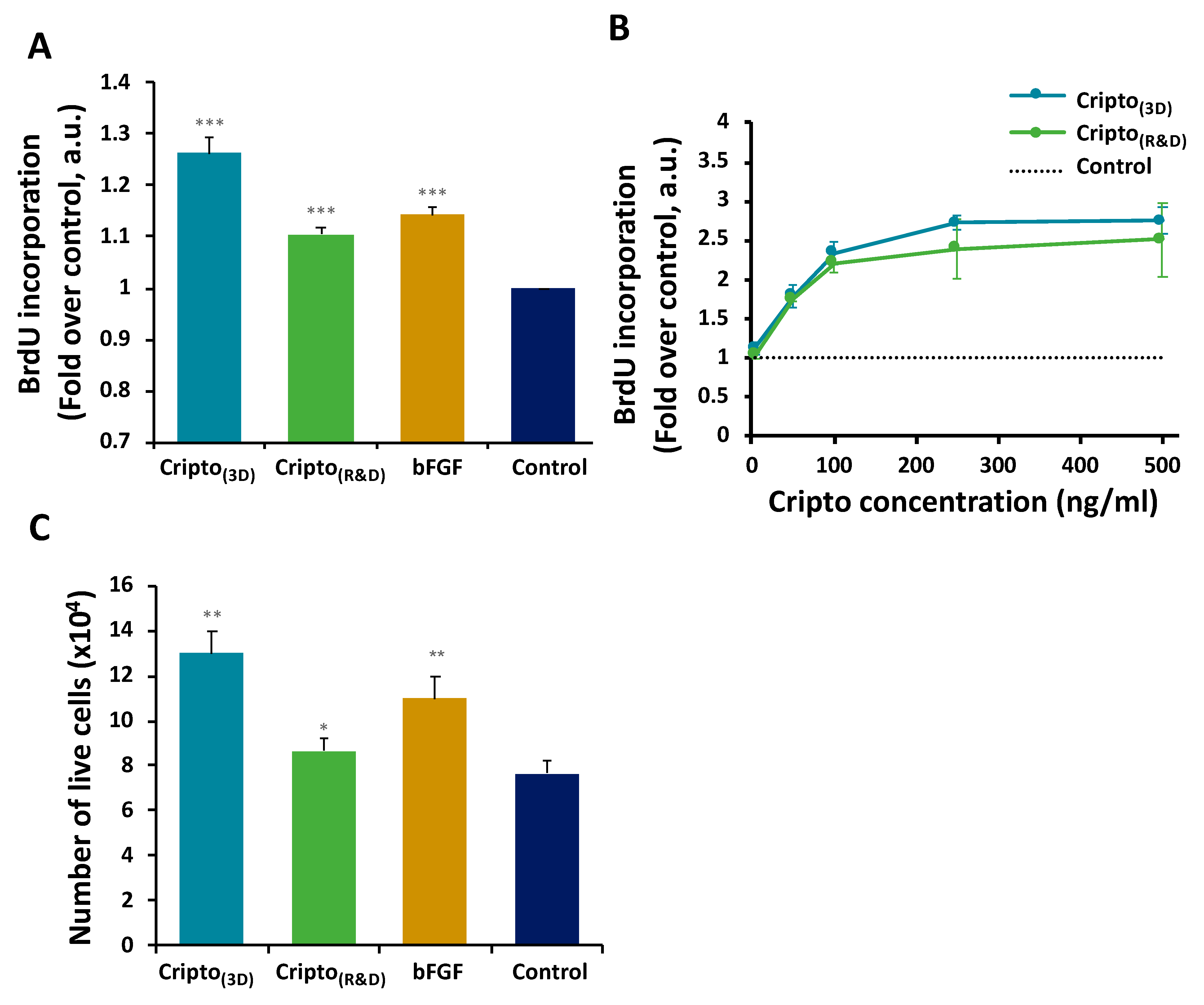{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Production of Recombinant Cripto in a 3D PF Microcarrier-Based System Using Stirred Suspension Bioreactors
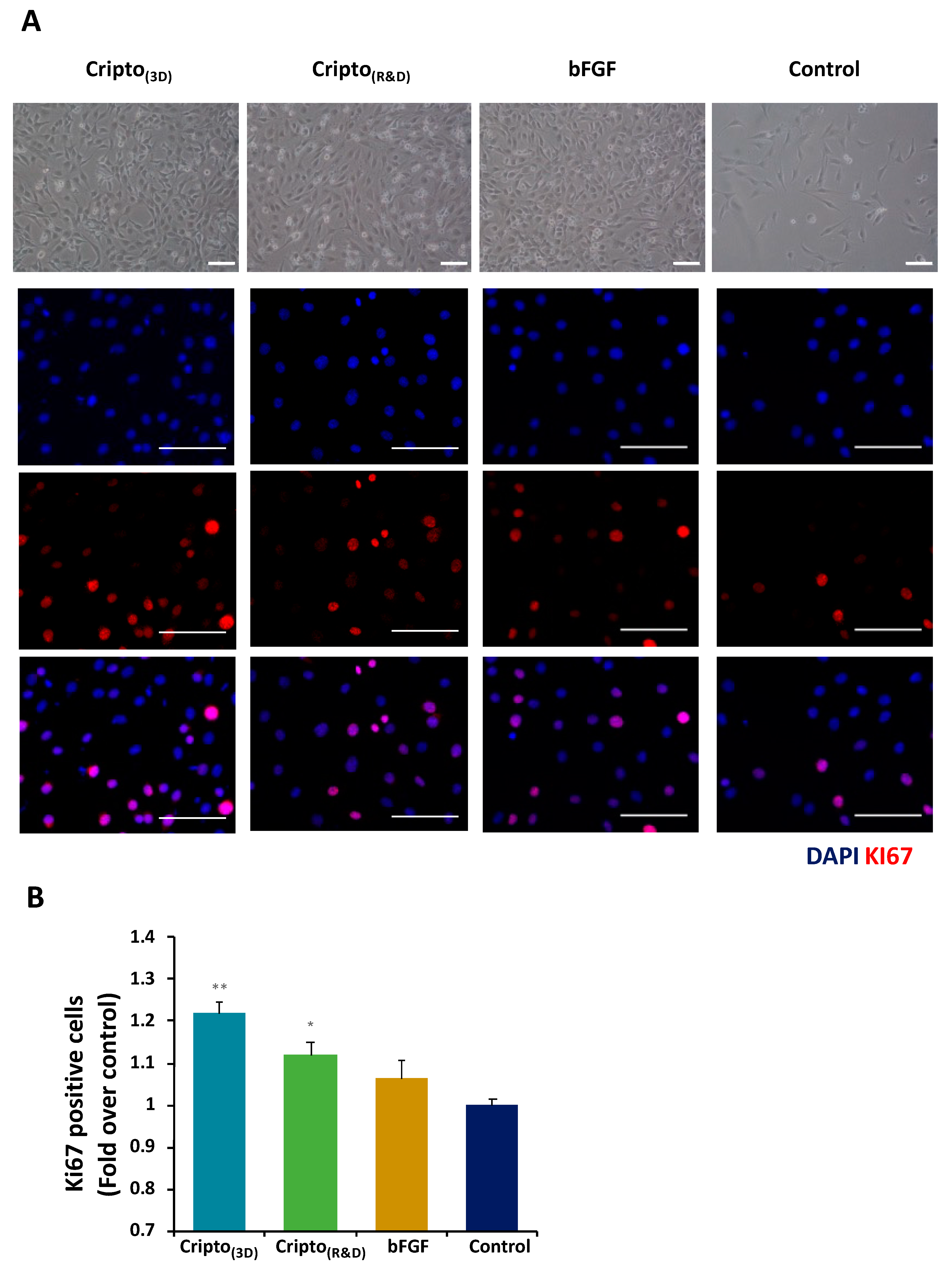
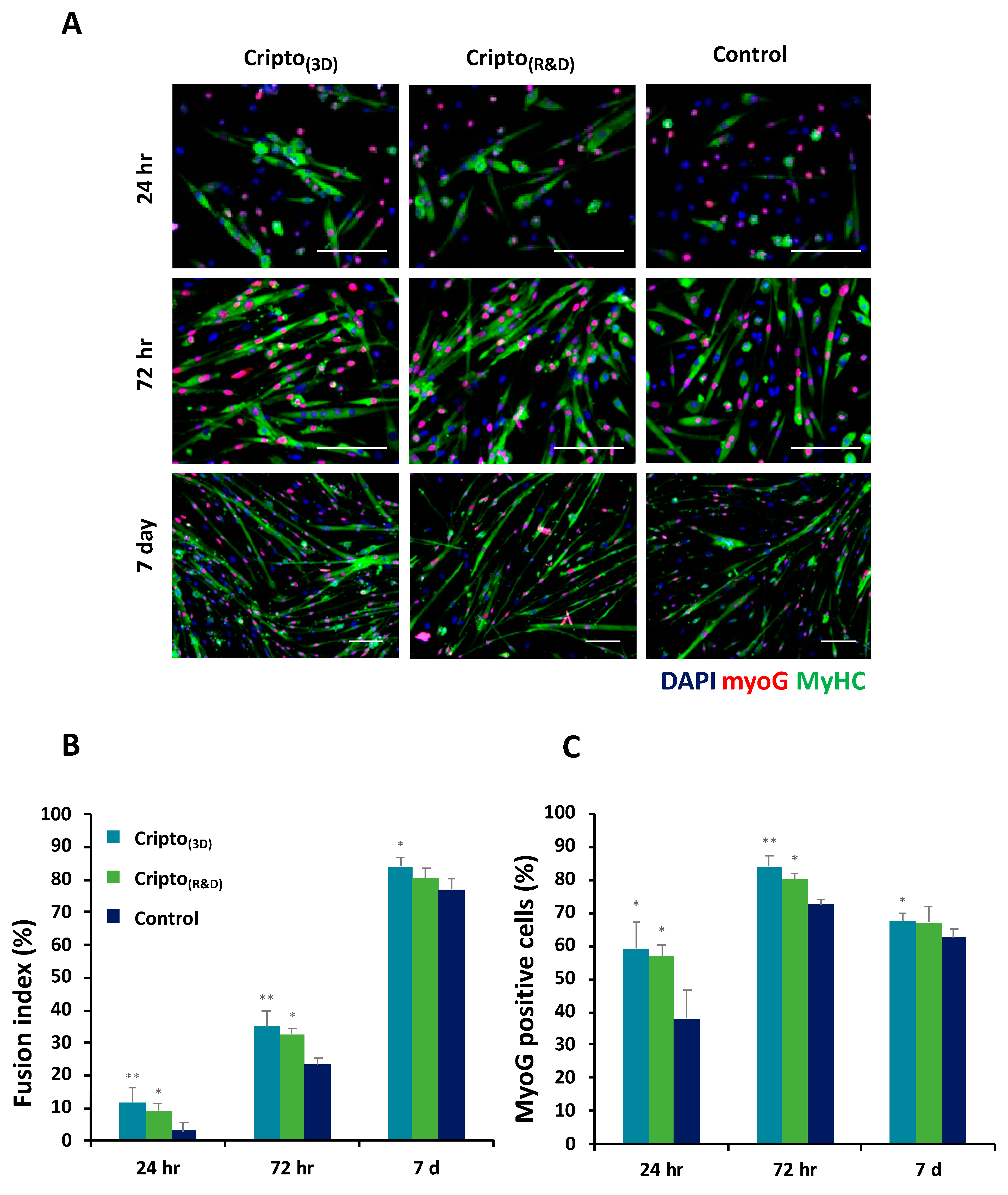
2.2. In Vitro Functional Activity of Purified Recombinant Cripto
2.3. Stability and Binding Capacity of Recombinant Cripto during Long-Term Storage
2.4. Discussion
3. Conclusions
4. Materials and Methods
4.1. Synthesis of PEG-Diacrylate (PEG-DA) and PEG-Fibrinogen (PF)
4.2. Cell Line Maintenance and Expansion in 2D Culture
4.3. Cell Seeding in 3D PF Microcarriers
4.4. Cell Viability and Imaging
4.5. Cultivation of Cells in Spinner Flasks
4.6. Cripto Production in 2D Cultivation System
4.7. Cripto Purification, Quantification, and Detection
4.8. BrdU Incorporation Cell Proliferation Assay
4.9. Ki67 Immunostaining Proliferation Assay
4.10. Muscle Satellite Cell Differentiation Assay
4.11. Stability and Functionality of Cripto over Time
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, M.M.; Mollet, M.; Hubert, R.S.; Kyung, Y.S.; Zhang, G.G. Industrial Production of Therapeutic Proteins: Cell Lines, Cell Culture, and Purification. In Handbook of Industrial Chemistry and Biotechnology; Springer: Cham, Switzerland, 2017; pp. 1639–1669. [Google Scholar] [CrossRef]
- Dimitrov, D.S. Therapeutic Proteins. Methods Mol. Biol. 2012, 899, 1–26. [Google Scholar] [PubMed]
- Dingermann, T. Recombinant Therapeutic Proteins: Production Platforms and Challenges. Biotechnol. J. 2008, 3, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, K.; Tiwari, M.; Tiwari, D.K.; Kerkar, S. Industrial Scale Production of Important Therapeutic Proteins Using Bacterial Expression System. In Microbial Products of Health, Environment and Agriculture; Springer: Cham, Switzerland, 2021; pp. 183–202. [Google Scholar] [CrossRef]
- Minchiotti, G.; Manco, G.; Parisi, S.; Lago, C.T.; Rosa, F.; Persico, M.G. Structure-Function Analysis of the EGF-CFC Family Member Cripto Identifies Residues Essential for Nodal Signalling. Development 2001, 128, 4501–4510. [Google Scholar] [CrossRef]
- Lubiniecki, A.S. Pharmaceutical Applications of Recombinant DNA-Modified Mammalian Cells. Dev. Ind. Microbiol. 1987, 28, 133–138. [Google Scholar]
- Bebbington, C.; Hentschel, C. The Expression of Recombinant DNA Products in Mammalian Cells. Trends Biotechnol. 1985, 3, 314–317. [Google Scholar] [CrossRef]
- Genzel, Y. Designing Cell Lines for Viral Vaccine Production: Where do We Stand? Biotechnol. J. 2015, 10, 728–740. [Google Scholar] [CrossRef]
- Chen, X.Y.; Chen, J.Y.; Tong, X.M.; Mei, J.G.; Chen, Y.F.; Mou, X.Z. Recent Advances in the Use of Microcarriers for Cell Cultures and Their Ex Vivo and In Vivo Applications. Biotechnol. Lett. 2020, 42, 1–10. [Google Scholar] [CrossRef]
- Barrett, P.N.; Terpening, S.J.; Snow, D.; Cobb, R.R.; Kistner, O. Vero Cell Technology for Rapid Development of Inactivated Whole Virus Vaccines for Emerging Viral Diseases. Expert Rev. Vaccines 2017, 16, 883–894. [Google Scholar] [CrossRef]
- Hu, X.; Xiao, C.; Huang, Z.; Guo, Z.; Zhang, Z.; Li, Z. Pilot Production of U-PA with Porous Microcarrier Cell Culture. Cytotechnology 2000, 33, 13–19. [Google Scholar] [CrossRef]
- Kumar, A.; Goel, A.S.; Payne, J.K.; Evans, C.; Mikolajczyk, S.D.; Kuus-Reichel, K.; Saedi, M.S. Large-Scale Propagation of Recombinant Adherent Cells That Secrete a Stable Form of Human Glandular Kallikrein, hK2. Protein Expr. Purif. 1999, 15, 62–68. [Google Scholar] [CrossRef]
- Chu, L.; Blumentals, I.; Maheshwari, G. Production of Recombinant Therapeutic Proteins by Mammalian Cells in Suspension Culture. Methods Mol. Biol. 2005, 308, 107–121. [Google Scholar] [PubMed]
- Leong, W.; Wang, D.A. Cell-laden Polymeric Microspheres for Biomedical Applications. Trends Biotechnol. 2015, 33, 653–666. [Google Scholar] [CrossRef]
- Tavassoli, H.; Alhosseini, S.N.; Tay, A.; Chan, P.P.Y.; Weng Oh, S.K.; Warkiani, M.E. Large-Scale Production of Stem Cells Utilizing Microcarriers: A Biomaterials Engineering Perspective from Academic Research to Commercialized Products. Biomaterials 2018, 181, 333–346. [Google Scholar] [CrossRef] [PubMed]
- YekrangSafakar, A.; Acun, A.; Choi, J.W.; Song, E.; Zorlutuna, P.; Park, K. Hollow Microcarriers for Large-Scale Expansion of Anchorage-Dependent Cells in a Stirred Bioreactor. Biotechnol. Bioeng. 2018, 115, 1717–1728. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruedinger, F.; Lavrentieva, A.; Blume, C.; Pepelanova, I.; Scheper, T. Hydrogels for 3D Mammalian Cell Culture: A Starting Guide for Laboratory Practice. Appl. Microbiol. Biotechnol. 2014, 99, 623–636. [Google Scholar] [CrossRef]
- Liu, X.; Tang, T.C.; Tham, E.; Yuk, H.; Lin, S.; Lu, T.K.; Zhao, X. Stretchable Living Materials and Devices with Hydrogel-Elastomer Hybrids Hosting Programmed Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 2200–2205. [Google Scholar] [CrossRef] [Green Version]
- Goldshmid, R.; Mironi-Harpaz, I.; Shachaf, Y.; Seliktar, D. A Method for Preparation of Hydrogel Microcapsules for Stem Cell Bioprocessing and Stem Cell Therapy. Methods 2015, 84, 35–43. [Google Scholar] [CrossRef]
- Cohen, N.; Toister, E.; Lati, Y.; Girshengorn, M.; Levin, L.; Silberstein, L.; Seliktar, D.; Epstein, E. Cell Encapsulation Utilizing PEG-Fibrinogen Hydrogel Supports Viability and Enhances Productivity under Stress Conditions. Cytotechnology 2018, 70, 1075–1083. [Google Scholar] [CrossRef]
- Franco, C.L.; Price, J.; West, J.L. Development and Optimization of a Dual-Photoinitiator, Emulsion-Based Technique for Rapid Generation of Cell-Laden Hydrogel Microspheres. Acta Biomater. 2011, 7, 3267–3276. [Google Scholar] [CrossRef]
- Zoratto, N.; Montanari, E.; Viola, M.; Wang, J.; Coviello, T.; Di Meo, C.; Matricardi, P. Strategies to Load Therapeutics into Polysaccharide-Based Nanogels with a Focus on Microfluidics: A Review. Carbohydr. Polym. 2021, 266, 118119. [Google Scholar] [CrossRef]
- Hamami, R.; Simaan-Yameen, H.; Gargioli, C.; Seliktar, D. Comparison of Four Different Preparation Methods for Making Injectable Microgels for Tissue Engineering and Cell Therapy. Regen. Eng. Transl. Med. 2022, 8, 615–629. [Google Scholar] [CrossRef]
- Song, W.; Lima, A.C.; Mano, J.F. Bioinspired Methodology to Fabricate Hydrogel Spheres for Multi-Applications Using Superhydrophobic Substrates. Soft Matter 2010, 6, 5868–5871. [Google Scholar] [CrossRef]
- Oliveira, M.B.; Kossover, O.; Mano, J.F.; Seliktar, D. Injectable PEGylated Fibrinogen Cell-Laden Microparticles Made with a Continuous Solvent- and Oil-Free Preparation Method. Acta Biomater. 2015, 13, 78–87. [Google Scholar] [CrossRef]
- Wang, T.; Lacík, I.; Brissová, M.; Anilkumar, A.V.; Prokop, A.; Hunkele, D.; Green, R.; Snahrokhi, K.; Powers, A.C. An Encapsulation System for the Immunoisolation of Pancreatic Islets. Nat. Biotechnol. 1997, 15, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Heathman, T.R.J.; Stolzing, A.; Fabian, C.; Rafiq, Q.A.; Coopman, K.; Nienow, A.W.; Kara, B.; Hewitt, C.J. Scalability and Process Transfer of Mesenchymal Stromal Cell Production from Monolayer to Microcarrier Culture Using Human Platelet Lysate. Cytotherapy 2016, 18, 523–535. [Google Scholar] [CrossRef] [Green Version]
- Tsai, A.C.; Jeske, R.; Chen, X.; Yuan, X.; Li, Y. Influence of Microenvironment on Mesenchymal Stem Cell Therapeutic Potency: From Planar Culture to Microcarriers. Front. Bioeng. Biotechnol. 2020, 8, 640. [Google Scholar] [CrossRef]
- Alves, P.M.; Moreira, J.L.; Rodrigues, J.M.; Aunins, J.G.; Carrondo, M.J.T. Two-Dimensional Versus Three-Dimensional Culture Systems: Effects on Growth and Productivity of BHK Cells. Biotechnol. Bioeng. 1996, 52, 429–432. [Google Scholar] [CrossRef]
- Skardal, A.; Sarker, S.F.; Crabbé, A.; Nickerson, C.A.; Prestwich, G.D. The Generation of 3-D Tissue Models Based on Hyaluronan Hydrogel-Coated Microcarriers within a Rotating Wall Vessel Bioreactor. Biomaterials 2010, 31, 8426–8435. [Google Scholar] [CrossRef] [PubMed]
- Abranches, E.; Bekman, E.; Henrique, D.; Cabral, J.M.S. Expansion of Mouse Embryonic Stem Cells on Microcarriers. Biotechnol. Bioeng. 2007, 96, 1211–1221. [Google Scholar] [CrossRef]
- Goldshmid, R.; Seliktar, D. Hydrogel Modulus Affects Proliferation Rate and Pluripotency of Human Mesenchymal Stem Cells Grown in Three-Dimensional Culture. ACS Biomater. Sci. Eng. 2017, 3, 3433–3446. [Google Scholar] [CrossRef]
- Alfred, R.; Radford, J.; Fan, J.; Boon, K.; Krawetz, R.; Rancourt, D.; Kallos, M.S. Efficient Suspension Bioreactor Expansion of Murine Embryonic Stem Cells on Microcarriers in Serum-Free Medium. Biotechnol. Prog. 2011, 27, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Badenes, S.M.; Fernandes, T.G.; Rodrigues, C.A.V.; Diogo, M.M.; Cabral, J.M.S. Microcarrier-Based Platforms for In Vitro Expansion and Differentiation of Human Pluripotent Stem Cells in Bioreactor Culture Systems. J. Biotechnol. 2016, 234, 71–82. [Google Scholar] [CrossRef]
- Wang, J.; Yu, Y.; Guo, J.; Lu, W.; Wei, Q.; Zhao, Y. The Construction and Application of Three-Dimensional Biomaterials. Adv. Biosyst. 2020, 4, e1900238. [Google Scholar] [CrossRef] [PubMed]
- Song, S.H.; Lee, J.H.; Yoon, J.; Park, W. Functional Microparticle R&D for IVD and Cell Therapeutic Technology: Large-Scale Commercialized Products. Biochip J. 2019, 13, 95–104. [Google Scholar]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Annabi, N.; Tamayol, A.; Uquillas, J.A.; Akbari, M.; Bertassoni, L.E.; Cha, C.; Camci-Unal, G.; Dokmeci, M.R.; Peppas, N.A.; Khademhosseini, A. 25th Anniversary Article: Rational Design and Applications of Hydrogels in Regenerative Medicine. Adv. Mater. 2014, 26, 85–124. [Google Scholar] [CrossRef]
- Seliktar, D. Designing Cell-Compatible Hydrogels for Biomedical Applications. Science 2012, 336, 1124–1128. [Google Scholar] [CrossRef]
- Robb, K.P.; Fitzgerald, J.C.; Barry, F.; Viswanathan, S. Mesenchymal Stromal Cell Therapy: Progress in Manufacturing and Assessments of Potency. Cytotherapy 2019, 21, 289–306. [Google Scholar] [CrossRef]
- Almany, L.; Seliktar, D. Biosynthetic Hydrogel Scaffolds Made from Fibrinogen and Polyethylene Glycol for 3D Cell Cultures. Biomaterials 2005, 26, 2467–2477. [Google Scholar] [CrossRef]
- Mironi-Harpaz, I.; Wang, D.Y.; Venkatraman, S.; Seliktar, D. Photopolymerization of Cell-Encapsulating Hydrogels: Crosslinking Efficiency Versus Cytotoxicity. Acta Biomater. 2012, 8, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Dikovsky, D.; Bianco-Peled, H.; Seliktar, D. The Effect of Structural Alterations of PEG-Fibrinogen Hydrogel Scaffolds on 3-D Cellular Morphology and Cellular Migration. Biomaterials 2006, 27, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Frisman, I.; Seliktar, D.; Bianco-Peled, H. Nanostructuring PEG-Fibrinogen Hydrogels to Control Cellular Morphogenesis. Biomaterials 2011, 32, 7839–7846. [Google Scholar] [CrossRef]
- Zeng, Q.; Gao, Y.; Zhou, Y. Understanding the Role of Cripto-1 in Cancer Progression and Therapeutic Strategies. Clin. Transl. Oncol. 2022, 1–10. [Google Scholar] [CrossRef]
- Strizzi, L.; Bianco, C.; Normanno, N.; Salomon, D. Cripto-1: A Multifunctional Modulator During Embryogenesis and Oncogenesis. Oncogene 2005, 24, 5731–5741. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.M.; Schier, A.F. The EGF-CFC Gene Family in Vertebrate Development. Trends Genet. 2000, 16, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Kemaladewi, D.U.; ‘t Hoen, P.A.; ’ten Dijke, P.; van Ommen, G.J.; Hoogaars, W.M. TGF-β Signaling in Duchenne Muscular Dystrophy. Future Neurol. 2012, 7, 209–224. [Google Scholar] [CrossRef]
- Bianco, C.; Salomon, D.S. Targeting the Embryonic Gene Cripto-1 in Cancer and Beyond. Expert Opin. Ther. Pat. 2010, 20, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Guardiola, O.; Lafuste, P.; Brunelli, S.; Iaconis, S.; Touvier, T.; Mourikis, P.; De Bock, K.; Lonardo, E.; Andolfi, G.; Bouché, A.; et al. Cripto Regulates Skeletal Muscle Regeneration and Modulates Satellite Cell Determination by Antagonizing Myostatin. Proc. Natl. Acad. Sci. USA 2012, 109, E3231–E3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurm, F.M. Production of Recombinant Protein Therapeutics in Cultivated Mammalian Cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Ahmed, S.; Chauhan, V.M.; Ghaemmaghami, A.M.; Aylott, J.W. New Generation of Bioreactors that Advance Extracellular Matrix Modelling and Tissue Engineering. Biotechnol. Lett. 2019, 41, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Harrison, S.T.L.; Tai, S.L. Advances in Bioreactor Systems for the Production of Biologicals in Mammalian Cells. ChemBioEng Rev. 2022, 9, 42–62. [Google Scholar] [CrossRef]
- Prezioso, C.; Iaconis, S.; Andolfi, G.; Zentilin, L.; Iavarone, F.; Guardiola, O.; Minchiotti, G. Conditional Cripto Overexpression in Satellite Cells Promotes Myogenic Commitment and Enhances Early Regeneration. Front. Cell Dev. Biol. 2015, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Iavarone, F.; Guardiola, O.; Scagliola, A.; Andolfi, G.; Esposito, F.; Serrano, A.; Perdiguero, E.; Brunelli, S.; Muñoz-Cánoves, P.; Minchiotti, G. Cripto Shapes Macrophage Plasticity and Restricts EndMT in Injured and Diseased Skeletal Muscle. EMBO Rep. 2020, 21, e49075. [Google Scholar] [CrossRef]
- Sart, S.; Agathos, S.N.; Li, Y. Engineering Stem Cell Fate with Biochemical and Biomechanical Properties of Microcarriers. Biotechnol. Prog. 2013, 29, 1354–1366. [Google Scholar] [CrossRef] [PubMed]
- Yom-Tov, O.; Seliktar, D.; Bianco-Peled, H. A Modified Emulsion Gelation Technique to Improve Buoyancy of Hydrogel Tablets for Floating Drug Delivery Systems. Mater. Sci. Eng. C 2015, 55, 335–342. [Google Scholar] [CrossRef]
- Yom-Tov, O.; Neufeld, L.; Seliktar, D.; Bianco-Peled, H. A Novel Design of Injectable Porous Hydrogels with In Situ Pore Formation. Acta Biomater. 2014, 10, 4236–4246. [Google Scholar] [CrossRef]
- Bryant, S.J.; Nuttelman, C.R.; Anseth, K.S. Cytocompatibility of UV and Visible Light Photoinitiating Systems on Cultured NIH/3T3 Fibroblasts In Vitro. J. Biomater. Sci. Polym. Ed. 2012, 11, 439–457. [Google Scholar] [CrossRef]
- Yosef, A.; Kossover, O.; Mironi-Harpaz, I.; Mauretti, A.; Melino, S.; Mizrahi, J.; Seliktar, D. Fibrinogen-Based Hydrogel Modulus and Ligand Density Effects on Cell Morphogenesis in Two-Dimensional and Three-Dimensional Cell Cultures. Adv. Healthc. Mater. 2019, 8, 1801436. [Google Scholar] [CrossRef]
- Dikovsky, D.; Bianco-Peled, H.; Seliktar, D. Defining the Role of Matrix Compliance and Proteolysis in Three-Dimensional Cell Spreading and Remodeling. Biophys. J. 2008, 94, 2914–2925. [Google Scholar] [CrossRef] [Green Version]
- Loubière, C.; Delafosse, A.; Guedon, E.; Toye, D.; Chevalot, I.; Olmos, E. Optimization of the Impeller Design for Mesenchymal Stem Cell Culture on Microcarriers in Bioreactors. Chem. Eng. Technol. 2019, 42, 1702–1708. [Google Scholar] [CrossRef]
- Cermola, F.; D’Aniello, C.; Tatè, R.; De Cesare, D.; Martinez-Arias, A.; Minchiotti, G.; Patriarca, E.J. Gastruloid Development Competence Discriminates Different States of Pluripotency. Stem Cell Rep. 2021, 16, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Afify, S.M.; Hassan, G.; Nawara, H.M.; Zahra, M.H.; Xu, Y.; Alam, M.J.; Saitoh, K.; Mansour, H.; Abu Quora, H.A.; Sheta, M.; et al. Optimization of Production and Characterization of a Recombinant Soluble Human Cripto-1 Protein Inhibiting Self-Renewal of Cancer Stem Cells. J. Cell. Biochem. 2022, 123, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Vinther, L.; Lademann, U.; Andersen, E.V.; Højrup, P.; Thaysen-Andersen, M.; Krogh, B.O.; Viuff, B.; Brünner, N.; Stenvang, J.; Moreira, J.M.A. Purification and Characterization of Bioactive His6-Tagged Recombinant Human Tissue Inhibitor of Metalloproteinases-1 (TIMP-1) Protein Expressed at High Yields in Mammalian Cells. Protein Expr. Purif. 2014, 101, 157–164. [Google Scholar] [CrossRef]
- Seno, M.; Desantis, M.; Kannan, S.; Bianco, C.; Tada, H.; Kim, N.; Kosaka, M.; Gullick, W.J.; Yamada, H.; Salomon, D.S. Purification and Characterization of a Recombinant Human Cripto-1 Protein. Growth Factors 1998, 15, 215–229. [Google Scholar] [CrossRef]
- Minchiotti, G.; Parisi, S.; Liguori, G.; Signore, M.; Lania, G.; Adamson, E.D.; Lago, C.T.; Persico, M.G. Membrane-Anchorage of Cripto Protein by Glycosylphosphatidylinositol and Its Distribution during Early Mouse Development. Mech. Dev. 2000, 90, 133–142. [Google Scholar] [CrossRef]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef] [Green Version]
- Bianco, C.; Adkins, H.B.; Wechselberger, C.; Seno, M.; Normanno, N.; De Luca, A.; Sun, Y.; Khan, N.; Kenney, N.; Ebert, A.; et al. Cripto-1 Activates Nodal- and ALK4-Dependent and -Independent Signaling Pathways in Mammary Epithelial Cells. Mol. Cell. Biol. 2002, 22, 2586–2597. [Google Scholar] [CrossRef] [Green Version]
- Frokjaer, S.; Otzen, D.E. Protein Drug Stability: A Formulation Challenge. Nat. Rev. Drug Discov. 2005, 4, 298–306. [Google Scholar] [CrossRef]
- Vicente, T.; Roldão, A.; Peixoto, C.; Carrondo, M.J.T.; Alves, P.M. Large-Scale Production and Purification of VLP-Based Vaccines. J. Invertebr. Pathol. 2011, 107, S42–S48. [Google Scholar] [CrossRef] [PubMed]
- Gonen-Wadmany, M.; Oss-Ronen, L.; Seliktar, D. Protein–Polymer Conjugates for Forming Photopolymerizable Biomimetic Hydrogels for Tissue Engineering. Biomaterials 2007, 28, 3876–3886. [Google Scholar] [CrossRef]
- Casalino, L.; Comes, S.; Lambazzi, G.; De Stefano, B.; Filosa, S.; De Falco, S.; De Cesare, D.; Minchiotti, G.; Patriarca, E.J. Control of Embryonic Stem Cell Metastability by L-Proline Catabolism. J. Mol. Cell Biol. 2011, 3, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunez, R. DNA Measurement and Cell Cycle Analysis by Flow Cytometry. Curr. Issues Mol. Biol. 2001, 3, 67–70. [Google Scholar] [PubMed]
- Syverud, B.C.; Lee, J.D.; VanDusen, K.W.; Larkin, L.M. Isolation and Purification of Satellite Cells for Skeletal Muscle Tissue Engineering. J. Regen. Med. 2014, 3, 117. [Google Scholar]
- Beckerman, M.; Harel, C.; Michael, I.; Klip, A.; Bilan, P.J.; Gallagher, E.J.; LeRoith, D.; Lewis, E.C.; Karnieli, E.; Levenberg, S. GLUT4-Overexpressing Engineered Muscle Constructs as a Therapeutic Platform to Normalize Glycemia in Diabetic Mice. Sci. Adv. 2021, 7, eabg3947. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lev, R.; Bar-Am, O.; Lati, Y.; Guardiola, O.; Minchiotti, G.; Seliktar, D. Biomanufacturing Recombinantly Expressed Cripto-1 Protein in Anchorage-Dependent Mammalian Cells Growing in Suspension Bioreactors within a Three-Dimensional Hydrogel Microcarrier. Gels 2023, 9, 243. https://doi.org/10.3390/gels9030243
Lev R, Bar-Am O, Lati Y, Guardiola O, Minchiotti G, Seliktar D. Biomanufacturing Recombinantly Expressed Cripto-1 Protein in Anchorage-Dependent Mammalian Cells Growing in Suspension Bioreactors within a Three-Dimensional Hydrogel Microcarrier. Gels. 2023; 9(3):243. https://doi.org/10.3390/gels9030243
Chicago/Turabian StyleLev, Rachel, Orit Bar-Am, Yoni Lati, Ombretta Guardiola, Gabriella Minchiotti, and Dror Seliktar. 2023. "Biomanufacturing Recombinantly Expressed Cripto-1 Protein in Anchorage-Dependent Mammalian Cells Growing in Suspension Bioreactors within a Three-Dimensional Hydrogel Microcarrier" Gels 9, no. 3: 243. https://doi.org/10.3390/gels9030243