Uniparental Inheritance and Recombination as Strategies to Avoid Competition and Combat Muller’s Ratchet among Mitochondria in Natural Populations of the Fungus Amanita phalloides


Abstract
:1. Introduction
2. Materials and Methods
2.1. Collecting Mushrooms, and Sequencing of Genomes and a Transcriptome
2.2. Reference Mitochondrial Genome Assembly and Annotation
2.3. Assembling the Mitochondrial Genomes of the Remaining Samples, Haplotype Network Reconstruction, and Nuclear Phylogeny Reconstruction
2.4. Comparing Mitochondrial and Nuclear Genomes
2.5. Testing for Recombination among Mitochondrial Genomes
2.6. Testing for Biparental Inheritance of Mitochondria
3. Results
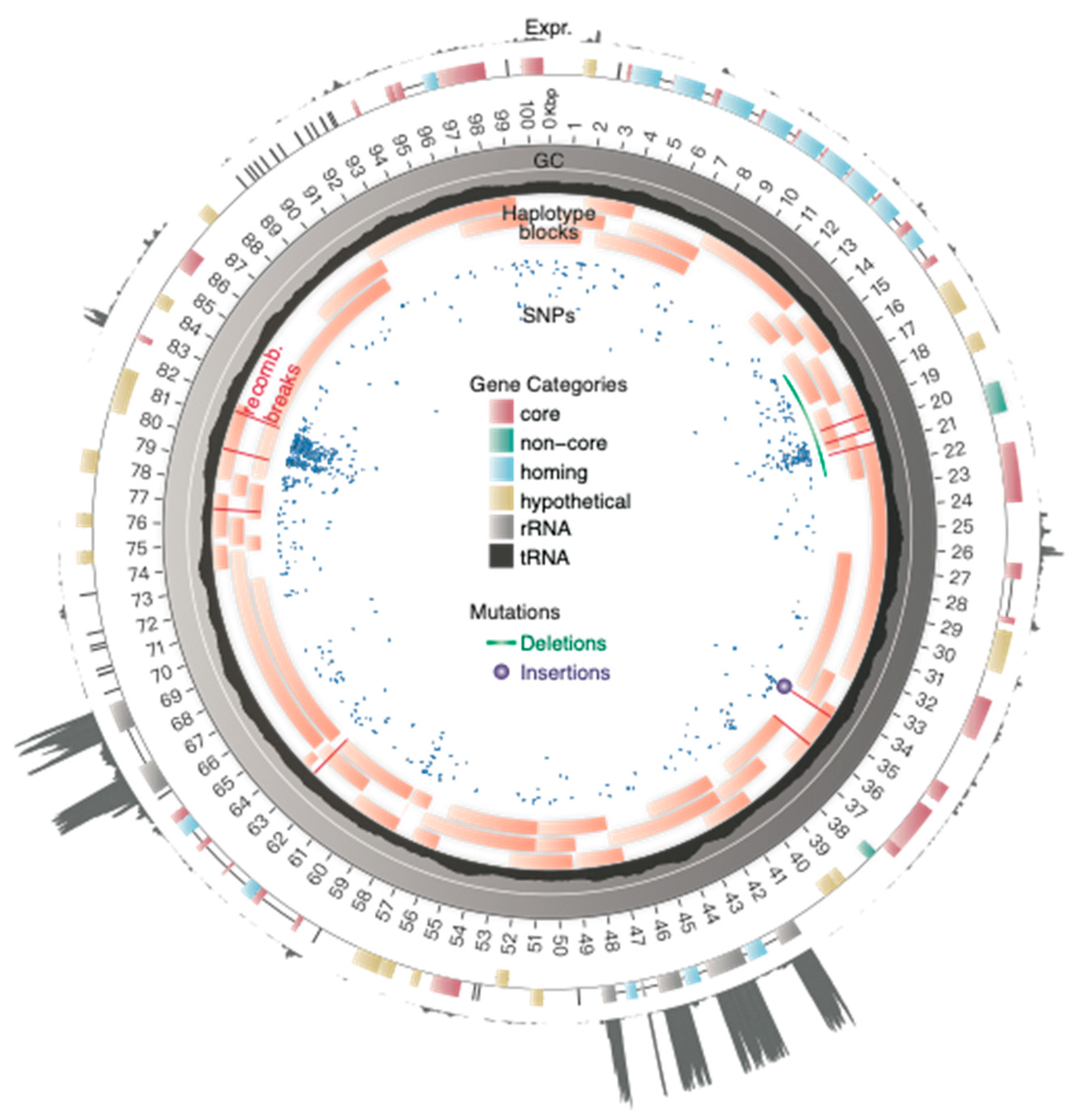
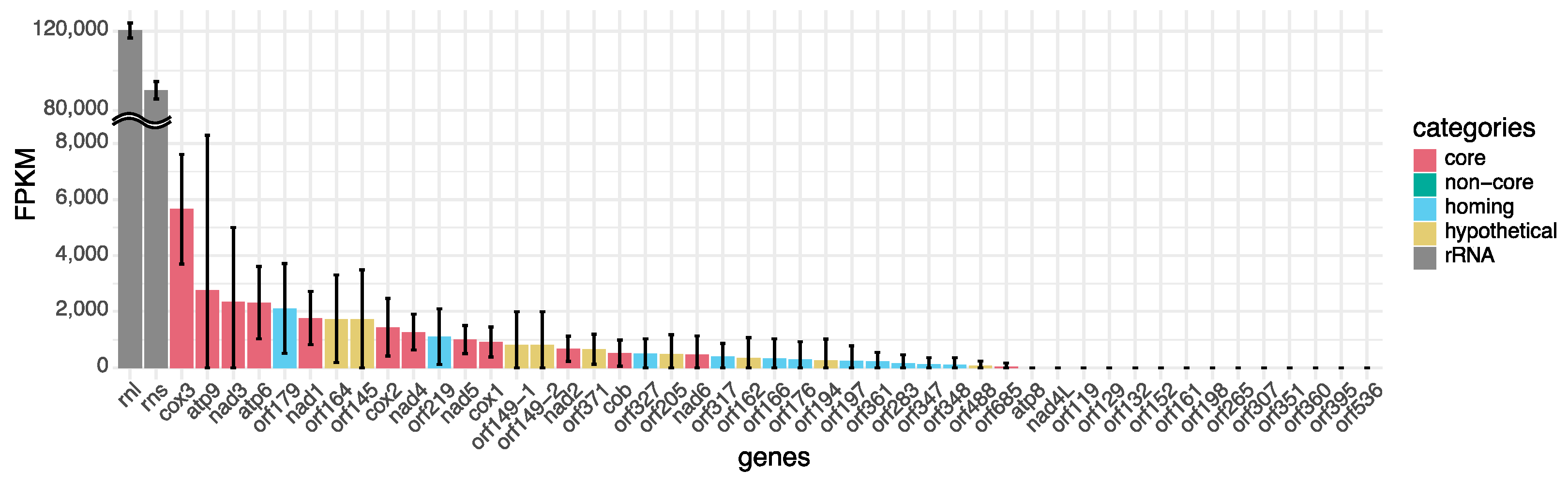
3.1. Reference Genome Architecture and Gene Expression
3.2. Structural Diversity
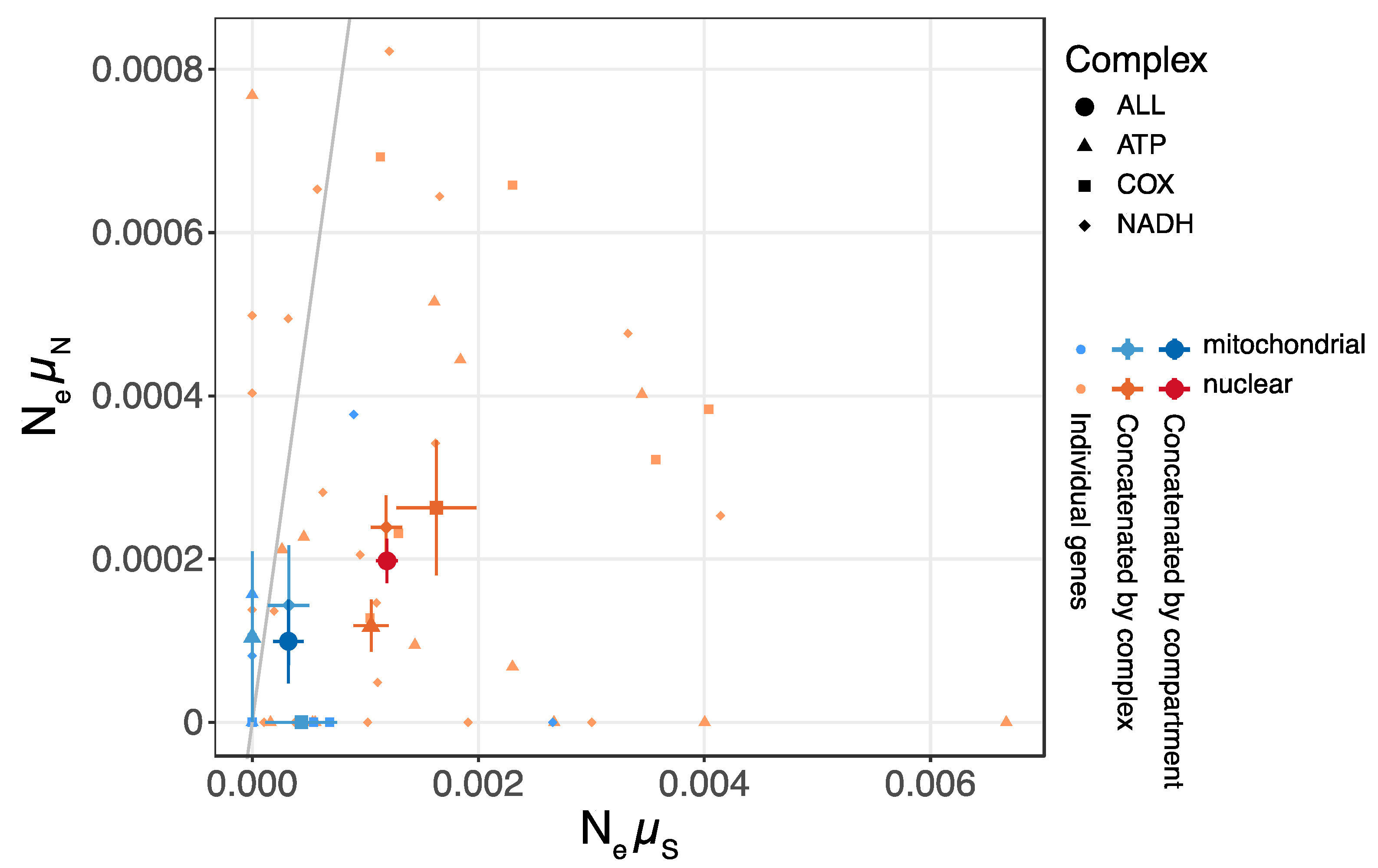
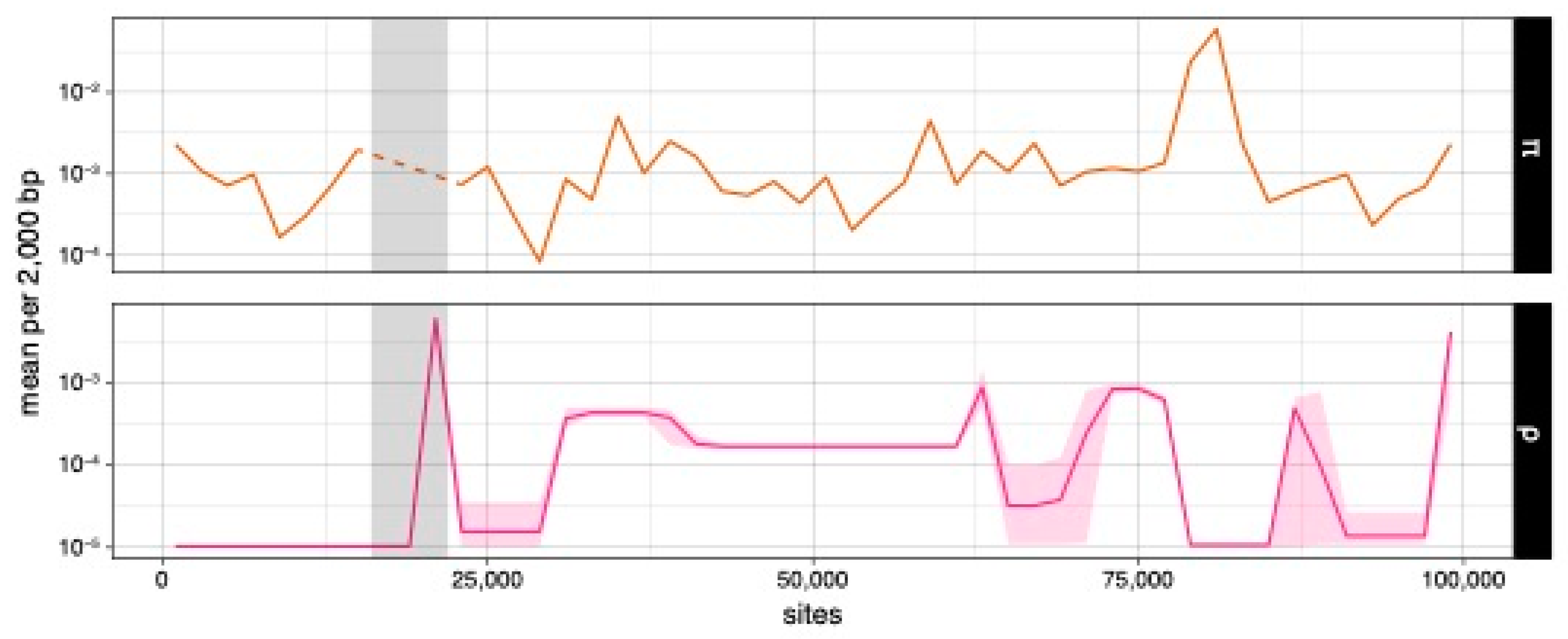
3.3. Nucleotide Diversity and Efficacy of Selection in Mitochondrial and Nuclear Genomes
3.4. Detecting Mitochondrial Recombination and Estimating Rates
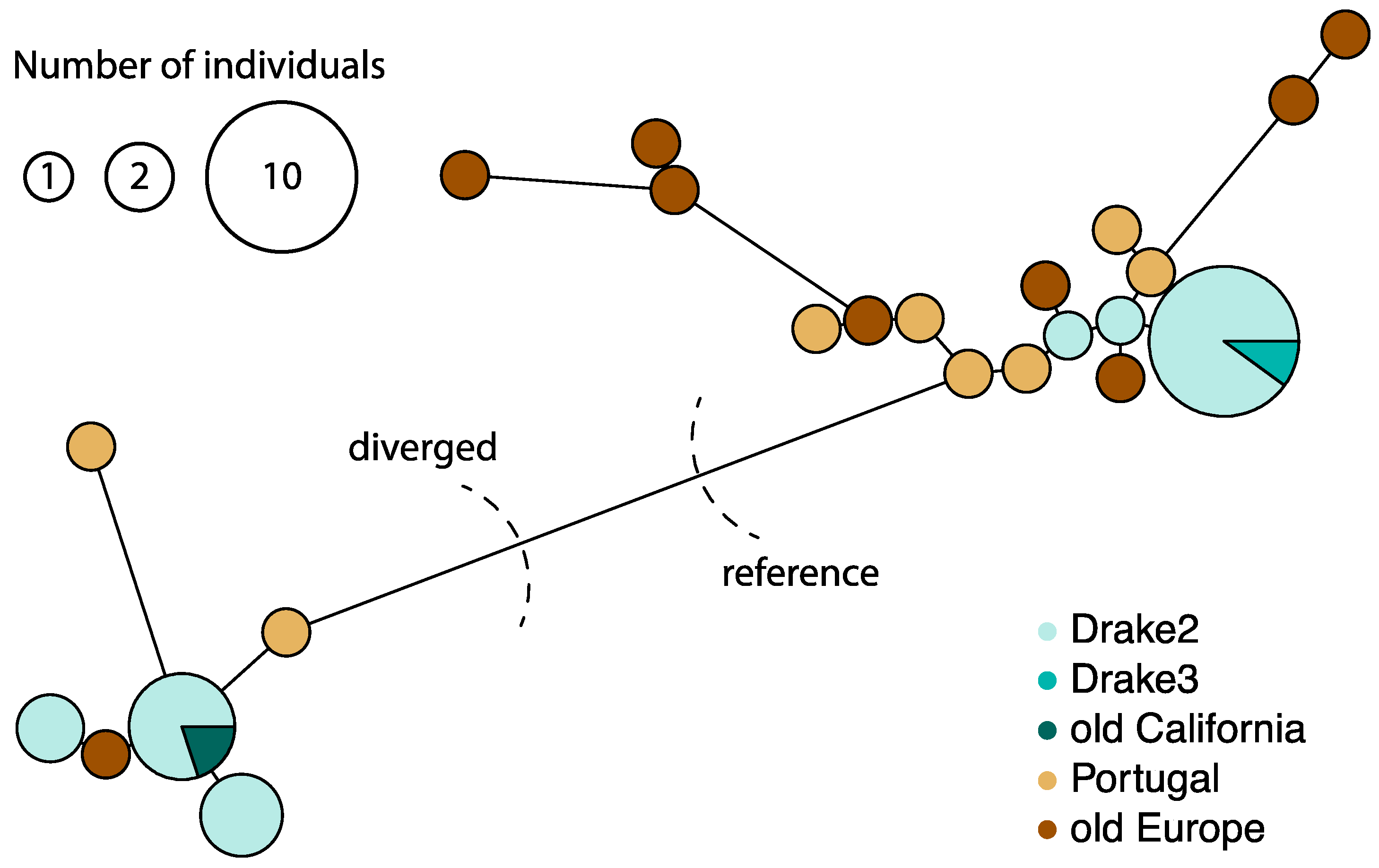
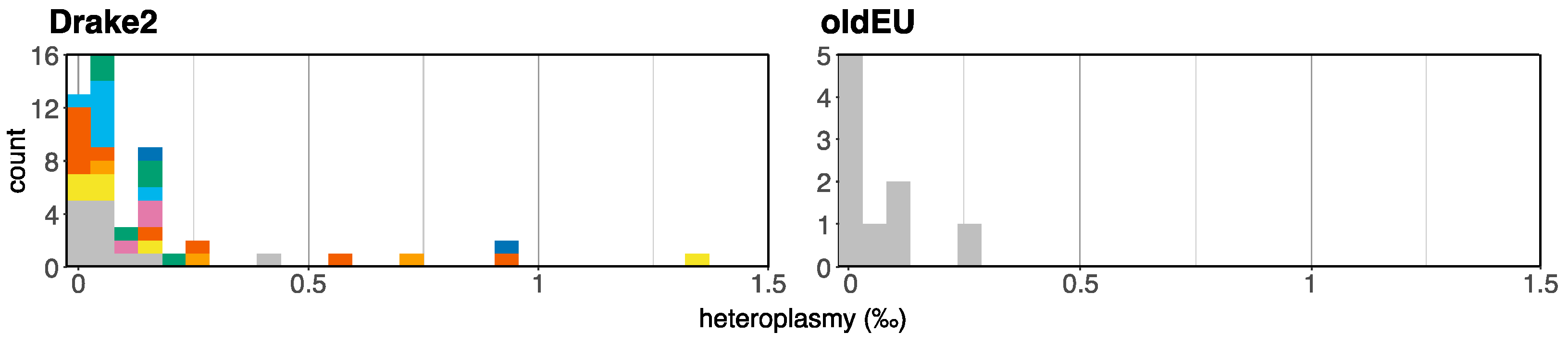
3.5. Testing for Biparental Inheritance of Mitochondria
4. Discussion
4.1. Mitochondrial Genome Architecture and Structural Diversity
4.2. Rare Recombination Contributes Lower Nucleotide Diversity and Selection Efficacy in Mitochondria as Compared with Nuclei
4.3. Uniparental Inheritance as an Alternative Strategy to Combat Muller’s Ratchet
4.4. Mitochondrial Genomes Provide Evidence for a 28-Year-Old Homokaryotic Individual
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [Green Version]
- Birky, C.W., Jr. The Inheritance of Genes in Mitochondria and Chloroplasts: Laws, Mechanisms, and Models. Annu. Rev. Genet. 2001, 35, 125–148. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Sung, W.; Morris, K.; Coffey, N.; Landry, C.R.; Dopman, E.B.; Dickinson, W.J.; Okamoto, K.; Kulkarni, S.; Hartl, D.L.; et al. A Genome-Wide View of the Spectrum of Spontaneous Mutations in Yeast. Proc. Natl. Acad. Sci. USA 2008, 105, 9272–9277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denver, D.R.; Morris, K.; Lynch, M.; Thomas, W.K. High Mutation Rate and Predominance of Insertions in the Caenohabditis elegans Nuclear Genome. Nature 2004, 430, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Sandor, S.; Zhang, Y.; Xu, J. Fungal Mitochondrial Genomes and Genetic Polymorphisms. Appl. Microbiol. Biotechnol. 2018, 102, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates, Inc.: Sunderland, MA, USA, 2007. [Google Scholar]
- Hill, W.G.; Robertson, A. The Effect of Linkage on Limits to Artificial Selection. Genet. Res. 1966, 8, 269–294. [Google Scholar] [CrossRef]
- Felsenstein, J. The Evolution Advantage of Recombination. Genetics 1974, 78, 737–756. [Google Scholar] [CrossRef]
- Muller, H.J. The Relation of Recombination to Mutational Advance. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1964, 1, 2–9. [Google Scholar] [CrossRef]
- Bentley, K.E.; Mandel, J.R.; McCauley, D.E. Paternal Leakage and Heteroplasmy of Mitochondrial Genomes in Silene vulgaris: Evidence from Experimental Crosses. Genetics 2010, 185, 961–968. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Mitochondrial Inheritance in Yeast. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Radzvilavicius, A.L.; Kokko, H.; Christie, J.R. Mitigating Mitochondrial Genome Erosion without Recombination. Genetics 2017, 207, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Bergstrom, C.T.; Pritchard, J. Germline Bottlenecks and the Evolutionary Maintenance of Mitochondrial Genomes. Genetics 1998, 149, 2135–2146. [Google Scholar] [CrossRef]
- Christie, J.R.; Beekman, M. Uniparental Inheritance Promotes Adaptive Evolution in Cytoplasmic Genomes. Mol. Biol. Evol. 2017, 34, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.R.; Zeyl, C.; Cooke, E. Conflicting Levels of Selection in the Accumulation of Mitochondrial Defects in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2002, 99, 3690–3694. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, P. Mitochondrial Inheritance in Basidiomycete Fungi. Fungal Biol. Rev. 2015, 29, 209–219. [Google Scholar] [CrossRef]
- Casselton, L.A.; Condit, A. A Mitochondrial Mutant of Coprinus lagopus. J. Gen. Microbiol. 1972, 72, 521–527. [Google Scholar] [CrossRef] [Green Version]
- May, G.; Taylor, J.W. Patterns of Mating and Mitochondrial DNA Inheritance in the Agaric Basidiomycete Coprinus cinereus. Genetics 1988, 118, 213–220. [Google Scholar] [CrossRef]
- Specht, C.A.; Novotny, C.P.; Ullrich, R.C. Mitochondrial DNA of Schizophyllum commune: Restriction Map, Genetic Map, and Mode of Inheritance. Curr. Genet. 1992, 22, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Seefelder, S. Mitochondrial DNA and Its Inheritance in Pleurotus ostreatus and P. pulmonarius. Bot. Acta 1995, 108, 334–343. [Google Scholar] [CrossRef]
- Barroso, G.; Labarère, J. Genetic Evidence for Nonrandom Sorting of Mitochondria in the Basidiomycete Agrocybe aegerita. Appl. Environ. Microbiol. 1997, 63, 4686–4691. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.; Fukumasa-Nakai, Y. Mitochondrial DNA Inheritance in Sexual Crosses of Pleurotus ostreatus. Curr. Genet. 1996, 30, 549–552. [Google Scholar] [CrossRef]
- Wang, P.; Sha, T.; Zhang, Y.; Cao, Y.; Mi, F.; Liu, C.; Yang, D.; Tang, X.; He, X.; Dong, J.; et al. Frequent Heteroplasmy and Recombination in the Mitochondrial Genomes of the Basidiomycete Mushroom Thelephora ganbajun. Sci. Rep. 2017, 7, 1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista-Ferreira, J.L.C.; Economou, A.; Casselton, L.A. Mitochondrial Genetics of Coprinus: Recombination of Mitochondrial Genomes. Curr. Genet. 1983, 7, 405–407. [Google Scholar] [CrossRef]
- Saville, B.J.; Kohli, Y.; Anderson, J.B. MtDNA Recombination in a Natural Population. Proc. Natl. Acad. Sci. USA 1998, 95, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Zhang, Y.; Yu, Z.; Mi, F.; Liu, C.; Tang, X.; Long, Y.; He, X.; Wang, P.; Xu, J. Structure, Gene Flow, and Recombination among Geographic Populations of a Russula virescens Ally from Southwestern China. PLoS ONE 2013, 8, e73174. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, Y.; Pun, N. Mitochondrial Recombination in Natural Populations of the Button Mushroom Agaricus bisporus. Fungal Genet. Biol. 2013, 55, 92–97. [Google Scholar] [CrossRef]
- Li, M.; Liang, J.; Li, Y.; Feng, B.; Yang, Z.L.; James, T.Y.; Xu, J. Genetic Diversity of Dahongjun, the Commercially Important “‘big Red Mushroom’” from Southern China. PLoS ONE 2010, 5, e10684. [Google Scholar] [CrossRef] [Green Version]
- Pino-Bodas, R.; Laakso, I.; Stenroos, S. Genetic Variation and Factors Affecting the Genetic Structure of the Lichenicolous Fungus Heterocephalacria bachmannii (Filobasidiales, Basidiomycota). PLoS ONE 2017, 12, e0189603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Megarioti, A.H.; Kouvelis, V.N. The Coevolution of Fungal Mitochondrial Introns and Their Homing Endonucleases (GIY-YIG and LAGLIDADG). Genome Biol. Evol. 2020, 12, 1337–1354. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, B.S.; Stoddard, B.L. Homing Endonucleases: Structural and Functional Insight into the Catalysts of Intron/Intein Mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef]
- Jacquier, A.; Dujon, B. An Intron-Encoded Protein Is Active in a Gene Conversion Process That Spreads an Intron into a Mitochondrial Gene. Cell 1985, 41, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, B.L. Homing Endonucleases from Mobile Group i Introns: Discovery to Genome Engineering. Mob. DNA 2014, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Argast, G.M.; Stephens, K.M.; Emond, M.J.; Monnat, R.J. I-Ppol and I-CreI Homing Site Sequence Degeneracy Determined by Random Mutagenesis and Sequential in Vitro Enrichment. J. Mol. Biol. 1998, 280, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Pringle, A.; Vellinga, E.C. Last Chance to Know? Using Literature to Explore the Biogeography and Invasion Biology of the Death Cap Mushroom Amanita phalloides (Vaill. Ex Fr. :Fr.) Link. Biol. Invasions 2006, 8, 1131–1144. [Google Scholar] [CrossRef]
- Brandenburg, W.E.; Ward, K.J. Mushroom Poisoning Epidemiology in the United States. Mycologia 2018, 110, 637–641. [Google Scholar] [CrossRef]
- Li, Q.; He, X.; Ren, Y.; Xiong, C.; Jin, X.; Peng, L.; Huang, W. Comparative Mitogenome Analysis Reveals Mitochondrial Genome Differentiation in Ectomycorrhizal and Asymbiotic Amanita Species. Front. Microbiol. 2020, 11, 1382. [Google Scholar] [CrossRef]
- Wang, Y.-W.; McKeon, M.C.; Elmore, H.; Hess, J.; Golan, J.; Gage, H.; Mao, W.; Harrow, L.; Gonçalves, S.C.; Hull, C.M.; et al. Invasive Californian Death Caps Develop Mushrooms and Sporulate Unisexually and Bisexually. bioRxiv 2023. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De Novo Assembly of Organelle Genomes from Whole Genome Data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Lang, B.F.; Burger, G. MFannot. Available online: https://megasun.bch.umontreal.ca/apps/mfannot/ (accessed on 6 March 2020).
- Pritchard, A.E.; Sable, C.L.; Venuti, S.E.; Cummings, D.J. Analysis of NADH Dehydrogenase Proteins, ATPase Subunit 9, Cytochrome b, and Ribosomal Protein L14 Encoded in the Mitochondrial DNA of Paramecium. Nucleic Acids Res. 1990, 18, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chan, P.P.; Lowe, T.M. TRNAscan-SE: Searching for TRNA Genes in Genomic Sequences. In Gene Prediction: Methods and Protocols; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 1–14. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching during Cell Differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E. Pegas: An R Package for Population Genetics with an Integrated-Modular Approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Flutre, T.; Duprat, E.; Feuillet, C.; Quesneville, H. Considering Transposable Element Diversification in de Novo Annotation Approaches. PLoS ONE 2011, 6, e16526. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Patterson, M.; Garg, S.; O Fischer, S.; Pisanti, N.; Klau, G.; Schöenhuth, A.; Marschall, T. WhatsHap: Fast and Accurate Read-Based Phasing. bioRxiv 2016. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Akey, J.M.; Zhang, K.; Chakraborty, R.; Jin, L. Distribution of Recombination Crossovers and the Origin of Haplotype Blocks: The Interplay of Population History, Recombination, and Mutation. Am. J. Hum. Genet. 2002, 71, 1227–1234. [Google Scholar] [CrossRef] [Green Version]
- Maynard Smith, J. Analyzing the Mosaic Structure of Genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar]
- Piganeau, G.; Eyre-Walker, A. A Reanalysis of the Indirect Evidence for Recombination in Human Mitochondrial DNA. Heredity 2004, 92, 282–288. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A Simple and Robust Statistical Test for Detecting the Presence of Recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [Green Version]
- Piganeau, G.; Gardner, M.; Eyre-Walker, A. A Broad Survey of Recombination in Animal Mitochondria. Mol. Biol. Evol. 2004, 21, 2319–2325. [Google Scholar] [CrossRef] [Green Version]
- Posada, D. Evaluation of Methods for Detecting Recombination from DNA Sequences: Empirical Data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A Genetic Algorithm for Recombination Detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. Bioinformatics 2005, 5, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Auton, A.; McVean, G. Recombination Rate Estimation in the Presence of Hotspots. Genome Res. 2007, 17, 1219–1227. [Google Scholar] [CrossRef] [Green Version]
- Pyle, A.; Hudson, G.; Wilson, I.J.; Coxhead, J.; Smertenko, T.; Herbert, M.; Santibanez-Koref, M.; Chinnery, P.F. Extreme-Depth Re-Sequencing of Mitochondrial DNA Finds No Evidence of Paternal Transmission in Humans. PLoS Genet. 2015, 11, e1005040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Piganeau, G.; Eyre-Walker, A. Evidence for Variation in the Effective Population Size of Animal Mitochondrial DNA. PLoS ONE 2009, 4, e4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, P.W. Gametic Disequilibrium Measures: Proceed with Caution. Genetics 1987, 117, 331–341. [Google Scholar] [CrossRef]
- Eyre-Walker, A. Do Mitochondria Recombine in Humans? Philos. Trans. R. Soc. B Biol. Sci. 2000, 355, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, J. Mitochondrial Genome Polymorphisms in the Human Pathogenic Fungus Cryptococcus neoformans. Front. Microbiol. 2020, 11, 706. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhang, X.; Xie, B.; Lin, L.; Hsiang, T.; Lin, X.; Lin, Y.; Zhang, X.; Ma, Y.; Miao, W.; et al. Intra-Specific Comparison of Mitochondrial Genomes Reveals Host Gene Fragment Exchange via Intron Mobility in Tremella fuciformis. BMC Genom. 2020, 21, 426. [Google Scholar] [CrossRef]
- Burt, A.; Koufopanou, V. Homing Endonuclease Genes: The Rise and Fall and Rise Again of a Selfish Element. Curr. Opin. Genet. Dev. 2004, 14, 609–615. [Google Scholar] [CrossRef]
- Lee, H.H.; Ke, H.M.; Lin, C.Y.I.; Lee, T.J.; Chung, C.L.; Tsai, I.J.; Stajich, J. Evidence of Extensive Intraspecific Noncoding Reshuffling in a 169-Kb Mitochondrial Genome of a Basidiomycetous Fungus. Genome Biol. Evol. 2019, 11, 2774–2788. [Google Scholar] [CrossRef] [Green Version]
- Krasich, R.; Copeland, W.C. DNA Polymerases in the Mitochondria: A Critical Review of the Evidence. Front. Biosci. Landmark 2017, 22, 692–709. [Google Scholar] [CrossRef] [Green Version]
- Charlesworth, D.; Morgan, M.T.; Charlesworth, B. Mutation Accumulation in Finite Outbreeding and Inbreeding Populations. Genet. Res. 1993, 61, 39–56. [Google Scholar] [CrossRef] [Green Version]
- Fritsch, E.S.; Chabbert, C.D.; Klaus, B.; Steinmetz, L.M. A Genome–Wide Map of Mitochondrial DNA Recombination in Yeast. Genetics 2014, 198, 755–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlesworth, B.; Morgan, M.T.; Charlesworth, D. The Effect of Deleterious Mutations on Neutral Molecular Variation. Genetics 1993, 134, 1289–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-W.; Ané, C.C. KIMGENS: A Novel Method to Estimate Kinship in Organisms with Mixed Haploid Diploid Genetic Systems Robust to Population Structure. Bioinformatics 2022, 38, 3044–3050. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Whole Alignment | 70~95 kbp | |
|---|---|---|
| LDr2 10 kbp * | 7/10 | - |
| LDr2 20 kbp * | 4/5 | - |
| LDD’ 10 kbp * | 1/10 | - |
| LDD’ 20 kbp * | 3/5 | - |
| Maximum χ2 ** | 0 | 0 |
| PHI ** | - | 0.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-W.; Elmore, H.; Pringle, A. Uniparental Inheritance and Recombination as Strategies to Avoid Competition and Combat Muller’s Ratchet among Mitochondria in Natural Populations of the Fungus Amanita phalloides. J. Fungi 2023, 9, 476. https://doi.org/10.3390/jof9040476
Wang Y-W, Elmore H, Pringle A. Uniparental Inheritance and Recombination as Strategies to Avoid Competition and Combat Muller’s Ratchet among Mitochondria in Natural Populations of the Fungus Amanita phalloides. Journal of Fungi. 2023; 9(4):476. https://doi.org/10.3390/jof9040476
Chicago/Turabian StyleWang, Yen-Wen, Holly Elmore, and Anne Pringle. 2023. "Uniparental Inheritance and Recombination as Strategies to Avoid Competition and Combat Muller’s Ratchet among Mitochondria in Natural Populations of the Fungus Amanita phalloides" Journal of Fungi 9, no. 4: 476. https://doi.org/10.3390/jof9040476