
CeGAL: Redefining a Widespread Fungal-Specific Transcription Factor Family Using an In Silico Error-Tracking Approach
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of SGD Sequences
2.2. Collection of UniprotKB Sequences
2.3. Construction of BLAST Databases of Proteins with Full Domain Architecture
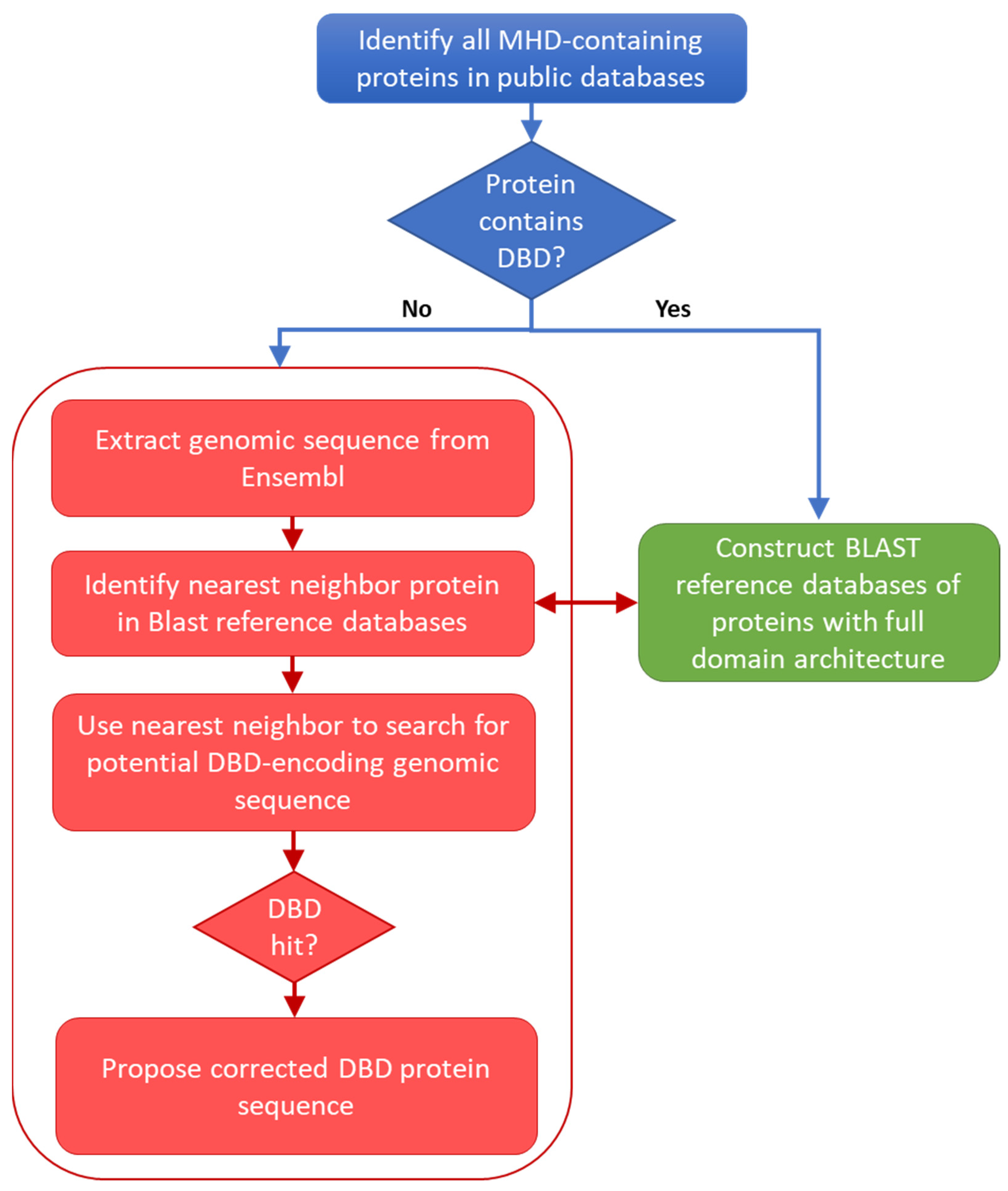
2.4. Extraction of Genomic Sequences
2.5. Identification of Nearest-Neighbor Reference Sequences
2.6. Identification of Missing DBD Sequences
3. Results
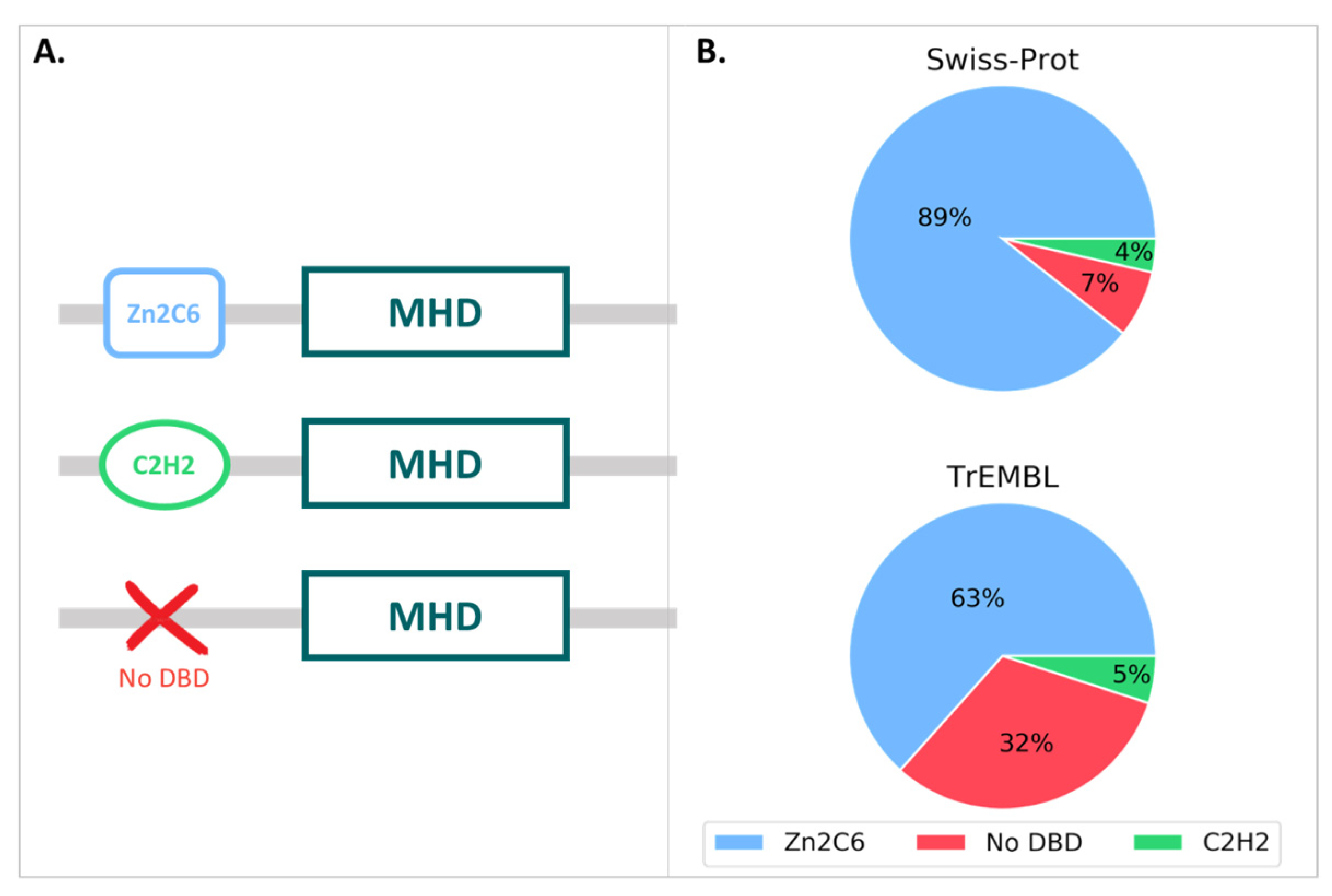
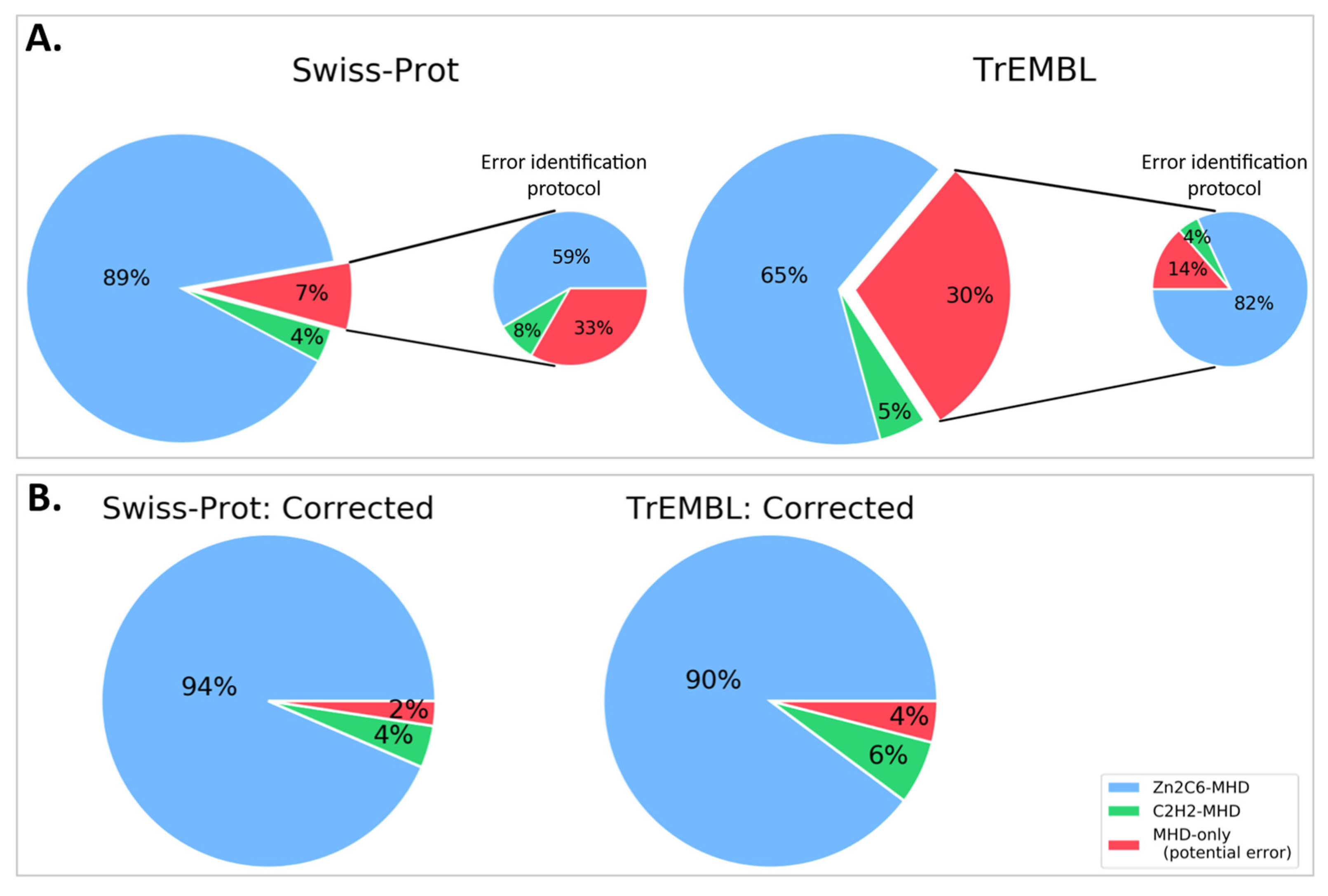
3.1. MHD-Containing Proteins in the SGD Database
3.2. MHD-Containing Proteins in the UniProt Database
3.3. Manual Analysis of the 12 Swiss-Prot MHD-Only Sequences
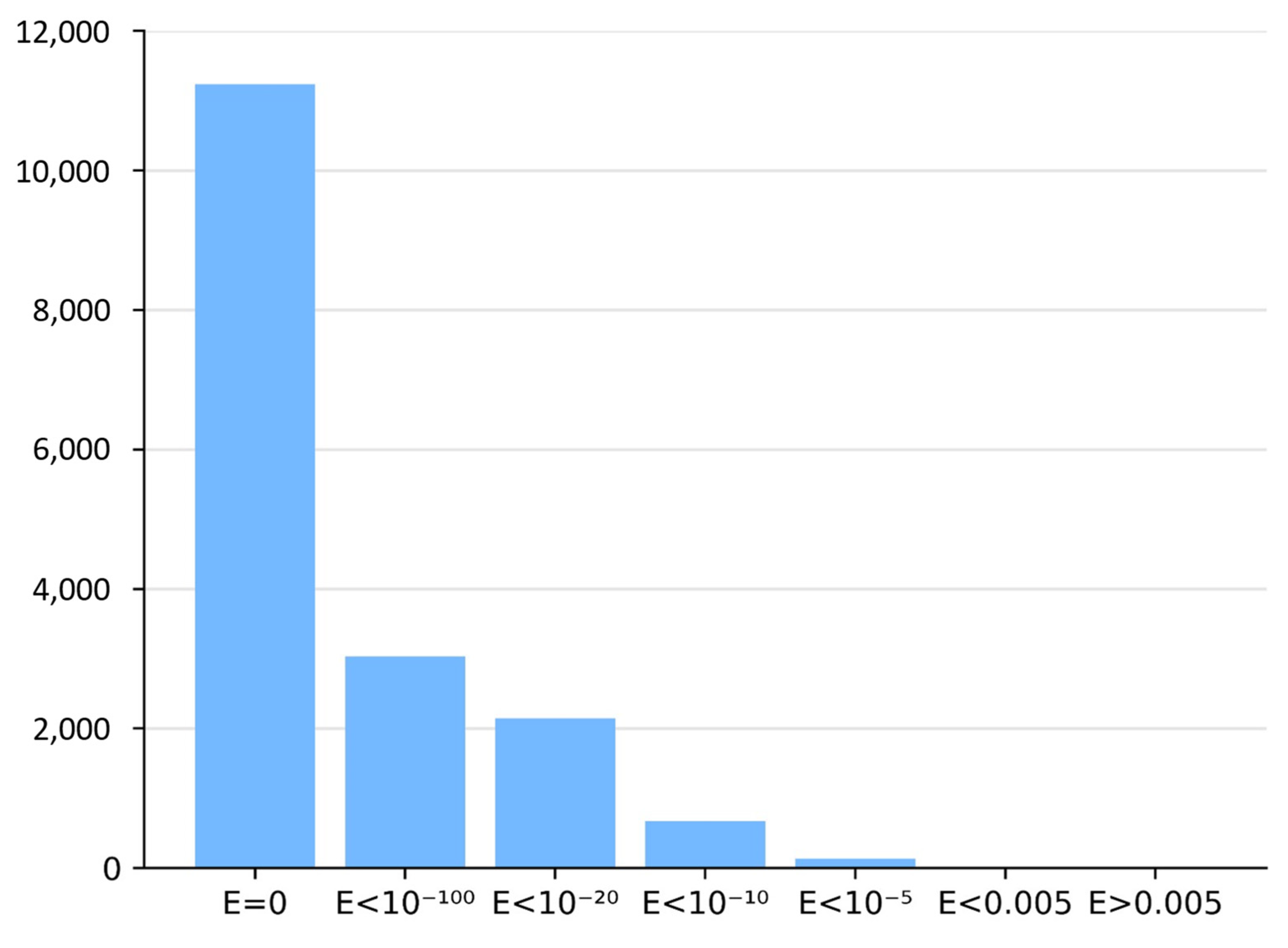
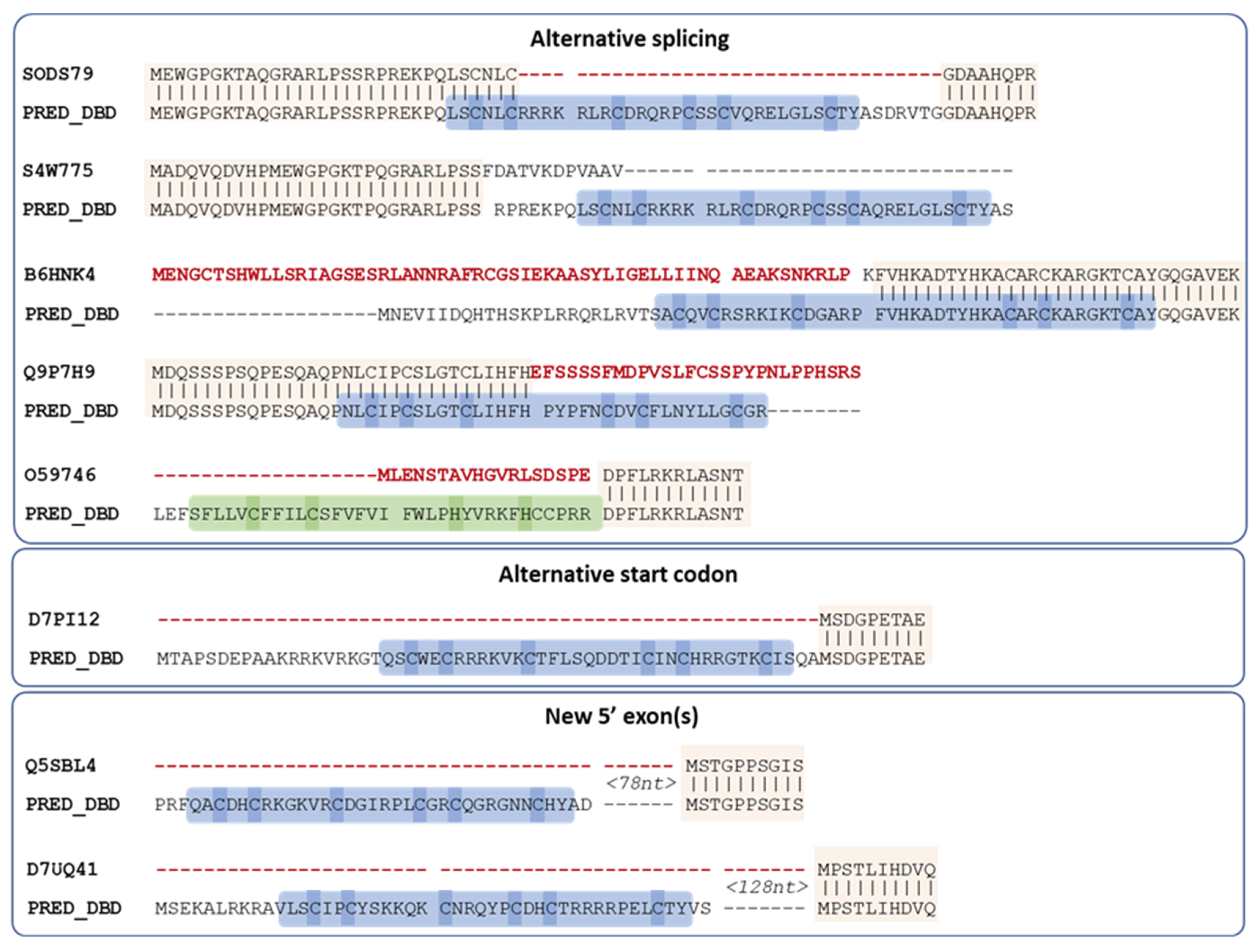
3.4. Automatic Analysis of 16,760 TrEMBL MHD-Only Sequences
3.5. Reassessment of Domain Pairs in MHD-Containing Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lehninger, A.; Nelson, D.; Cox, M. Principles of Biochemistry, 2nd ed.; Worth: New York, NY, USA, 1993. [Google Scholar]
- Schjerling, P.; Holmberg, S. Comparative Amino Acid Sequence Analysis of the C6 Zinc Cluster Family of Transcriptional Regulators. Nucleic Acids Res. 1996, 24, 4599–4607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallee, B.L.; Coleman, J.E.; Auld, D.S. Zinc Fingers, Zinc Clusters, and Zinc Twists in DNA-Binding Protein Domains. Proc. Natl. Acad. Sci. USA 1991, 88, 999–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacPherson, S.; Larochelle, M.; Turcotte, B. A Fungal Family of Transcriptional Regulators: The Zinc Cluster Proteins. Microbiol. Mol. Biol. Rev. 2006, 70, 583–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelest, E. Transcription Factors in Fungi. FEMS Microbiol. Lett. 2008, 286, 145–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelest, E. Transcription Factors in Fungi: TFome Dynamics, Three Major Families, and Dual-Specificity TFs. Front. Genet. 2017, 8, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tianqiao, S.; Xiong, Z.; You, Z.; Dong, L.; Jiaoling, Y.; Junjie, Y.; Mina, Y.; Huijuan, C.; Mingli, Y.; Xiayan, P.; et al. Genome-Wide Identification of Zn2Cys6 Class Fungal-Specific Transcription Factors (ZnFTFs) and Functional Analysis of UvZnFTF1 in Ustilaginoidea Virens. Rice Sci. 2021, 28, 567–578. [Google Scholar] [CrossRef]
- Todd, R.B.; Zhou, M.; Ohm, R.A.; Leeggangers, H.A.; Visser, L.; de Vries, R.P. Prevalence of Transcription Factors in Ascomycete and Basidiomycete Fungi. BMC Genom. 2014, 15, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, E.; Singh, K.B.; Oliver, R.P.; Tan, K. Transcription Factor Control of Virulence in Phytopathogenic Fungi. Mol. Plant Pathol. 2021, 22, 858–881. [Google Scholar] [CrossRef]
- Piskacek, M.; Havelka, M.; Rezacova, M.; Knight, A. The 9aaTAD Transactivation Domains: From Gal4 to P53. PLoS ONE 2016, 11, e0162842. [Google Scholar] [CrossRef] [Green Version]
- Todd, R.B.; Andrianopoulos, A. Evolution of a Fungal Regulatory Gene Family: The Zn(II)2Cys6 Binuclear Cluster DNA Binding Motif. Fungal Genet. Biol. 1997, 21, 388–405. [Google Scholar] [CrossRef]
- Poch, O. Conservation of a Putative Inhibitory Domain in the GAL4 Family Members. Gene 1997, 184, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, J.J.; Sorger, P.K.; Harrison, S.C. Crystal Structure of the Yeast Inner Kinetochore Subunit Cep3p. Structure 2007, 15, 1422–1430. [Google Scholar] [CrossRef] [Green Version]
- Purvis, A.; Singleton, M.R. Insights into Kinetochore–DNA Interactions from the Structure of Cep3Δ. EMBO Rep. 2008, 9, 56–62. [Google Scholar] [CrossRef]
- Näär, A.M.; Thakur, J.K. Nuclear Receptor-like Transcription Factors in Fungi. Genes Dev. 2009, 23, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Turcotte, B.; Liang, X.B.; Robert, F.; Soontorngun, N. Transcriptional Regulation of Nonfermentable Carbon Utilization in Budding Yeast. FEMS Yeast Res. 2009, 10, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollapour, M.; Piper, P.W. Activity of the Yeast Zinc-Finger Transcription Factor War1 Is Lost with Alanine Mutation of Two Putative Phosphorylation Sites in the Activation Domain. Yeast Chichester Engl. 2012, 29, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Tanwar, D.K.; Penha, E.D.S.; Wolf, Y.I.; Koonin, E.V.; Basu, M.K. Grammar of Protein Domain Architectures. Proc. Natl. Acad. Sci. USA 2019, 116, 3636–3645. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro Protein Families and Domains Database: 20 Years On. Nucleic Acids Res. 2021, 49, D344–D354. [Google Scholar] [CrossRef]
- Zhang, W.-Q.; Gui, Y.-J.; Short, D.P.G.; Li, T.-G.; Zhang, D.-D.; Zhou, L.; Liu, C.; Bao, Y.-M.; Subbarao, K.V.; Chen, J.-Y.; et al. Verticillium Dahliae Transcription Factor VdFTF1 Regulates the Expression of Multiple Secreted Virulence Factors and Is Required for Full Virulence in Cotton. Mol. Plant Pathol. 2018, 19, 841–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etxebeste, O. Transcription Factors in the Fungus Aspergillus Nidulans: Markers of Genetic Innovation, Network Rewiring and Conflict between Genomics and Transcriptomics. J. Fungi 2021, 7, 600. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.R.; Wong, E.D.; Nash, R.S.; Aleksander, S.; Alexander, M.; Douglass, E.; Karra, K.; Miyasato, S.R.; Simison, M.; Skrzypek, M.S.; et al. New Data and Collaborations at the Saccharomyces Genome Database: Updated Reference Genome, Alleles, and the Alliance of Genome Resources. Genetics 2021, 220, iyab224. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.S.; Eddy, S.R.; Portugaly, E. Hidden Markov Model Speed Heuristic and Iterative HMM Search Procedure. BMC Bioinform. 2010, 11, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 50, D161–D164. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-K.; Ehrlich, K.C. Genome-Wide Analysis of the Zn(II)2Cys6 Zinc Cluster-Encoding Gene Family in Aspergillus Flavus. Appl. Microbiol. Biotechnol. 2013, 97, 4289–4300. [Google Scholar] [CrossRef] [PubMed]
- Galagan, J.E.; Henn, M.R.; Ma, L.-J.; Cuomo, C.A.; Birren, B. Genomics of the Fungal Kingdom: Insights into Eukaryotic Biology. Genome Res. 2005, 15, 1620–1631. [Google Scholar] [CrossRef] [Green Version]
- International Society for Biocuration. Biocuration: Distilling Data into Knowledge. PLoS Biol. 2018, 16, e2002846. [Google Scholar] [CrossRef]
- Gabrielsen, A.M. Openness and Trust in Data-Intensive Science: The Case of Biocuration. Med. Health Care Philos. 2020, 23, 497–504. [Google Scholar] [CrossRef]
- Chen, Q.; Britto, R.; Erill, I.; Jeffery, C.J.; Liberzon, A.; Magrane, M.; Onami, J.; Robinson-Rechavi, M.; Sponarova, J.; Zobel, J.; et al. Quality Matters: Biocuration Experts on the Impact of Duplication and Other Data Quality Issues in Biological Databases. Genom. Proteom. Bioinform. 2020, 18, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L. Next-Generation Genome Annotation: We Still Struggle to Get It Right. Genome Biol. 2019, 20, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ejigu, G.F.; Jung, J. Review on the Computational Genome Annotation of Sequences Obtained by Next-Generation Sequencing. Biology 2020, 9, 295. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Frankish, A.; Flicek, P. Progress, Challenges, and Surprises in Annotating the Human Genome. Annu. Rev. Genom. Hum. Genet. 2020, 21, 55–79. [Google Scholar] [CrossRef] [PubMed]
- Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. A Benchmark Study of Ab Initio Gene Prediction Methods in Diverse Eukaryotic Organisms. BMC Genom. 2020, 21, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poux, S.; Magrane, M.; Arighi, C.N.; Bridge, A.; O’Donovan, C.; Laiho, K. Expert Curation in UniProtKB: A Case Study on Dealing with Conflicting and Erroneous Data. Database J. Biol. Databases Curation 2014, 2014, bau016. [Google Scholar] [CrossRef] [Green Version]
- Prosdocimi, F.; Linard, B.; Pontarotti, P.; Poch, O.; Thompson, J.D. Controversies in Modern Evolutionary Biology: The Imperative for Error Detection and Quality Control. BMC Genom. 2012, 13, 5. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Scalzitti, N.; Jeannin-Girardon, A.; Collet, P.; Poch, O.; Thompson, J.D. Understanding the Causes of Errors in Eukaryotic Protein-Coding Gene Prediction: A Case Study of Primate Proteomes. BMC Bioinform. 2020, 21, 513. [Google Scholar] [CrossRef]
- Zhang, D.; Guelfi, S.; Garcia-Ruiz, S.; Costa, B.; Reynolds, R.H.; D’Sa, K.; Liu, W.; Courtin, T.; Peterson, A.; Jaffe, A.E.; et al. Incomplete Annotation Has a Disproportionate Impact on Our Understanding of Mendelian and Complex Neurogenetic Disorders. Sci. Adv. 2020, 6, eaay8299. [Google Scholar] [CrossRef]
- Nagy, A.; Patthy, L. MisPred: A Resource for Identification of Erroneous Protein Sequences in Public Databases. Database 2013, 2013, bat053. [Google Scholar] [CrossRef]
- Evans, T.; Loose, M. AlignWise: A Tool for Identifying Protein-Coding Sequence and Correcting Frame-Shifts. BMC Bioinform. 2015, 16, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drăgan, M.-A.; Moghul, I.; Priyam, A.; Bustos, C.; Wurm, Y. GeneValidator: Identify Problems with Protein-Coding Gene Predictions. Bioinformatics 2016, 32, 1559–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, C.; Berzuini, C.; Bashton, M.; Gough, J.; Teichmann, S.A. Supra-Domains: Evolutionary Units Larger than Single Protein Domains. J. Mol. Biol. 2004, 336, 809–823. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, W.A.; Chen, K.; Hou, T.; Wang, W. On the Detection of Functionally Coherent Groups of Protein Domains with an Extension to Protein Annotation. BMC Bioinform. 2007, 8, 390. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, J.; Zaverucha, G.; Vaquero, C.; Carbone, A. Improvement in Protein Domain Identification Is Reached by Breaking Consensus, with the Agreement of Many Profiles and Domain Co-Occurrence. PLoS Comput. Biol. 2016, 12, e1005038. [Google Scholar] [CrossRef]
- Menichelli, C.; Gascuel, O.; Bréhélin, L. Improving Pairwise Comparison of Protein Sequences with Domain Co-Occurrence. PLoS Comput. Biol. 2018, 14, e1005889. [Google Scholar] [CrossRef] [Green Version]
- Monteiro, P.T.; Oliveira, J.; Pais, P.; Antunes, M.; Palma, M.; Cavalheiro, M.; Galocha, M.; Godinho, C.P.; Martins, L.C.; Bourbon, N.; et al. YEASTRACT+: A Portal for Cross-Species Comparative Genomics of Transcription Regulation in Yeasts. Nucleic Acids Res. 2020, 48, D642–D649. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Si, J.; Zhao, R.; Wu, R. An Overview of the Prediction of Protein DNA-Binding Sites. Int. J. Mol. Sci. 2015, 16, 5194–5215. [Google Scholar] [CrossRef]
- Lambert, S.A.; Yang, A.W.H.; Sasse, A.; Cowley, G.; Albu, M.; Caddick, M.X.; Morris, Q.D.; Weirauch, M.T.; Hughes, T.R. Similarity Regression Predicts Evolution of Transcription Factor Sequence Specificity. Nat. Genet. 2019, 51, 981–989. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.D.; Miyasato, S.R.; Aleksander, S.; Karra, K.; Nash, R.S.; Skrzypek, M.S.; Weng, S.; Engel, S.R.; Cherry, J.M. Saccharomyces Genome Database Update: Server Architecture, Pan-Genome Nomenclature, and External Resources. Genetics 2023, iyac191. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain Annotation | S288C Reference Strain | 47 Other Strains |
|---|---|---|
| Zn2C6–MHD | 44 (100%) | 1540 (86%) |
| MHD-only | 0 (0%) | 253 (14%) |
| Total | 44 | 1793 |
| Error Type | Probable Cause of Error | MHD-Only |
|---|---|---|
| Genome sequence error | Frameshift | 190 (75%) |
| 2 or more scaffolds | 9 (4%) | |
| Gene prediction error | Wrong start codon | 46 (18%) |
| Undetermined | Undetermined | 8 (3%) |
| Total | 253 (100%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayer, C.; Vogt, A.; Uslu, T.; Scalzitti, N.; Chennen, K.; Poch, O.; Thompson, J.D. CeGAL: Redefining a Widespread Fungal-Specific Transcription Factor Family Using an In Silico Error-Tracking Approach. J. Fungi 2023, 9, 424. https://doi.org/10.3390/jof9040424
Mayer C, Vogt A, Uslu T, Scalzitti N, Chennen K, Poch O, Thompson JD. CeGAL: Redefining a Widespread Fungal-Specific Transcription Factor Family Using an In Silico Error-Tracking Approach. Journal of Fungi. 2023; 9(4):424. https://doi.org/10.3390/jof9040424
Chicago/Turabian StyleMayer, Claudine, Arthur Vogt, Tuba Uslu, Nicolas Scalzitti, Kirsley Chennen, Olivier Poch, and Julie D. Thompson. 2023. "CeGAL: Redefining a Widespread Fungal-Specific Transcription Factor Family Using an In Silico Error-Tracking Approach" Journal of Fungi 9, no. 4: 424. https://doi.org/10.3390/jof9040424