

Sources of Antifungal Drugs

Abstract
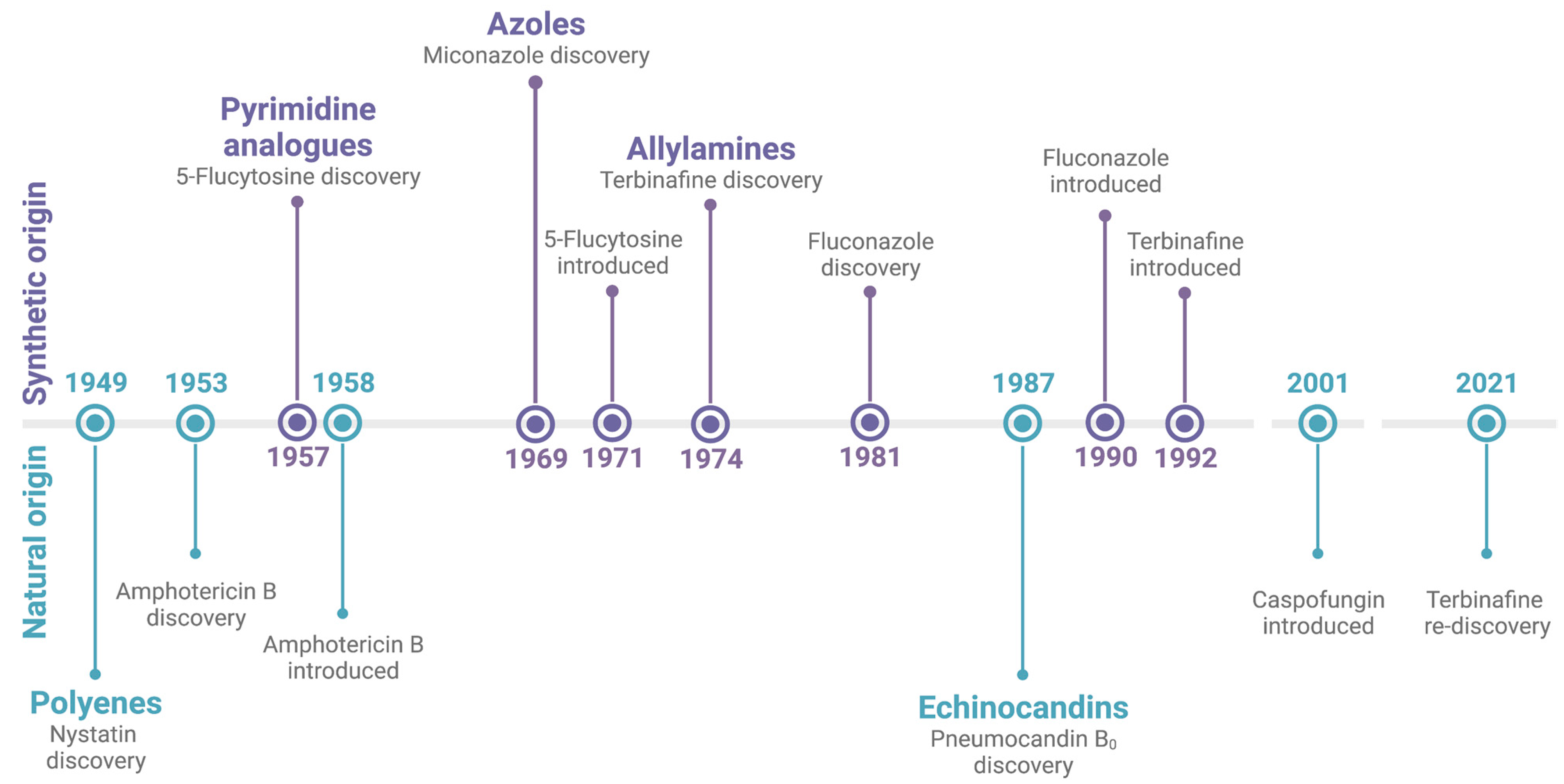
:1. Introduction
2. Natural Products
3. Synthetic Compounds
4. Non-Traditional Antifungal Options
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Alcazar-Fuoli, L.; Mellado, E. Current status of antifungal resistance and its impact on clinical practice. Br. J. Haematol. 2014, 166, 471–484. [Google Scholar] [CrossRef]
- Armstrong-James, D.; Meintjes, G.; Brown, G.D. A neglected epidemic: Fungal infections in HIV/AIDS. Trends Microbiol. 2014, 22, 120–127. [Google Scholar] [CrossRef]
- Benedict, K.; Jackson, B.R.; Chiller, T.; Beer, K.D. Estimation of Direct Healthcare Costs of Fungal Diseases in the United States. Clin. Infect. Dis. 2019, 68, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Douglas, L.J. Candida biofilms and their role in infection. Trends Microbiol. 2003, 11, 30–36. [Google Scholar] [CrossRef]
- Ramage, G.; Martinez, J.P.; Lopez-Ribot, J.L. Candida biofilms on implanted biomaterials: A clinically significant problem. FEMS Yeast Res. 2006, 6, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Sanglard, D. Emerging Threats in Antifungal-Resistant Fungal Pathogens. Front. Med. 2016, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Wingard, J.R.; Kubilis, P.; Lee, L.; Yee, G.; White, M.; Walshe, L.; Bowden, R.; Anaissie, E.; Hiemenz, J.; Lister, J. Clinical significance of nephrotoxicity in patients treated with amphotericin B for suspected or proven aspergillosis. Clin. Infect. Dis. 1999, 29, 1402–1407. [Google Scholar] [CrossRef]
- Lemke, A.; Kiderlen, A.F.; Kayser, O. Amphotericin B. Appl. Microbiol. Biotechnol. 2005, 68, 151–162. [Google Scholar] [CrossRef]
- Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Amphotericin biosynthesis in Streptomyces nodosus: Deductions from analysis of polyketide synthase and late genes. Chem. Biol. 2001, 8, 713–723. [Google Scholar] [CrossRef]
- Anderson, T.M.; Clay, M.C.; Cioffi, A.G.; Diaz, K.A.; Hisao, G.S.; Tuttle, M.D.; Nieuwkoop, A.J.; Comellas, G.; Maryum, N.; Wang, S.; et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol. 2014, 10, 400–406. [Google Scholar] [CrossRef]
- Mesa-Arango, A.C.; Trevijano-Contador, N.; Roman, E.; Sanchez-Fresneda, R.; Casas, C.; Herrero, E.; Arguelles, J.C.; Pla, J.; Cuenca-Estrella, M.; Zaragoza, O. The production of reactive oxygen species is a universal action mechanism of Amphotericin B against pathogenic yeasts and contributes to the fungicidal effect of this drug. Antimicrob. Agents Chemother. 2014, 58, 6627–6638. [Google Scholar] [CrossRef] [Green Version]
- Polak, A.; Scholer, H.J. Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy 1975, 21, 113–130. [Google Scholar] [CrossRef]
- Waldorf, A.R.; Polak, A. Mechanisms of action of 5-fluorocytosine. Antimicrob. Agents Chemother. 1983, 23, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Louie, A.; Liu, W.; Miller, D.A.; Sucke, A.C.; Liu, Q.F.; Drusano, G.L.; Mayers, M.; Miller, M.H. Efficacies of high-dose fluconazole plus amphotericin B and high-dose fluconazole plus 5-fluorocytosine versus amphotericin B, fluconazole, and 5-fluorocytosine monotherapies in treatment of experimental endocarditis, endophthalmitis, and pyelonephritis due to Candida albicans. Antimicrob. Agents Chemother. 1999, 43, 2831–2840. [Google Scholar] [CrossRef] [Green Version]
- Perfect, J.R.; Dismukes, W.E.; Dromer, F.; Goldman, D.L.; Graybill, J.R.; Hamill, R.J.; Harrison, T.S.; Larsen, R.A.; Lortholary, O.; Nguyen, M.H.; et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of america. Clin. Infect. Dis. 2010, 50, 291–322. [Google Scholar] [CrossRef] [Green Version]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J.; et al. Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef] [Green Version]
- Maertens, J.A. History of the development of azole derivatives. Clin. Microbiol. Infect. 2004, 10 (Suppl. S1), 1–10. [Google Scholar] [CrossRef] [Green Version]
- Woolley, D.W. Some Biological Effects Produced by Benzimidazole and Their Reversal by Purines. J. Biol. Chem. 1944, 152, 225–232. [Google Scholar] [CrossRef]
- Sheehan, D.J.; Hitchcock, C.A.; Sibley, C.M. Current and emerging azole antifungal agents. Clin. Microbiol. Rev. 1999, 12, 40–79. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.; Krysan, D.J. Drug resistance and tolerance in fungi. Nat. Rev. Microbiol. 2020, 18, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Vanden Bossche, H.; Koymans, L. Cytochromes P450 in fungi. Mycoses 1998, 41 (Suppl. S1), 32–38. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.E. Pharmacology of the allylamines. J. Am. Acad. Dermatol. 1990, 23, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Jessup, C.J.; Ryder, N.S.; Ghannoum, M.A. An evaluation of the in vitro activity of terbinafine. Med. Mycol. 2000, 38, 155–159. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed, S.E.; Abdelaziz, N.A.; Osman, H.H.; El-Housseiny, G.S.; Aleissawy, A.E.; Aboshanab, K.M. Lysinibacillus Isolate MK212927: A Natural Producer of Allylamine Antifungal ‘Terbinafine’. Molecules 2021, 27, 201. [Google Scholar] [CrossRef]
- Balkovec, J.M.; Hughes, D.L.; Masurekar, P.S.; Sable, C.A.; Schwartz, R.E.; Singh, S.B. Discovery and development of first in class antifungal caspofungin (CANCIDAS(R))—A case study. Nat. Prod. Rep. 2014, 31, 15–34. [Google Scholar] [CrossRef]
- Nyfeler, R.; Keller-Schierlein, W. Stoffwechselprodukte von Mikroorganismen 143. Mitteilung. Echinocandin B, ein neuartiges Polypeptid-Antibioticum aus Aspergillus nidulans var. echinulatus: Isolierung und Bausteine. [Metabolites of microorganisms. 143. Echinocandin B, a novel polypeptide-antibiotic from Aspergillus nidulans var. echinulatus: Isolation and structural components]. Helv. Chim. Acta 1974, 57, 2459–2477. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Zagaliotis, P.; Walsh, T.J. Novel antifungal agents in clinical trials. F1000Res 2021, 10, 507. [Google Scholar] [CrossRef]
- Fera, M.T.; La Camera, E.; De Sarro, A. New triazoles and echinocandins: Mode of action, in vitro activity and mechanisms of resistance. Expert Rev. Anti-Infect. Ther. 2009, 7, 981–998. [Google Scholar] [CrossRef]
- Eagle, H. A Paradoxical Zone Phenomenon in the Bactericidal Action of Penicillin in vitro. Science 1948, 107, 44–45. [Google Scholar] [CrossRef]
- Vanstraelen, K.; Lagrou, K.; Maertens, J.; Wauters, J.; Willems, L.; Spriet, I. The Eagle-like effect of echinocandins: What’s in a name? Expert. Rev. Anti-Infect. Ther. 2013, 11, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Capoor, M.R.; Bal, A.M. Echinocandins. In Comprehensive Pharmacology; Kenakin, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 363–371. [Google Scholar] [CrossRef]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017, 16, 603–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Daele, R.; Spriet, I.; Wauters, J.; Maertens, J.; Mercier, T.; Van Hecke, S.; Bruggemann, R. Antifungal drugs: What brings the future? Med. Mycol. 2019, 57, S328–S343. [Google Scholar] [CrossRef] [Green Version]
- Hoenigl, M.; Sprute, R.; Egger, M.; Arastehfar, A.; Cornely, O.A.; Krause, R.; Lass-Florl, C.; Prattes, J.; Spec, A.; Thompson, G.R., 3rd; et al. The Antifungal Pipeline: Fosmanogepix, Ibrexafungerp, Olorofim, Opelconazole, and Rezafungin. Drugs 2021, 81, 1703–1729. [Google Scholar] [CrossRef]
- Georgacopoulos, O.; Nunnally, N.S.; Ransom, E.M.; Law, D.; Birch, M.; Lockhart, S.R.; Berkow, E.L. In Vitro Activity of Novel Antifungal Olorofim against Filamentous Fungi and Comparison to Eight Other Antifungal Agents. J. Fungi 2021, 7, 378. [Google Scholar] [CrossRef]
- Oliver, J.D.; Sibley, G.E.M.; Beckmann, N.; Dobb, K.S.; Slater, M.J.; McEntee, L.; du Pre, S.; Livermore, J.; Bromley, M.J.; Wiederhold, N.P.; et al. F901318 represents a novel class of antifungal drug that inhibits dihydroorotate dehydrogenase. Proc. Natl. Acad. Sci. USA 2016, 113, 12809–12814. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, M.; Moloney, M.; Soltow, Q.A.; Pillar, C.M.; Shaw, K.J. Evaluation of Resistance Development to the Gwt1 Inhibitor Manogepix (APX001A) in Candida Species. Antimicrob. Agents Chemother. 2019, 64, e01387-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, K.J.; Ibrahim, A.S. Fosmanogepix: A Review of the First-in-Class Broad Spectrum Agent for the Treatment of Invasive Fungal Infections. J. Fungi 2020, 6, 239. [Google Scholar] [CrossRef]
- Hoy, S.M. Oteseconazole: First Approval. Drugs 2022, 82, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Warrilow, A.G.; Hull, C.M.; Parker, J.E.; Garvey, E.P.; Hoekstra, W.J.; Moore, W.R.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob. Agents Chemother. 2014, 58, 7121–7127. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.C.; Hawkins, N.J.; Sanglard, D.; Gurr, S.J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 2018, 360, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Ademe, M.; Girma, F. Candida auris: From Multidrug Resistance to Pan-Resistant Strains. Infect. Drug Resist. 2020, 13, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, A.G. Responsibilities and Opportunities of the Private Practitioner in Preventive Medicine. Can. Med. Assoc. J. 1929, 20, 11–13. [Google Scholar] [PubMed]
- Schatz, A.; Bugle, E.; Waksman, S.A. Streptomycin, a Substance Exhibiting Antibiotic Activity Against Gram-Positive and Gram-Negative Bacteria. Exp. Biol. Med. 1944, 55, 66–69. [Google Scholar] [CrossRef]
- Waksman, S.A.; Lechevalier, H.A.; Schaffner, C.P. Candicidin and other polyenic antifungal antibiotics. Bull. World Health Organ. 1965, 33, 219–226. [Google Scholar] [PubMed]
- Hazen, E.L.; Brown, R. Fungicidin, an antibiotic produced by a soil actinomycete. Proc. Soc. Exp. Biol. Med. 1951, 76, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J.; Clardy, J. Natural products: Beyond grind and find. Nat. Chem. 2009, 1, 261–263. [Google Scholar] [CrossRef]
- Roemer, T.; Xu, D.; Singh, S.B.; Parish, C.A.; Harris, G.; Wang, H.; Davies, J.E.; Bills, G.F. Confronting the challenges of natural product-based antifungal discovery. Chem. Biol. 2011, 18, 148–164. [Google Scholar] [CrossRef]
- Nishikawa, J.L.; Boeszoermenyi, A.; Vale-Silva, L.A.; Torelli, R.; Posteraro, B.; Sohn, Y.J.; Ji, F.; Gelev, V.; Sanglard, D.; Sanguinetti, M.; et al. Inhibiting fungal multidrug resistance by disrupting an activator-Mediator interaction. Nature 2016, 530, 485–489. [Google Scholar] [CrossRef] [Green Version]
- Pierce, C.G.; Chaturvedi, A.K.; Lazzell, A.L.; Powell, A.T.; Saville, S.P.; McHardy, S.F.; Lopez-Ribot, J.L. A Novel Small Molecule Inhibitor of Candida albicans Biofilm Formation, Filamentation and Virulence with Low Potential for the Development of Resistance. NPJ Biofilms Microbiomes 2015, 1, 15012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vila, T.; Lopez-Ribot, J.L. Screening the Pathogen Box for Identification of Candida albicans Biofilm Inhibitors. Antimicrob. Agents Chemother. 2017, 61, e02006–e02016. [Google Scholar] [CrossRef] [Green Version]
- Pouliot, M.; Jeanmart, S. Pan Assay Interference Compounds (PAINS) and Other Promiscuous Compounds in Antifungal Research. J. Med. Chem. 2016, 59, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Claessen, D.; Rozen, D.E.; Kuipers, O.P.; Sogaard-Andersen, L.; van Wezel, G.P. Bacterial solutions to multicellularity: A tale of biofilms, filaments and fruiting bodies. Nat. Rev. Microbiol. 2014, 12, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Chandra, J.; Kuhn, D.M.; Mukherjee, P.K.; Hoyer, L.L.; McCormick, T.; Ghannoum, M.A. Biofilm formation by the fungal pathogen Candida albicans: Development, architecture, and drug resistance. J. Bacteriol. 2001, 183, 5385–5394. [Google Scholar] [CrossRef] [Green Version]
- Desai, J.V.; Mitchell, A.P.; Andes, D.R. Fungal biofilms, drug resistance, and recurrent infection. Cold Spring Harb. Perspect. Med. 2014, 4, a019729. [Google Scholar] [CrossRef] [Green Version]
- Mathe, L.; Van Dijck, P. Recent insights into Candida albicans biofilm resistance mechanisms. Curr. Genet. 2013, 59, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, J.; Mukherjee, P.K.; Leidich, S.D.; Faddoul, F.F.; Hoyer, L.L.; Douglas, L.J.; Ghannoum, M.A. Antifungal resistance of candidal biofilms formed on denture acrylic in vitro. J. Dent. Res. 2001, 80, 903–908. [Google Scholar] [CrossRef]
- Hawser, S.P.; Douglas, L.J. Resistance of Candida albicans biofilms to antifungal agents in vitro. Antimicrob. Agents Chemother. 1995, 39, 2128–2131. [Google Scholar] [CrossRef]
- Ramage, G.; Wickes, B.L.; Lopez-Ribot, J.L. Biofilms of Candida albicans and their associated resistance to antifungal agents. Am. Clin. Lab. 2001, 20, 42–44. [Google Scholar] [PubMed]
- Nett, J.; Lincoln, L.; Marchillo, K.; Massey, R.; Holoyda, K.; Hoff, B.; VanHandel, M.; Andes, D. Putative role of beta-1,3 glucans in Candida albicans biofilm resistance. Antimicrob. Agents Chemother. 2007, 51, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taff, H.T.; Mitchell, K.F.; Edward, J.A.; Andes, D.R. Mechanisms of Candida biofilm drug resistance. Future Microbiol. 2013, 8, 1325–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, L.M.; Fuerst, P. The future of high-throughput screening. J. Biomol. Screen. 2008, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, C.C.; Britt, J.R.; Evans, J.R.; Akee, R.K.; Whitt, J.A.; Trinh, S.K.; Harris, M.J.; Thompson, J.R.; Ewing, T.L.; Shipley, S.M.; et al. NCI Program for Natural Product Discovery: A Publicly-Accessible Library of Natural Product Fractions for High-Throughput Screening. ACS Chem. Biol. 2018, 13, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Abba, Y.; Hassim, H.; Hamzah, H.; Noordin, M.M. Antiviral Activity of Resveratrol against Human and Animal Viruses. Adv. Virol. 2015, 2015, 184241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, M.R.; Parks, R.J. Curcumin as an Antiviral Agent. Viruses 2020, 12, 1242. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.M.; Dahlin, J.L.; Bisson, J.; Graham, J.; Pauli, G.F.; Walters, M.A. The Essential Medicinal Chemistry of Curcumin. J. Med. Chem. 2017, 60, 1620–1637. [Google Scholar] [CrossRef]
- Vestergaard, M.; Ingmer, H. Antibacterial and antifungal properties of resveratrol. Int. J. Antimicrob. Agents 2019, 53, 716–723. [Google Scholar] [CrossRef]
- Dziekan, J.M.; Yu, H.; Chen, D.; Dai, L.; Wirjanata, G.; Larsson, A.; Prabhu, N.; Sobota, R.M.; Bozdech, Z.; Nordlund, P. Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci. Transl. Med. 2019, 11, eaau3174. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Huang, L.; Li, J.; Fan, Q.; Long, Y.; Li, Y.; Zhou, B. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS ONE 2010, 5, e9582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, E.W. Trading molecules and tracking targets in symbiotic interactions. Nat. Chem. Biol. 2008, 4, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bills, G.; Li, Y.; Chen, L.; Yue, Q.; Niu, X.M.; An, Z. New insights into the echinocandins and other fungal non-ribosomal peptides and peptaibiotics. Nat. Prod. Rep. 2014, 31, 1348–1375. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.A. Horizontal gene transfer in fungi. FEMS Microbiol. Lett. 2012, 329, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, M.A.; Messer, S.A.; Rhomberg, P.R.; Borroto-Esoda, K.; Castanheira, M. Differential Activity of the Oral Glucan Synthase Inhibitor SCY-078 against Wild-Type and Echinocandin-Resistant Strains of Candida Species. Antimicrob. Agents Chemother. 2017, 61, e00161-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Meier-Kolthoff, J.P.; Klenk, H.P.; Clement, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, Physiology, and Natural Products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Liu, G.; Chater, K.F.; Chandra, G.; Niu, G.; Tan, H. Molecular regulation of antibiotic biosynthesis in streptomyces. Microbiol. Mol. Biol. Rev. 2013, 77, 112–143. [Google Scholar] [CrossRef] [Green Version]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces? Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef]
- Bagnis, C.I.; Deray, G. Amphotericin B nephrotoxicity. Saudi J. Kidney Dis. Transpl. 2002, 13, 481–491. [Google Scholar]
- Ullmann, A.J. Nephrotoxicity in the setting of invasive fungal diseases. Mycoses 2008, 51 (Suppl. S1), 25–30. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.M.; Lancaster, A.K.; Scherz-Shouval, R.; Whitesell, L.; Lindquist, S. Fitness trade-offs restrict the evolution of resistance to amphotericin B. PLoS Biol. 2013, 11, e1001692. [Google Scholar] [CrossRef]
- Hargreaves, P.L.; Nguyen, T.S.; Ryan, R.O. Spectroscopic studies of amphotericin B solubilized in nanoscale bilayer membranes. Biochim. Biophys. Acta 2006, 1758, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahn, U.; Hagenmaier, H.; Hohne, H.; Konig, W.A.; Wolf, G.; Zahner, H. Stoffwechselprodukte von mikroorganismen. 154. Mitteilung. Nikkomycin, ein neuer hemmstoff der chitinsynthese bei pilzen. Arch. Microbiol. 1976, 107, 143–160. [Google Scholar] [CrossRef]
- Suzuki, S.; Isono, K.; Nagatsu, J.; Mizutani, T.; Kawashima, Y.; Mizuno, T. A New Antibiotic, Polyoxin A. J. Antibiot. 1965, 18, 131. [Google Scholar]
- Ruiz-Herrera, J.; San-Blas, G. Chitin synthesis as target for antifungal drugs. Curr. Drug Targets Infect. Disord. 2003, 3, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Khare, R.K.; Becker, J.M.; Naider, F.R. Synthesis and anticandidal properties of polyoxin L analogues containing alpha-amino fatty acids. J. Med. Chem. 1988, 31, 650–656. [Google Scholar] [CrossRef]
- Hector, R.F.; Pappagianis, D. Inhibition of chitin synthesis in the cell wall of Coccidioides immitis by polyoxin D. J. Bacteriol. 1983, 154, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.M.; Covert, N.L.; Shenbagamurthi, P.; Steinfeld, A.S.; Naider, F. Polyoxin D inhibits growth of zoopathogenic fungi. Antimicrob. Agents Chemother. 1983, 23, 926–929. [Google Scholar] [CrossRef] [Green Version]
- Chiou, C.C.; Mavrogiorgos, N.; Tillem, E.; Hector, R.; Walsh, T.J. Synergy, pharmacodynamics, and time-sequenced ultrastructural changes of the interaction between nikkomycin Z and the echinocandin FK463 against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2001, 45, 3310–3321. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.; Connolly, P.; Schnizlein-Bick, C.; Durkin, M.; Kohler, S.; Smedema, M.; Brizendine, E.; Hector, R.; Wheat, J. Comparison of nikkomycin Z with amphotericin B and itraconazole for treatment of histoplasmosis in a murine model. Antimicrob. Agents Chemother. 2000, 44, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Hector, R.F.; Zimmer, B.L.; Pappagianis, D. Evaluation of nikkomycins X and Z in murine models of coccidioidomycosis, histoplasmosis, and blastomycosis. Antimicrob. Agents Chemother. 1990, 34, 587–593. [Google Scholar] [CrossRef] [Green Version]
- Hector, R.F.; Braun, P.C. Synergistic action of nikkomycins X and Z with papulacandin B on whole cells and regenerating protoplasts of Candida albicans. Antimicrob. Agents Chemother. 1986, 29, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hector, R.F.; Schaller, K. Positive interaction of nikkomycins and azoles against Candida albicans in vitro and in vivo. Antimicrob. Agents Chemother. 1992, 36, 1284–1289. [Google Scholar] [CrossRef] [Green Version]
- Larwood, D.J. Nikkomycin Z-Ready to Meet the Promise? J. Fungi 2020, 6, 261. [Google Scholar] [CrossRef] [PubMed]
- Holden, W.M.; Fites, J.S.; Reinert, L.K.; Rollins-Smith, L.A. Nikkomycin Z is an effective inhibitor of the chytrid fungus linked to global amphibian declines. Fungal Biol. 2014, 118, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Nix, D.E.; Swezey, R.R.; Hector, R.; Galgiani, J.N. Pharmacokinetics of nikkomycin Z after single rising oral doses. Antimicrob. Agents Chemother. 2009, 53, 2517–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galgiani, J.N. Coccidioidomycosis: Changing perceptions and creating opportunities for its control. Ann. N. Y. Acad. Sci. 2007, 1111, 1–18. [Google Scholar] [CrossRef]
- Shubitz, L.F.; Trinh, H.T.; Perrill, R.H.; Thompson, C.M.; Hanan, N.J.; Galgiani, J.N.; Nix, D.E. Modeling nikkomycin Z dosing and pharmacology in murine pulmonary coccidioidomycosis preparatory to phase 2 clinical trials. J. Infect. Dis. 2014, 209, 1949–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldholmi, M.; Marchand, P.; Ourliac-Garnier, I.; Le Pape, P.; Ganesan, A. A Decade of Antifungal Leads from Natural Products: 2010–2019. Pharmaceuticals 2019, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Alferova, V.A.; Novikov, R.A.; Bychkova, O.P.; Rogozhin, E.A.; Shuvalov, M.V.; Prokhorenko, I.A.; Sadykova, V.S.; Kulko, A.B.; Dezhenkova, L.G.; Stepashkina, E.A.; et al. Astolides A and B, antifungal and cytotoxic naphthoquinone-derived polyol macrolactones from Streptomyces hygroscopicus. Tetrahedron 2018, 74, 7442–7449. [Google Scholar] [CrossRef]
- Ding, N.; Jiang, Y.; Han, L.; Chen, X.; Ma, J.; Qu, X.; Mu, Y.; Liu, J.; Li, L.; Jiang, C.; et al. Bafilomycins and Odoriferous Sesquiterpenoids from Streptomyces albolongus Isolated from Elephas maximus Feces. J. Nat. Prod. 2016, 79, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Perez-Victoria, I.; Oves-Costales, D.; Lacret, R.; Martin, J.; Sanchez-Hidalgo, M.; Diaz, C.; Cautain, B.; Vicente, F.; Genilloud, O.; Reyes, F. Structure elucidation and biosynthetic gene cluster analysis of caniferolides A-D, new bioactive 36-membered macrolides from the marine-derived Streptomyces caniferus CA-271066. Org. Biomol. Chem. 2019, 17, 2954–2971. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Iwata, F.; Yamada, S.; Katayama, M. Neomaclafungins A-I: Oligomycin-class macrolides from a marine-derived actinomycete. J. Nat. Prod. 2012, 75, 1974–1982. [Google Scholar] [CrossRef]
- Hammer, P.E.; Hill, D.S.; Lam, S.T.; Van Pee, K.H.; Ligon, J.M. Four genes from Pseudomonas fluorescens that encode the biosynthesis of pyrrolnitrin. Appl. Environ. Microbiol. 1997, 63, 2147–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulochana, M.B.; Jayachandra, S.Y.; Kumar, S.K.; Dayanand, A. Antifungal attributes of siderophore produced by the Pseudomonas aeruginosa JAS-25. J. Basic Microbiol. 2014, 54, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.H.; Thrane, C.; Christophersen, C.; Anthoni, U.; Sorensen, J. Structure, production characteristics and fungal antagonism of tensin—A new antifungal cyclic lipopeptide from Pseudomonas fluorescens strain 96.578. J. Appl. Microbiol. 2000, 89, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.K.; Jacobs, N.J.; Rajamani, S.; Krishnamurthy, M.; Cubillos-Ruiz, J.R.; Hogan, D.A. Antifungal mechanisms by which a novel Pseudomonas aeruginosa phenazine toxin kills Candida albicans in biofilms. Mol. Microbiol. 2010, 78, 1379–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.; Sass, G.; Swietnicki, W.; Stevens, D.A. Review of Potential Pseudomonas Weaponry, Relevant to the Pseudomonas-Aspergillus Interplay, for the Mycology Community. J. Fungi 2020, 6, 81. [Google Scholar] [CrossRef]
- Castaldi, S.; Masi, M.; Sautua, F.; Cimmino, A.; Isticato, R.; Carmona, M.; Tuzi, A.; Evidente, A. Pseudomonas fluorescens Showing Antifungal Activity against Macrophomina phaseolina, a Severe Pathogenic Fungus of Soybean, Produces Phenazine as the Main Active Metabolite. Biomolecules 2021, 11, 1728. [Google Scholar] [CrossRef]
- Li, D.; Tao, W.; Yu, D.; Li, S. Emulsifying Properties of Rhamnolipids and Their In Vitro Antifungal Activity against Plant Pathogenic Fungi. Molecules 2022, 27, 7746. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Wei, Z.; Guan, Z.; Cai, Y.; Liao, X. Antifungal Activity of Isolated Bacillus amyloliquefaciens SYBC H47 for the Biocontrol of Peach Gummosis. PLoS ONE 2016, 11, e0162125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Shao, D.; Jiang, C.; Shi, J.; Li, Q.; Huang, Q.; Rajoka, M.S.R.; Yang, H.; Jin, M. Biological activity of lipopeptides from Bacillus. Appl. Microbiol. Biotechnol. 2017, 101, 5951–5960. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Chou, H.P.; Huang, J.W.; Deng, W.L. Genomic and biochemical characterization of antifungal compounds produced by Bacillus subtilis PMB102 against Alternaria brassicicola. Microbiol. Res. 2021, 251, 126815. [Google Scholar] [CrossRef]
- Sarwar, A.; Brader, G.; Corretto, E.; Aleti, G.; Ullah, M.A.; Sessitsch, A.; Hafeez, F.Y. Qualitative analysis of biosurfactants from Bacillus species exhibiting antifungal activity. PLoS ONE 2018, 13, e0198107. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, F.; Reza Fazeli, M.; Akhavan Sepahi, A.; Noormohammadi, Z. Isolation, Screening and Identification of Native and New Bacillus subtilis with Strong Antifungal Compound against Fusarium oxysporum. Biocontrol Sci. 2022, 27, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Salazar, F.; Ortiz, A.; Sansinenea, E. A Strong Antifungal Activity of 7-O-Succinyl Macrolactin A vs Macrolactin A from Bacillus amyloliquefaciens ELI149. Curr. Microbiol. 2020, 77, 3409–3413. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Qin, Z.; Wu, S.; Zhao, P.; Zhen, C.; Gao, H. Antifungal Mechanism of Volatile Organic Compounds Produced by Bacillus subtilis CF-3 on Colletotrichum gloeosporioides Assessed Using Omics Technology. J. Agric. Food Chem. 2021, 69, 5267–5278. [Google Scholar] [CrossRef]
- Um, S.; Fraimout, A.; Sapountzis, P.; Oh, D.C.; Poulsen, M. The fungus-growing termite Macrotermes natalensis harbors bacillaene-producing Bacillus sp. that inhibit potentially antagonistic fungi. Sci. Rep. 2013, 3, 3250. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.R.; Park, J.S.; Jung, W.J. Antifungal evaluation of fengycin isoforms isolated from Bacillus amyloliquefaciens PPL against Fusarium oxysporum f. sp. lycopersici. Microb. Pathog. 2020, 149, 104509. [Google Scholar] [CrossRef]
- Wang, J.; Qiu, J.; Yang, X.; Yang, J.; Zhao, S.; Zhou, Q.; Chen, L. Identification of Lipopeptide Iturin A Produced by Bacillus amyloliquefaciens NCPSJ7 and Its Antifungal Activities against Fusarium oxysporum f. sp. niveum. Foods 2022, 11, 2996. [Google Scholar] [CrossRef] [PubMed]
- Ajesh, K.; Sudarslal, S.; Arunan, C.; Sreejith, K. Kannurin, a novel lipopeptide from Bacillus cereus strain AK1: Isolation, structural evaluation and antifungal activities. J. Appl. Microbiol. 2013, 115, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Tian, Z.; Luo, Y.; Cheng, Y.; Long, C.A. Antagonistic Activity and the Mechanism of Bacillus amyloliquefaciens DH-4 Against Citrus Green Mold. Phytopathology 2018, 108, 1253–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehghanifar, S.; Keyhanfar, M.; Emtiazi, G. Production and partial purification of thermostable bacteriocins from Bacillus pumilus ZED17 and DFAR8 strains with antifungal activity. Mol. Biol. Res. Commun. 2019, 8, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.; Chen, X.H.; Vater, J.; Franke, P.; Nicholson, G.; Borriss, R.; Sussmuth, R.D. Macrolactin is the polyketide biosynthesis product of the pks2 cluster of Bacillus amyloliquefaciens FZB42. J. Nat. Prod. 2007, 70, 1417–1423. [Google Scholar] [CrossRef]
- Wang, D.; Li, A.; Han, H.; Liu, T.; Yang, Q. A potent chitinase from Bacillus subtilis for the efficient bioconversion of chitin-containing wastes. Int. J. Biol. Macromol. 2018, 116, 863–868. [Google Scholar] [CrossRef]
- Prakash, J.; Arora, N.K. Novel metabolites from Bacillus safensis and their antifungal property against Alternaria alternata. Antonie Van Leeuwenhoek 2021, 114, 1245–1258. [Google Scholar] [CrossRef]
- Zhou, M.; Li, P.; Wu, S.; Zhao, P.; Gao, H. Bacillus subtilis CF-3 Volatile Organic Compounds Inhibit Monilinia fructicola Growth in Peach Fruit. Front. Microbiol. 2019, 10, 1804. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.G.; Steen, A.D.; Ladau, J.; Yin, J.; Crosby, L. Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 2018, 3, e00055-18. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.; Epstein, S.; D’Onofrio, A.; Ling, L.L. Uncultured microorganisms as a source of secondary metabolites. J. Antibiot. 2010, 63, 468–476. [Google Scholar] [CrossRef]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef]
- Davis, K.E.; Sangwan, P.; Janssen, P.H. Acidobacteria, Rubrobacteridae and Chloroflexi are abundant among very slow-growing and mini-colony-forming soil bacteria. Environ. Microbiol. 2011, 13, 798–805. [Google Scholar] [CrossRef] [PubMed]
- George, I.F.; Hartmann, M.; Liles, M.R.; Agathos, S.N. Recovery of as-yet-uncultured soil acidobacteria on dilute solid media. Appl. Environ. Microbiol. 2011, 77, 8184–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Donadio, S.; Monciardini, P.; Sosio, M. Polyketide synthases and nonribosomal peptide synthetases: The emerging view from bacterial genomics. Nat. Prod. Rep. 2007, 24, 1073–1109. [Google Scholar] [CrossRef]
- Baltz, R.H. Renaissance in antibacterial discovery from actinomycetes. Curr. Opin. Pharmacol. 2008, 8, 557–563. [Google Scholar] [CrossRef]
- Hover, B.M.; Kim, S.H.; Katz, M.; Charlop-Powers, Z.; Owen, J.G.; Ternei, M.A.; Maniko, J.; Estrela, A.B.; Molina, H.; Park, S.; et al. Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol. 2018, 3, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Nucci, M.; Marr, K.A. Emerging fungal diseases. Clin. Infect. Dis. 2005, 41, 521–526. [Google Scholar] [CrossRef] [Green Version]
- Heeres, J.; Backx, L.J.; Mostmans, J.H.; Van Cutsem, J. Antimycotic imidazoles. part 4. Synthesis and antifungal activity of ketoconazole, a new potent orally active broad-spectrum antifungal agent. J. Med. Chem. 1979, 22, 1003–1005. [Google Scholar] [CrossRef]
- Richardson, K.; Cooper, K.; Marriott, M.S.; Tarbit, M.H.; Troke, P.F.; Whittle, P.J. Discovery of fluconazole, a novel antifungal agent. Rev. Infect. Dis. 1990, 12 (Suppl. S3), S267–S271. [Google Scholar] [CrossRef] [PubMed]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Hert, J.; Irwin, J.J.; Laggner, C.; Keiser, M.J.; Shoichet, B.K. Quantifying biogenic bias in screening libraries. Nat. Chem. Biol. 2009, 5, 479–483. [Google Scholar] [CrossRef]
- McGilvray, A. Compound screening: Fresh hunting ground. Nature 2016, 533, S65–S67. [Google Scholar] [CrossRef] [Green Version]
- Desselle, M.R.; Neale, R.; Hansford, K.A.; Zuegg, J.; Elliott, A.G.; Cooper, M.A.; Blaskovich, M.A. Institutional profile: Community for Open Antimicrobial Drug Discovery—Crowdsourcing new antibiotics and antifungals. Future Sci. OA 2017, 3, FSO171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliver. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Hewitt, W.M.; Schwochert, J.; Haddad, T.D.; Townsend, C.E.; Etienne, L.; Lao, Y.; Limberakis, C.; Furukawa, A.; Mathiowetz, A.M.; et al. Nonclassical Size Dependence of Permeation Defines Bounds for Passive Adsorption of Large Drug Molecules. J. Med. Chem. 2017, 60, 1665–1672. [Google Scholar] [CrossRef] [Green Version]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef]
- Vij, R.; Troger, S.; Walt, C.; Haeckl, F.P.J.; Muller, R.; Kniemeyer, O.; Brakhage, A.; Hube, B.; Brunke, S. Antivirulence drug discovery to disarm Candida albicans with metabolites from myxobacteria. Med. Mycol. 2022, 60, 29–30. [Google Scholar] [CrossRef]
- Di Mambro, T.; Guerriero, I.; Aurisicchio, L.; Magnani, M.; Marra, E. The Yin and Yang of Current Antifungal Therapeutic Strategies: How Can We Harness Our Natural Defenses? Front. Pharmacol. 2019, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Fernandez de Ullivarri, M.; Arbulu, S.; Garcia-Gutierrez, E.; Cotter, P.D. Antifungal Peptides as Therapeutic Agents. Front. Cell. Infect. Microbiol. 2020, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, S.; Edgerton, M. How does it kill?: Understanding the candidacidal mechanism of salivary histatin 5. Eukaryot Cell 2014, 13, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, P.W.; Cheng, Y.L.; Hsieh, W.P.; Lan, C.Y. Responses of Candida albicans to the human antimicrobial peptide LL-37. J. Microbiol. 2014, 52, 581–589. [Google Scholar] [CrossRef]
- Bednarska, N.G.; van Eldere, J.; Gallardo, R.; Ganesan, A.; Ramakers, M.; Vogel, I.; Baatsen, P.; Staes, A.; Goethals, M.; Hammarstrom, P.; et al. Protein aggregation as an antibiotic design strategy. Mol. Microbiol. 2016, 99, 849–865. [Google Scholar] [CrossRef] [Green Version]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, S.; Ebel, F. Monoclonal Antibodies as Tools to Combat Fungal Infections. J. Fungi 2020, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Matthews, R.C.; Rigg, G.; Hodgetts, S.; Carter, T.; Chapman, C.; Gregory, C.; Illidge, C.; Burnie, J. Preclinical assessment of the efficacy of mycograb, a human recombinant antibody against fungal HSP90. Antimicrob. Agents Chemother. 2003, 47, 2208–2216. [Google Scholar] [CrossRef] [Green Version]
- Bugli, F.; Cacaci, M.; Martini, C.; Torelli, R.; Posteraro, B.; Sanguinetti, M.; Paroni Sterbini, F. Human monoclonal antibody-based therapy in the treatment of invasive candidiasis. Clin. Dev. Immunol. 2013, 2013, 403121. [Google Scholar] [CrossRef] [Green Version]
- Louie, A.; Stein, D.S.; Zack, J.Z.; Liu, W.; Conde, H.; Fregeau, C.; Vanscoy, B.D.; Drusano, G.L. Dose range evaluation of Mycograb C28Y variant, a human recombinant antibody fragment to heat shock protein 90, in combination with amphotericin B-desoxycholate for treatment of murine systemic candidiasis. Antimicrob. Agents Chemother. 2011, 55, 3295–3304. [Google Scholar] [CrossRef] [PubMed]
- Pachl, J.; Svoboda, P.; Jacobs, F.; Vandewoude, K.; van der Hoven, B.; Spronk, P.; Masterson, G.; Malbrain, M.; Aoun, M.; Garbino, J.; et al. A randomized, blinded, multicenter trial of lipid-associated amphotericin B alone versus in combination with an antibody-based inhibitor of heat shock protein 90 in patients with invasive candidiasis. Clin. Infect. Dis. 2006, 42, 1404–1413. [Google Scholar] [CrossRef]
- Charlier, C.; Hart, E.; Lefort, A.; Ribaud, P.; Dromer, F.; Denning, D.W.; Lortholary, O. Fluconazole for the management of invasive candidiasis: Where do we stand after 15 years? J. Antimicrob. Chemother. 2006, 57, 384–410. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.V.N.; Wang, R.; Specht, C.A.; Levitz, S.M. Vaccines for human fungal diseases: Close but still a long way to go. NPJ Vaccines 2021, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, K.; Masso-Silva, J.A.; Rivera, A.; Xue, C. A Heat-Killed Cryptococcus Mutant Strain Induces Host Protection against Multiple Invasive Mycoses in a Murine Vaccine Model. mBio 2019, 10, e02145-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, H.B.; Cobb, J.M.; Smith, C.E. Immunity to coccidioi-domycosis induced in mice by purified spherule, arthrospore, and mycelial vaccines. Trans. N. Y. Acad. Sci. 1960, 22, 436–449. [Google Scholar] [CrossRef]
- Hester, M.M.; Lee, C.K.; Abraham, A.; Khoshkenar, P.; Ostroff, G.R.; Levitz, S.M.; Specht, C.A. Protection of mice against experimental cryptococcosis using glucan particle-based vaccines containing novel recombinant antigens. Vaccine 2020, 38, 620–626. [Google Scholar] [CrossRef]
- Wong, L.P.; Woo, P.C.; Wu, A.Y.; Yuen, K.Y. DNA immunization using a secreted cell wall antigen Mp1p is protective against Penicillium marneffei infection. Vaccine 2002, 20, 2878–2886. [Google Scholar] [CrossRef]
- Tso, G.H.W.; Reales-Calderon, J.A.; Pavelka, N. The Elusive Anti-Candida Vaccine: Lessons From the Past and Opportunities for the Future. Front. Immunol. 2018, 9, 897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Uppuluri, P.; Mamouei, Z.; Alqarihi, A.; Elhassan, H.; French, S.; Lockhart, S.R.; Chiller, T.; Edwards, J.E., Jr.; Ibrahim, A.S. The NDV-3A vaccine protects mice from multidrug resistant Candida auris infection. PLoS Pathog. 2019, 15, e1007460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uppuluri, P.; Singh, S.; Alqarihi, A.; Schmidt, C.S.; Hennessey, J.P., Jr.; Yeaman, M.R.; Filler, S.G.; Edwards, J.E.; Ibrahim, A.S. Human Anti-Als3p Antibodies Are Surrogate Markers of NDV-3A Vaccine Efficacy Against Recurrent Vulvovaginal Candidiasis. Front. Immunol. 2018, 9, 1349. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.E., Jr.; Schwartz, M.M.; Schmidt, C.S.; Sobel, J.D.; Nyirjesy, P.; Schodel, F.; Marchus, E.; Lizakowski, M.; DeMontigny, E.A.; Hoeg, J.; et al. A Fungal Immunotherapeutic Vaccine (NDV-3A) for Treatment of Recurrent Vulvovaginal Candidiasis-A Phase 2 Randomized, Double-Blind, Placebo-Controlled Trial. Clin. Infect. Dis. 2018, 66, 1928–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Van Dijck, P.; Datta, A. Environmental sensing and signal transduction pathways regulating morphopathogenic determinants of Candida albicans. Microbiol. Mol. Biol. Rev. 2007, 71, 348–376. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guo, Y.; Jiang, C.; Chang, Q.; Zhang, S.; Luo, T.; Zhang, B.; Jia, X.; Hung, M.C.; Dong, C.; et al. JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat. Med. 2017, 23, 337–346. [Google Scholar] [CrossRef] [Green Version]
- Sulakvelidze, A.; Kutter, E. Bacteriophage Therapy in Humans. In Bacteriophages; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar] [CrossRef]
- Kotta-Loizou, I.; Coutts, R.H.A. Mycoviruses in Aspergilli: A Comprehensive Review. Front. Microbiol. 2017, 8, 1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Gupta, S.; Shrivastava, J.N. Presence of Virus like Particles in Human Pathogenic Fungi: Chrysosporium sps and Candida albicans. Indian J. Virol. 2011, 22, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Border, D.J. Electron microscopy of cells of Saccharomyces cerevisiae infected with double stranded RNA viruses from Aspergillus niger and Penicillium stoloniferum. Nat. New Biol. 1972, 236, 87–88. [Google Scholar] [CrossRef]
- Theuretzbacher, U.; Piddock, L.J.V. Non-traditional Antibacterial Therapeutic Options and Challenges. Cell Host Microbe 2019, 26, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Batista-Duharte, A.; Sendra, L.; Herrero, M.J.; Portuondo, D.L.; Tellez-Martinez, D.; Olivera, G.; Fernandez-Delgado, M.; Javega, B.; Herrera, G.; Martinez, A.; et al. Foxp3 Silencing with Antisense Oligonucleotide Improves Immunogenicity of an Adjuvanted Recombinant Vaccine against Sporothrix schenckii. Int. J. Mol. Sci. 2021, 22, 3470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Leibowitz, M.J.; Zhang, Y. Antisense oligonucleotides effectively inhibit the co-transcriptional splicing of a Candida group I intron in vitro and in vivo: Implications for antifungal therapeutics. FEBS Lett. 2009, 583, 734–738. [Google Scholar] [CrossRef]
- Gorlach, J.M.; McDade, H.C.; Perfect, J.R.; Cox, G.M. Antisense repression in Cryptococcus neoformans as a laboratory tool and potential antifungal strategy. Microbiology 2002, 148, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, D.; Azevedo, N.M.; Barbosa, A.; Almeida, C.; Rodrigues, M.E.; Henriques, M.; Silva, S. Application of 2′-OMethylRNA′ Antisense Oligomer to Control Candida albicans EFG1 Virulence Determinant. Mol. Ther. Nucleic Acids 2019, 18, 508–517. [Google Scholar] [CrossRef] [Green Version]
- Araujo, D.; Mil-Homens, D.; Henriques, M.; Silva, S. Anti-EFG1 2′-OMethylRNA oligomer inhibits Candida albicans filamentation and attenuates the candidiasis in Galleria mellonella. Mol. Ther. Nucleic Acids 2022, 27, 517–523. [Google Scholar] [CrossRef]
- Hawser, S.; Islam, K. Comparisons of the effects of fungicidal and fungistatic antifungal agents on the morphogenetic transformation of Candida albicans. J. Antimicrob. Chemother. 1999, 43, 411–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtappels, M.; Swinnen, E.; De Groef, L.; Wuyts, J.; Moons, L.; Lagrou, K.; Van Dijck, P.; Kucharikova, S. Antifungal Activity of Oleylphosphocholine on In Vitro and In Vivo Candida albicans Biofilms. Antimicrob. Agents Chemother. 2018, 62, e01767-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rex, J.H.; Fernandez Lynch, H.; Cohen, I.G.; Darrow, J.J.; Outterson, K. Designing development programs for non-traditional antibacterial agents. Nat. Commun. 2019, 10, 3416. [Google Scholar] [CrossRef]


{kind=link}
| Natural Products | Synthetic Compounds | ||
|---|---|---|---|
| Fungi-Derived | Bacteria-Derived | ||
| Most common hits | B-glucan synthase inhibitors (12% of hits) [50] | Polyenes (95% of hits) [47] | Azoles (15% of hits) [50] |
| Hit rates (%) | ~20% [50] | ~9% [50] | ~0.1–2% [51,52,53] |
| Common PAINs | Mycotoxins [50] | Ionophores [50] | Rhodanines [54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanreppelen, G.; Wuyts, J.; Van Dijck, P.; Vandecruys, P. Sources of Antifungal Drugs. J. Fungi 2023, 9, 171. https://doi.org/10.3390/jof9020171
Vanreppelen G, Wuyts J, Van Dijck P, Vandecruys P. Sources of Antifungal Drugs. Journal of Fungi. 2023; 9(2):171. https://doi.org/10.3390/jof9020171
Chicago/Turabian StyleVanreppelen, Giel, Jurgen Wuyts, Patrick Van Dijck, and Paul Vandecruys. 2023. "Sources of Antifungal Drugs" Journal of Fungi 9, no. 2: 171. https://doi.org/10.3390/jof9020171