
Terpenoid Biosynthesis Dominates among Secondary Metabolite Clusters in Mucoromycotina Genomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Candidate Secondary Metabolite Cluster Prediction
2.2. SMC-Encoded Protein Clustering and Annotation
2.3. SMC Similarity Network Construction and Visualization
2.4. Detection of Genes of Putative Bacterial Origin
3. Results and Discussion
3.1. Initial Screening
3.2. Limits of de Novo Prediction in the Absence of Reference Annotation
3.3. De Novo Predicted SMC Candidate Sequences Are Poorly Characterised by Synteny
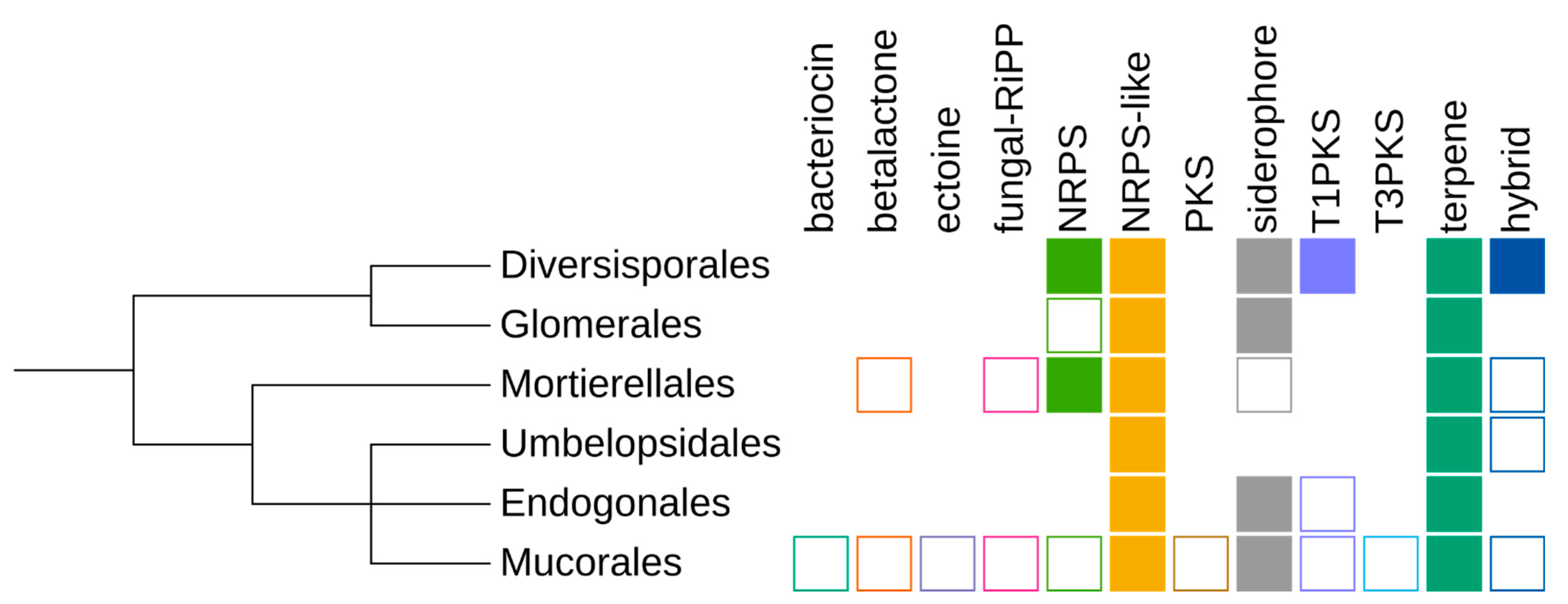
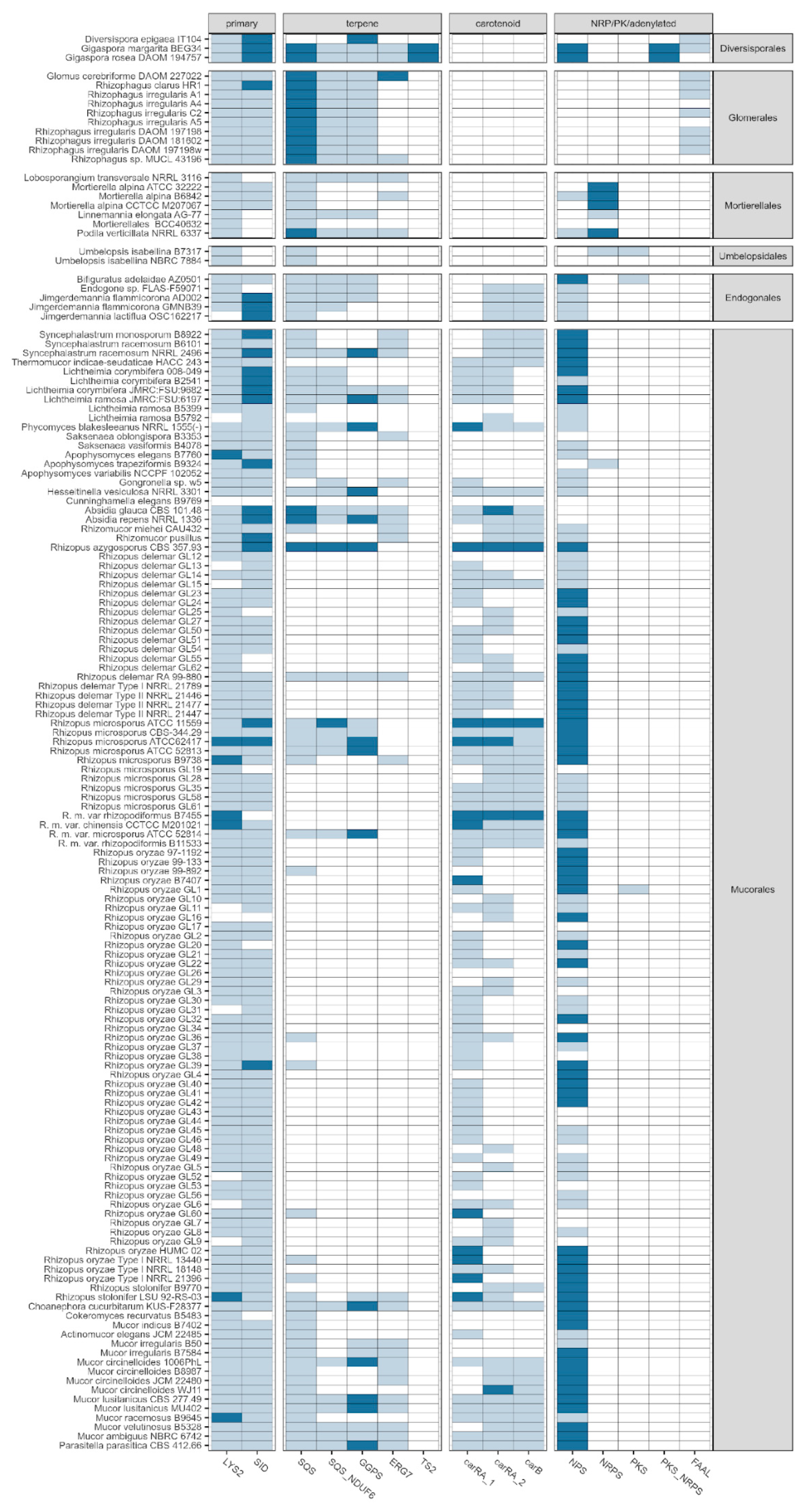
3.4. Taxonomic Distribution
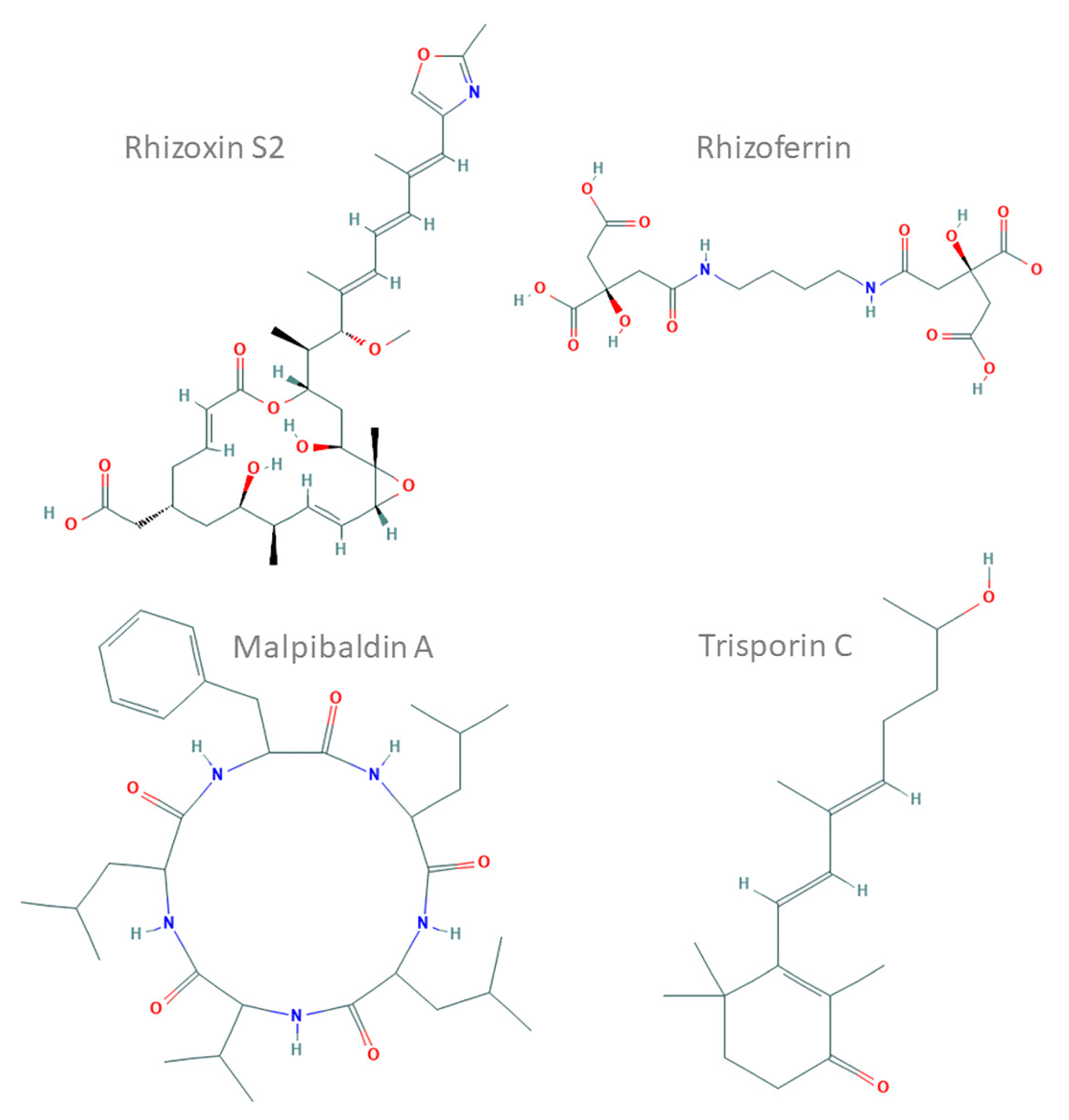
3.5. Cluster Types
3.6. Presence of Bacterial-Like SMCs
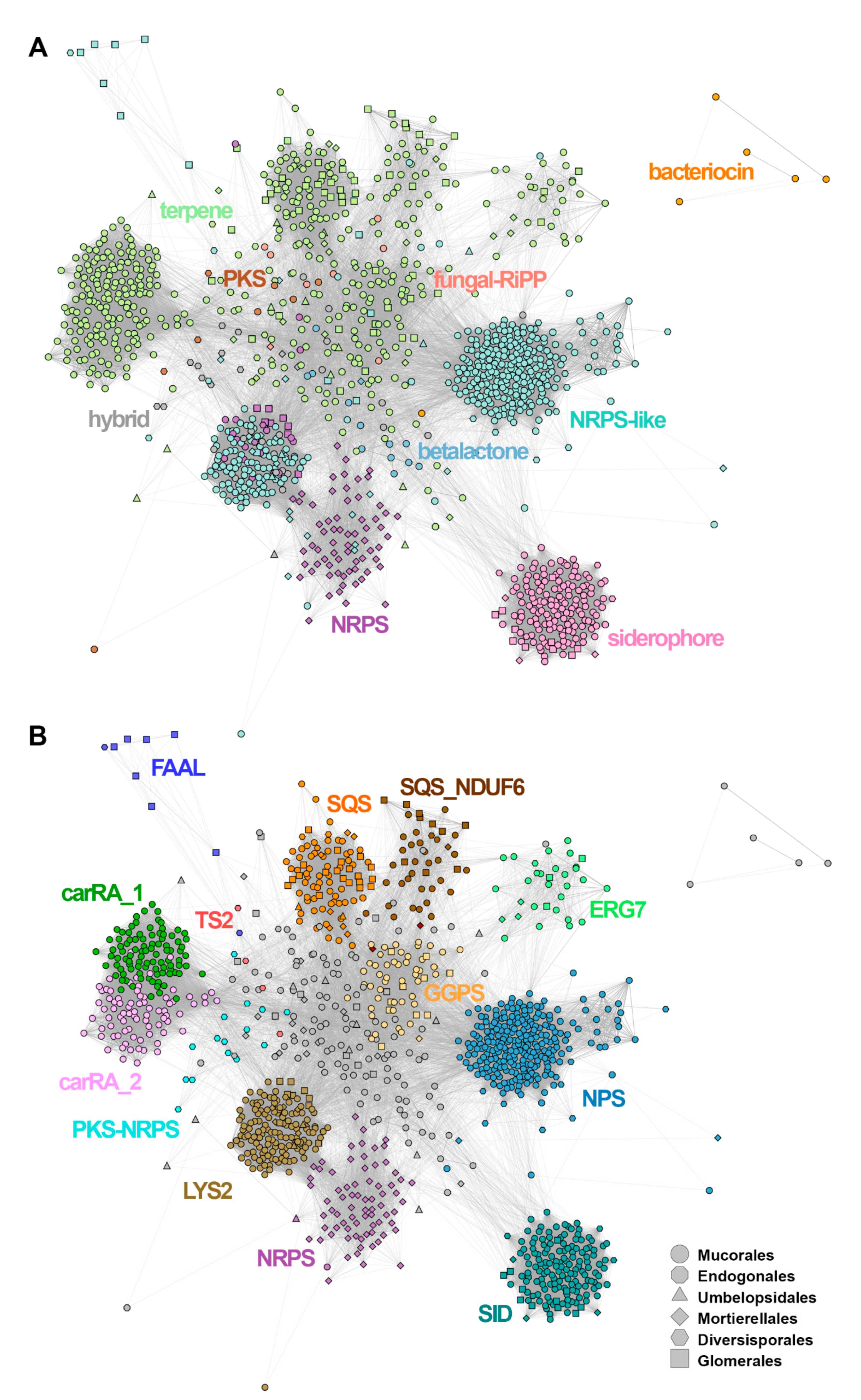
3.7. SMC Sequence Clustering
3.8. Predominance of Terpene Biosynthesis in Non-Dikarya Genomes
3.9. Heterogeneity of NRPS/NRPS-Like Homologues
3.10. Non-NRPS Siderophore Synthases Are Conserved in Early-Diverging Fungi
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Voigt, K.; Wolf, T.; Ochsenreiter, K.; Nagy, G.; Kaerger, K.; Shelest, E.; Papp, T. 15 Genetic and Metabolic Aspects of Primary and Secondary Metabolism of the Zygomycetes. Biochem. Mol. Biol. 2016, 361–385. [Google Scholar] [CrossRef]
- Shelest, E.; Voigt, K. 2 Genomics to Study Basal Lineage Fungal Biology: Phylogenomics Suggests a Common Origin. In Fungal Genomics; Springer: Berlin/Heidelberg, Germany, 2014; pp. 31–60. [Google Scholar] [CrossRef]
- Throckmorton, K.; Wiemann, P.; Keller, N. Evolution of Chemical Diversity in a Group of Non-Reduced Polyketide Gene Clusters: Using Phylogenetics to Inform the Search for Novel Fungal Natural Products. Toxins 2015, 3572–3607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides1. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef]
- Quin, M.B.; Flynn, C.M.; Schmidt-Dannert, C. Traversing the fungal terpenome. Nat. Prod. Rep. 2014, 1449–1473. [Google Scholar] [CrossRef] [PubMed]
- Renshaw, J.C.; Robson, G.D.; Trinci, A.P.; Wiebe, M.G.; Livens, F.R.; Collison, D.; Taylor, R.J. Fungal siderophores: Structures, functions and applications. Mycol. Res. 2002, 1123–1142. [Google Scholar] [CrossRef]
- Winkelmann, G. A search for glomuferrin: A potential siderophore of arbuscular mycorrhizal fungi of the genus Glomus. Biometals 2017, 30, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Prakash, H.; Rudramurthy, S.M.; Gandham, P.S.; Ghosh, A.K.; Kumar, M.M.; Badapanda, C.; Chakrabarti, A. Apophysomyces variabilis: Draft genome sequence and comparison of predictive virulence determinants with other medically important Mucorales. BMC Genom. 2017, 18, 736. [Google Scholar] [CrossRef] [Green Version]
- Carroll, C.S.; Grieve, C.L.; Murugathasan, I.; Bennet, A.J.; Czekster, C.M.; Liu, H.; Naismith, J.; Moore, M.M. The rhizoferrin biosynthetic gene in the fungal pathogen Rhizopus delemar is a novel member of the NIS gene family. Int. J. Biochem. Cell Biol. 2017, 89, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Škríba, A.; Patil, R.H.; Hubáček, P.; Dobiáš, R.; Palyzová, A.; Marešová, H.; Pluháček, T.; Havlíáek, V. Rhizoferrin Glycosylation in Rhizopus microsporus. J. Fungi 2020, 6, 89. [Google Scholar] [CrossRef]
- Baldeweg, F.; Warncke, P.; Fischer, D.; Gressler, M. Fungal Biosurfactants from Mortierella alpina. Org. Lett. 2019, 21, 1444–1448. [Google Scholar] [CrossRef]
- Wurlitzer, J.M.; Stanišić, A.; Wasmuth, I.; Jungmann, S.; Fischer, D.; Kries, H.; Gressler, M. Bacterial-like nonribosomal peptide synthetases produce cyclopeptides in the zygomycetous fungus. Appl. Environ. Microbiol. 2020, 87. [Google Scholar] [CrossRef] [PubMed]
- Paduch, R.; Trytek, M.; Król, S.K.; Kud, J.; Frant, M.; Kandefer-Szerszeń, M.; Fiedurek, J. Biological activity of terpene compounds produced by biotechnological methods. Pharm. Biol. 2016, 54, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebreton, A.; Meslet-Cladière, L.; Morin-Sardin, S.; Coton, E.; Jany, J.-L.; Barbier, G.; Corre, E. Comparative analysis of five Mucor species transcriptomes. Genomics 2019, 111, 1306–1314. [Google Scholar] [CrossRef]
- Venice, F.; Desirò, A.; Silva, G.; Salvioli, A.; Bonfante, P. The Mosaic Architecture of NRPS-PKS in the Arbuscular Mycorrhizal Fungus Gigaspora margarita Shows a Domain with Bacterial Signature. Front. Microbiol. 2020. [Google Scholar] [CrossRef]
- Lebreton, A.; Corre, E.; Jany, J.-L.; Brillet-Guéguen, L.; Pèrez-Arques, C.; Garre, V.; Monsoor, M.; Debuchy, R.; le Meur, C.; Coton, E.; et al. Comparative genomics applied to Mucor species with different lifestyles. BMC Genom. 2020, 21, 135. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.-S.; Li, X.-M.; Williams, K.; Proksch, P.; Ji, N.-Y.; Wang, B.-G. Rhizovarins A-F, Indole-Diterpenes from the Mangrove-Derived Endophytic Fungus Mucor irregularis QEN-189. J. Nat. Prod. 2016, 79, 2066–2074. [Google Scholar] [CrossRef]
- Partida-Martinez, L.P.; Hertweck, C. A gene cluster encoding rhizoxin biosynthesis in “Burkholderia rhizoxina”, the bacterial endosymbiont of the fungus Rhizopus microsporus. Chembiochem 2007, 8, 41–45. [Google Scholar] [CrossRef]
- Uehling, J.; Gryganskyi, A.; Hameed, K.; Tschaplinski, T.; Misztal, P.K.; Wu, S.; Desirò, A.; Vande Pol, N.; Du, Z.; Zienkiewicz, A.; et al. Comparative genomics of Mortierella elongata and its bacterial endosymbiont Mycoavidus cysteinexigens. Environ. Microbiol. 2017, 19, 2964–2983. [Google Scholar] [CrossRef]
- Hasan, H.A.H. Gibberellin and auxin-indole production by plant root-fungi and their biosynthesis under salinity-calcium interaction. Acta. Microbiol. Immunol. Hung. 2002, 49, 105–118. [Google Scholar] [CrossRef]
- Ozimek, E.; Jaroszuk-Ściseł, J.; Bohacz, J.; Korniłłowicz-Kowalska, T.; Tyśkiewicz, R.; Słomka, A.; Nowak, A.; Hanaka, A. Synthesis of Indoleacetic Acid, Gibberellic Acid and ACC-Deaminase by Mortierella Strains Promote Winter Wheat Seedlings Growth under Different Conditions. Int. J. Mol. Sci. 2018, 19, 3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, D.; Wang, L.; Han, M.; Wang, J.; Song, H.; Yan, X.; Duan, X.; Dong, J. Effects of an Endophytic Fungus Umbelopsis dimorpha on the Secondary Metabolites of Host–Plant Kadsura angustifolia. Front. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Fed-erhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic. Acids. Res. 2020, 48, D9–D16. [Google Scholar]
- Blin, K.; Kim, H.U.; Medema, M.H.; Weber, T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2019, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Yang, M.; Derbyshire, M.K.; Yamashita, R.A.; Marchler-Bauer, A. NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinform. 2020, 69, e90. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.; Tosatto, S.C.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Koczyk, G.; Dawidziuk, A.; Popiel, D. The Distant Siblings-A Phylogenomic Roadmap Illuminates the Origins of Extant Diversity in Fungal Aromatic Polyketide Biosynthesis. Genome. Biol. Evol. 2015, 7, 3132–3154. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Robinson, S.L.; Terlouw, B.R.; Smith, M.D.; Pidot, S.J.; Stinear, T.P.; Medema, M.H.; Wackett, L.P. Global analysis of adenylate-forming enzymes reveals?-lactone biosynthesis pathway in pathogenic Nocardia. J. Bio. Chem. 2020, 295, 14826–14839. [Google Scholar] [CrossRef]
- Dress, A.W.M.; Flamm, C.; Fritzsch, G.; Grünewald, S.; Kruspe, M.; Prohaska, S.J.; Stadler, P.F. Noisy: Identification of problematic columns in multiple sequence alignments. Algorithms Mol. Biol. 2008, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Takano, E.; Breitling, R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 2013, 30, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Rancurel, C.; Legrand, L.; Danchin, E.G.J. Alienness: Rapid Detection of Candidate Horizontal Gene Transfers across the Tree of Life. Genes 2017, 8, 248. [Google Scholar] [CrossRef]
- Tabima, J.F.; Trautman, I.A.; Chang, Y.; Wang, Y.; Mondo, S.; Kuo, A.; Salamov, A.; Grigoriev, I.V.; Stajich, J.E.; Spatafora, J.W. Phylogenomic Analyses of Non-Dikarya Fungi Supports Horizontal Gene Transfer Driving Diversification of Secondary Metabolism in the Amphibian Gastrointestinal Symbiont, Basidiobolus. Genes Genomes Genet. 2020, 10, 3417–3433. [Google Scholar]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Venice, F.; Ghignone, S.; Salvioli di Fossalunga, A.; Amselem, J.; Novero, M.; Xianan, X.; Toro, K.S.; Morin, E.; Lipzen, A.; Grigoriev, I.V. At the nexus of three kingdoms: The genome of the mycorrhizal fungus Gigaspora margarita provides insights into plant, endobacterial and fungal interactions. Environ. Microbiol. 2020, 22, 122–141. [Google Scholar] [CrossRef]
- Gryganskyi, A.; Muszewska, A. Whole genome sequencing and the Zygomycota. Fungal Genom. Biol. 2014, 4. [Google Scholar]
- Trivedi, O.A.; Arora, P.; Sridharan, V.; Tickoo, R.; Mohanty, D.; Gokhale, R.S. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature 2004, 428, 441–445. [Google Scholar] [CrossRef]
- Wei, G.; Eberl, F.; Chen, X.; Zhang, C.; Unsicker, S.B.; Köllner, T.G.; Gershenzon, J.; Chen, F. Evolution of isoprenyl diphosphate synthase-like terpene synthases in fungi. Sci. Rep. 2020, 10, 14944. [Google Scholar] [CrossRef]
- Britton, G.; Liaaen-Jensen, S.; Pfander, H. Carotenoids: Handbook; Springer Science & Business Media: Berlin, Germany, 2004. [Google Scholar]
- Lin, L.; Xu, J. Fungal Pigments and Their Roles Associated with Human Health. J. Fungi 2020, 6, 280. [Google Scholar] [CrossRef]
- Avalos, J.; Carmen Limón, M. Biological roles of fungal carotenoids. Curr. Genet. 2015, 61, 309–324. [Google Scholar] [CrossRef]
- Strobel, I.; Breitenbach, J.; Scheckhuber, C.Q.; Osiewacz, H.D.; Sandmann, G. Carotenoids and carotenogenic genes in Podospora anserina: Engineering of the carotenoid composition extends the life span of the mycelium. Curr. Genet. 2009, 55, 175–184. [Google Scholar] [CrossRef]
- Idnurm, A.; Rodríguez-Romero, J.; Corrochano, L.M.; Sanz, C.; Iturriaga, E.A.; Eslava, A.P.; Heitman, J. The Phycomyces madA gene encodes a blue-light photoreceptor for phototropism and other light responses. Proc. Natl. Acad. Sci. USA 2006, 103, 4546–4551. [Google Scholar] [CrossRef] [Green Version]
- Sahadevan, Y.; Richter-Fecken, M.; Kaerger, K.; Voigt, K.; Boland, W. Early and late trisporoids differentially regulate β-carotene production and gene transcript Levels in the mucoralean fungi Blakeslea trispora and Mucor mucedo. Appl. Environ. Microbiol. 2013, 79, 7466–7475. [Google Scholar] [CrossRef] [Green Version]
- Corrochano, L.M.; Kuo, A.; Marcet-Houben, M.; Polaino, S.; Salamov, A.; Villalobos-Escobedo, J.M.; Grimwood, J.; Álvarez, M.I.; Avalos, J.; Bauer, D. Expansion of Signal Transduction Pathways in Fungi by Extensive Genome Duplication. Curr. Biol. 2016, 26, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Sandmann, G. Molecular evolution of carotenoid biosynthesis from bacteria to plants. Physiol. Plant. 2002, 431–440. [Google Scholar] [CrossRef]
- Mohammadian Fazli, M.; Soleimani, N.; Mehrasbi, M.; Darabian, S.; Mohammadi, J.; Ramazani, A. Highly cadmium tolerant fungi: Their tolerance and removal potential. J. Environ. Health Sci. Eng. 2015, 13, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Luo, N.; Zhang, L.J.; Zhao, H.M.; Li, Y.W.; Cai, Q.Y.; Wong, M.H.; Mo, C.H. Do arbuscular mycorrhizal fungi affect cadmium uptake kinetics, subcellular distribution and chemical forms in rice? Sci. Total. Environ. 2016, 571, 1183–1190. [Google Scholar] [CrossRef]
- Sundari, S.K.; Adholeya, A. Growth profile of ectomycorrhizal fungal mycelium: Emphasis on substrate pH influence. Antonie Van Leeuwenhoek 2003, 83, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Vassaux, A.; Meunier, L.; Vandenbol, M.; Baurain, D.; Fickers, P.; Jacques PLeclère, V. Nonribosomal peptides in fungal cell factories: From genome mining to optimized heterologous production. Biotechnol. Adv. 2019, 37, 107449. [Google Scholar] [CrossRef] [PubMed]
- Kalb, D.; Lackner, G.; Hoffmeister, D. Functional and Phylogenetic Divergence of Fungal Adenylate-Forming Reductases. Appl. Environ. Microbiol. 2014, 6175–6183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, M.K.; Nouri, E.; Courty, P.-E.; Reinhardt, D. Diet of Arbuscular Mycorrhizal Fungi: Bread and Butter? Trends Plant. Sci. 2017, 22, 652–660. [Google Scholar] [CrossRef] [Green Version]
- Matelska, D.; Steczkiewicz, K.; Ginalski, K. Comprehensive classification of the PIN domain-like superfamily. Nucleic Acids Res. 2017, 45, 6995–7020. [Google Scholar] [CrossRef]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, nature and utility of universal iron chelator—Siderophore: A review. Microbiol. Res. 2018, 212–213, 103–111. [Google Scholar] [CrossRef]
- Carroll, C.S.; Moore, M.M. Ironing out siderophore biosynthesis: A review of non-ribosomal peptide synthetase (NRPS)-independent siderophore synthetases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 356–381. [Google Scholar] [CrossRef]
- Bushley, K.E.; Turgeon, B.G. Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol. Biol. 2010, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muszewska, A.; Okrasińska, A.; Steczkiewicz, K.; Drgas, O.; Orłowska, M.; Perlińska-Lenart, U.; Aleksandrzak-Piekarczyk, T.; Szatraj, K.; Zielenkiewicz, U.; Piłsyk, S. Metabolic Potential, Ecology and Presence of Associated Bacteria Is Reflected in Genomic Diversity of Mucoromycotina. Front. Microbiol. 2021, 12, 239. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koczyk, G.; Pawłowska, J.; Muszewska, A. Terpenoid Biosynthesis Dominates among Secondary Metabolite Clusters in Mucoromycotina Genomes. J. Fungi 2021, 7, 285. https://doi.org/10.3390/jof7040285
Koczyk G, Pawłowska J, Muszewska A. Terpenoid Biosynthesis Dominates among Secondary Metabolite Clusters in Mucoromycotina Genomes. Journal of Fungi. 2021; 7(4):285. https://doi.org/10.3390/jof7040285
Chicago/Turabian StyleKoczyk, Grzegorz, Julia Pawłowska, and Anna Muszewska. 2021. "Terpenoid Biosynthesis Dominates among Secondary Metabolite Clusters in Mucoromycotina Genomes" Journal of Fungi 7, no. 4: 285. https://doi.org/10.3390/jof7040285