
The Endometrial Microbiota—16S rRNA Gene Sequence Signatures in Healthy, Pregnant and Endometritis Dairy Cows
 , , and
, , and 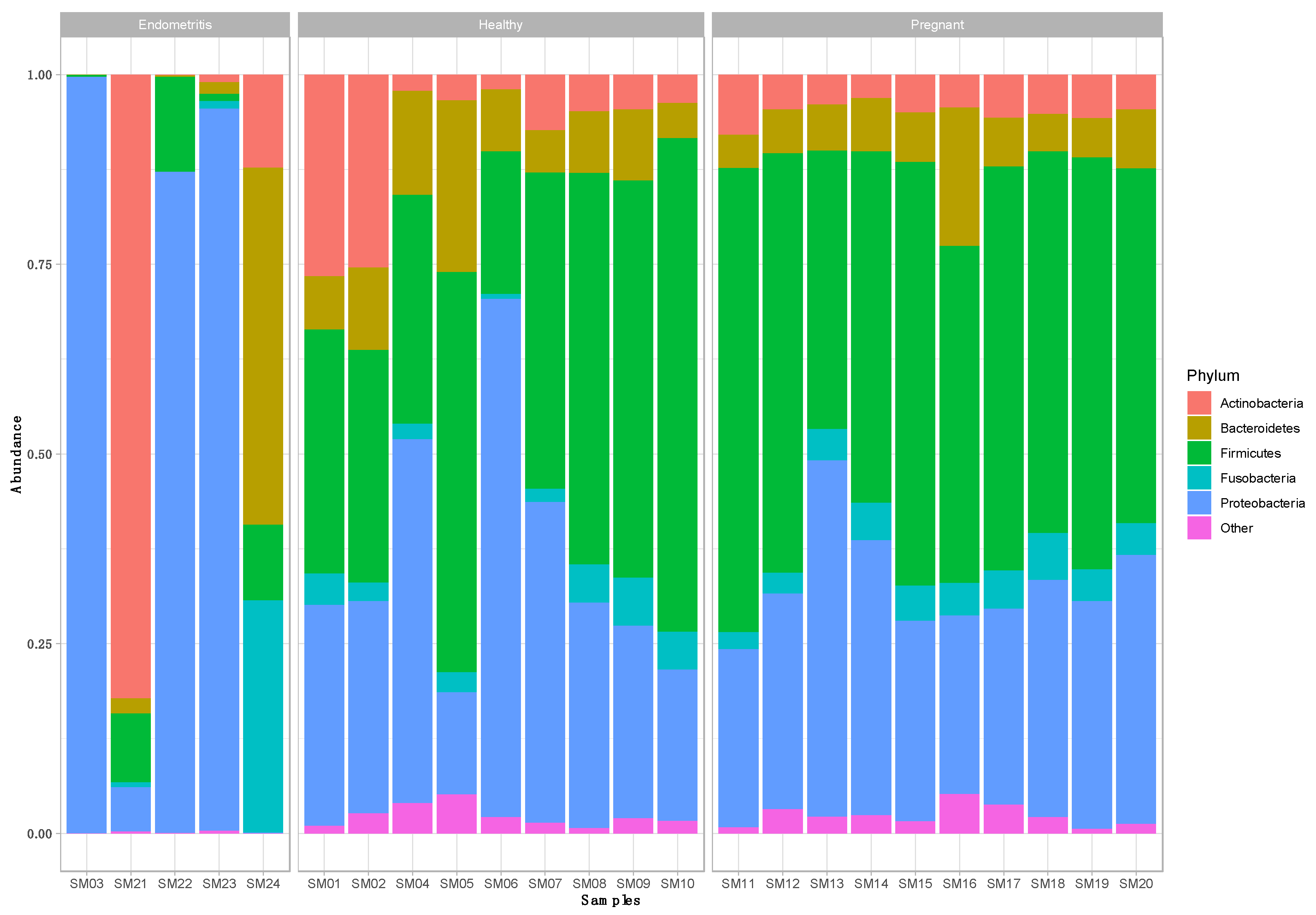{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction
2.3. 16S rRNA Gene Amplification and Sequencing
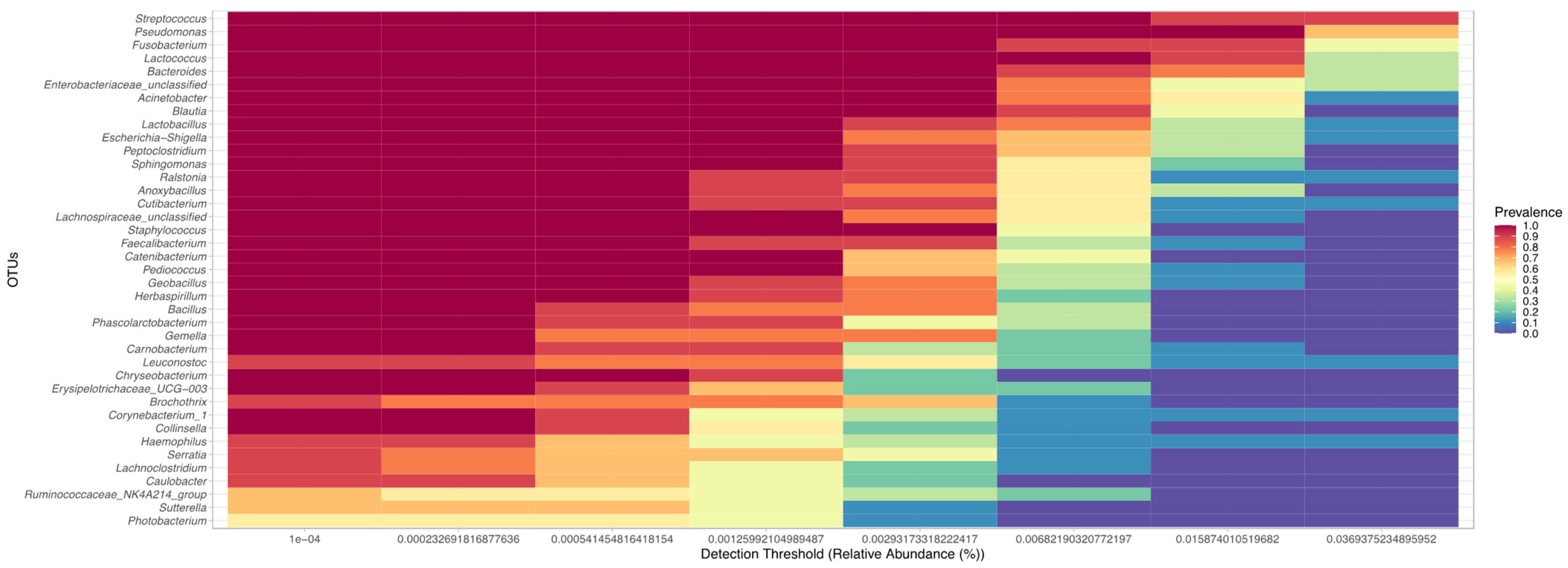
2.4. Data Analysis
3. Results
3.1. Taxonomic Composition of the Uterine Bacterial Community in Clinically Healthy Dairy Cows (n = 9)
3.2. Taxonomic Composition of the Uterine Bacterial Community in Dairy Cows with Endometritis (n = 5)
3.3. Taxonomic Composition of the Uterine Bacterial Community in Pregnant Dairy Cows (n = 10)
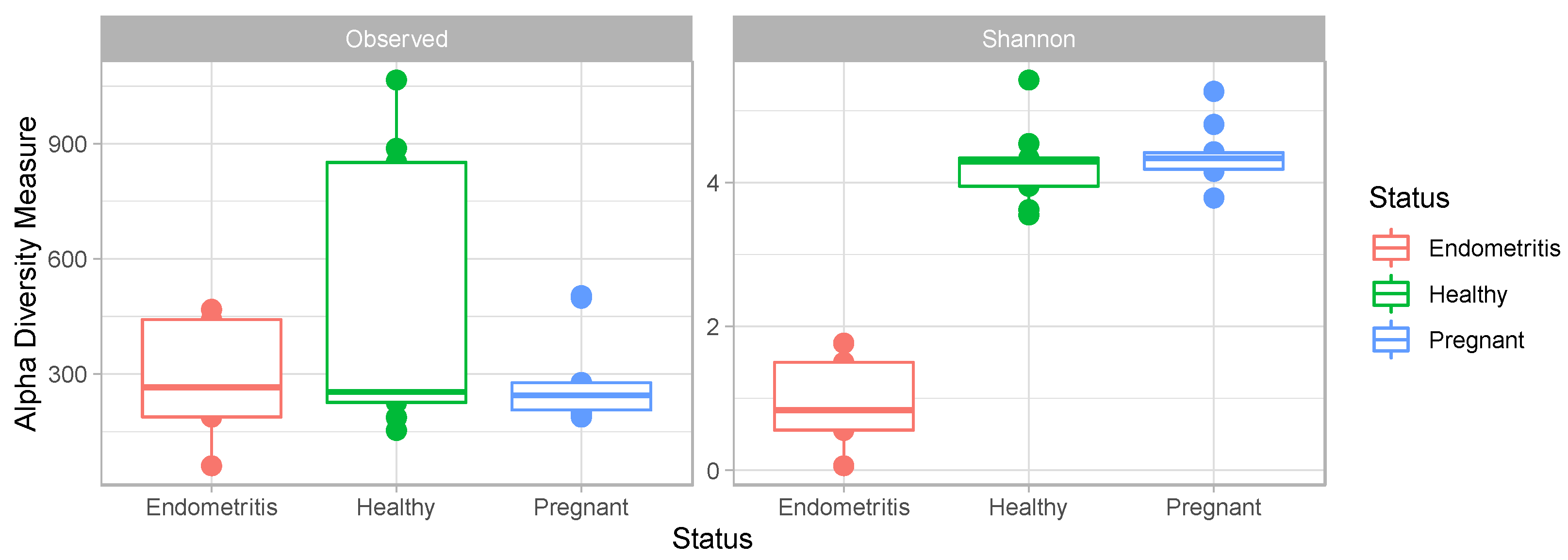
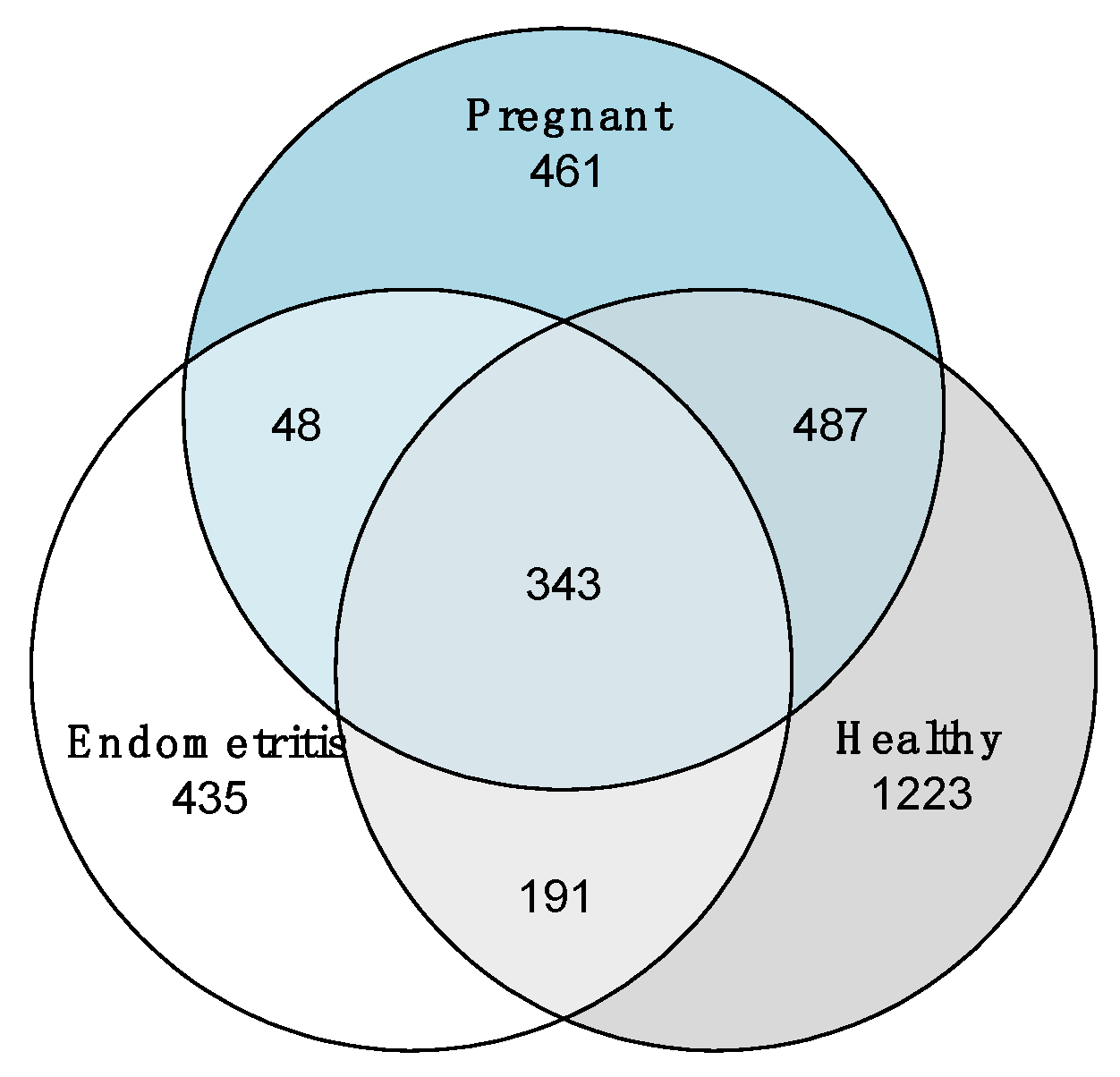
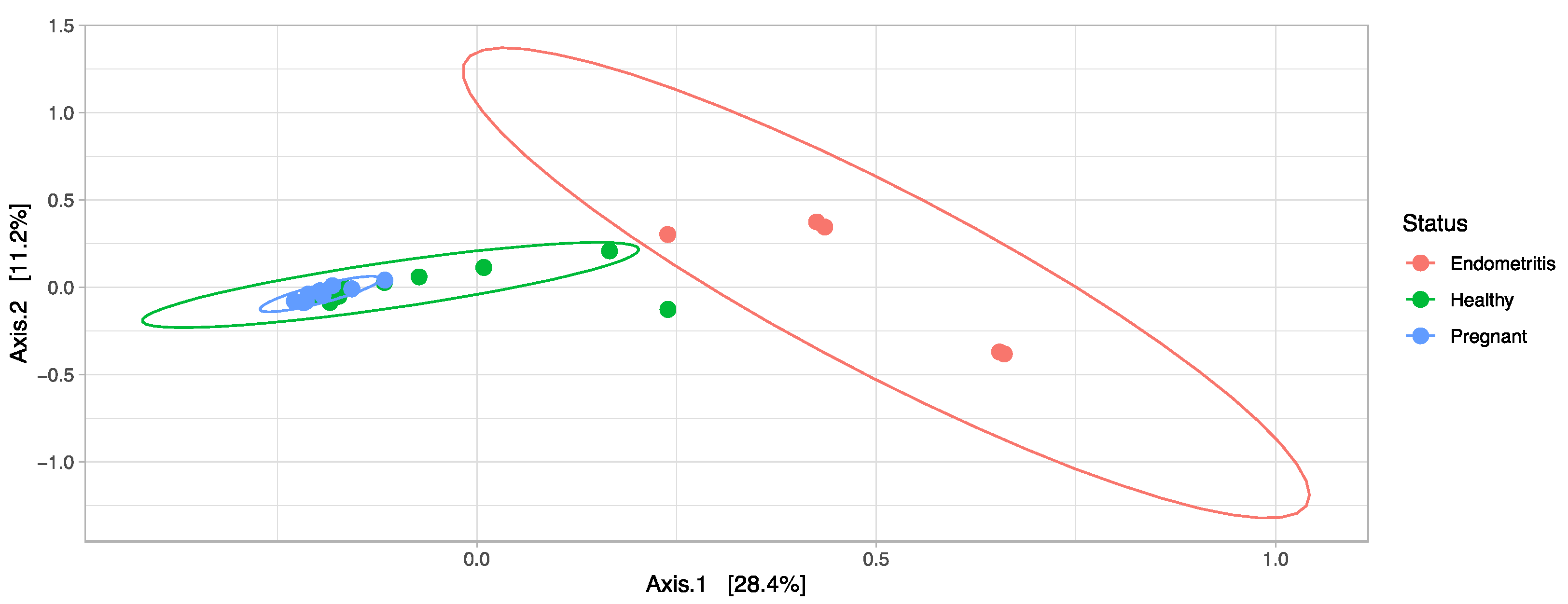
3.4. Comparison of the Uterine Bacterial Composition between Pregnant, Clinically Healthy and Endometritic Cows
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, M.; Lewis, G.S.; LeBlanc, S.; Gilbert, R.O. Defining Postpartum Uterine Disease in Cattle. Theriogenology 2006, 65, 1516–1530. [Google Scholar] [CrossRef]
- Lima, F.S.; Vieira-Neto, A.; Snodgrass, J.A.; de Vries, A.; Santos, J.E.P. Economic Comparison of Systemic Antimicrobial Therapies for Metritis in Dairy Cows. J. Dairy Sci. 2019, 102, 7345–7358. [Google Scholar] [CrossRef]
- Galvão, K.N.; Santos, J.E.P. Recent Advances in the Immunology and Uterine Microbiology of Healthy Cows and Cows That Develop Uterine Disease. Turk. J. Vet. Anim. Sci. 2014, 38, 577–588. [Google Scholar] [CrossRef]
- Kitaya, K.; Takeuchi, T.; Mizuta, S.; Matsubayashi, H.; Ishikawa, T. Endometritis: New Time, New Concepts. Fertil. Steril. 2018, 110, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, M. The Postpartum Uterus. Vet. Clin. N. Am. Food Anim. Pract. 2004, 20, 569–591. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.M.A.M.A.; Gilbert, R.O.; Bicalho, R.C.C. Metagenomic Analysis of the Uterine Bacterial Microbiota in Healthy and Metritic Postpartum Dairy Cows. J. Dairy Sci. 2011, 94, 291–302. [Google Scholar] [CrossRef]
- Appiah, M.O.; Wang, J.; Lu, W. Microflora in the Reproductive Tract of Cattle: A Review. Agriculture 2020, 10, 232. [Google Scholar] [CrossRef]
- Westermann, S.; Drillich, M.; Kaufmann, T.; Madoz, L.; Heuwieser, W. A Clinical Approach to Determine False Positive Findings of Clinical Endometritis by Vaginoscopy by the Use of Uterine Bacteriology and Cytology in Dairy Cows. Theriogenology 2010, 74, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Suthar, V.; Plöntzke, J.; Heuwieser, W. Relationship between Bacteriological Findings in the Second and Fourth Weeks Postpartum and Uterine Infection in Dairy Cows Considering Bacteriological Results. J. Dairy Sci. 2012, 95, 7105–7114. [Google Scholar] [CrossRef]
- Santos, T.M.A.; Bicalho, R.C. Diversity and Succession of Bacterial Communities in the Uterine Fluid of Postpartum Metritic, Endometritic and Healthy Dairy Cows. PLoS ONE 2012, 7, e53048. [Google Scholar] [CrossRef] [Green Version]
- Ault, T.B.; Clemmons, B.A.; Reese, S.T.; Dantas, F.G.; Franco, G.A.; Smith, T.P.L.; Edwards, J.L.; Myer, P.R.; Pohler, K.G. Uterine and Vaginal Bacterial Community Diversity Prior to Artificial Insemination between Pregnant and Nonpregnant Postpartum Cows. J. Anim. Sci. 2019, 97, 4298–4304. [Google Scholar] [CrossRef]
- Ault, T.B.; Clemmons, B.A.; Reese, S.T.; Dantas, F.G.; Franco, G.A.; Smith, T.P.L.; Edwards, J.L.; Myer, P.R.; Pohler, K.G. Bacterial Taxonomic Composition of the Postpartum Cow Uterus and Vagina Prior to Artificial Insemination. J. Anim. Sci. 2019, 97, 4305–4313. [Google Scholar] [CrossRef]
- Jeon, S.J.; Cunha, F.; Daetz, R.; Bicalho, R.C.; Lima, S.; Galvão, K.N. Ceftiofur Reduced Fusobacterium Leading to Uterine Microbiota Alteration in Dairy Cows with Metritis. Anim. Microbiome 2021, 3, s42523-s021. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.G.; Ericsson, A.C.A.; Poock, S.E.; Melendez, P.; Lucy, M.C. Hot Topic: 16S RRNA Gene Sequencing Reveals the Microbiome of the Virgin and Pregnant Bovine Uterus. J. Dairy Sci. 2017, 100, 4953–4960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassenaar, T.M.; Panigrahi, P. Is a Foetus Developing in a Sterile Environment? Lett. Appl. Microbiol. 2014, 59, 572–579. [Google Scholar] [CrossRef]
- Benner, M.; Ferwerda, G.; Joosten, I.; van der Molen, R.G. How Uterine Microbiota Might Be Responsible for a Receptive, Fertile Endometrium. Hum. Reprod. Update 2018, 24, 393–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardos, J.; Fiorentino, D.; Longman, R.E.; Paidas, M. Immunological Role of the Maternal Uterine Microbiome in Pregnancy: Pregnancies Pathologies and Alterated Microbiota. Front. Immunol. 2020, 10, 2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvão, K.N.; Bicalho, R.C.; Jeon, S.J. Symposium Review: The Uterine Microbiome Associated with the Development of Uterine Disease in Dairy Cows. J. Dairy Sci. 2019, 102, 11786–11797. [Google Scholar] [CrossRef] [PubMed]
- Karstrup, C.C.; Klitgaard, K.; Jensen, T.K.; Agerholm, J.S.; Pedersen, H.G. Presence of Bacteria in the Endometrium and Placentomes of Pregnant Cows. Theriogenology 2017, 99, 41–47. [Google Scholar] [CrossRef]
- MacIntyre, D.M.; Lim, H.C.; Ryan, K.; Kimmins, S.; Small, J.A.; MacLaren, L.A. Implantation-Associated Changes in Bovine Uterine Expression of Integrins and Extracellular Matrix. Biol. Reprod. 2002, 66, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasimanickam, R.; Duffield, T.; Foster, R.; Gartley, C.; Leslie, K.; Walton, J.; Johnson, W. Endometrial Cytology and Ultrasonography for the Detection of Subclinical Endometritis in Postpartum Dairy Cows. Theriogenology 2004, 62, 9–23. [Google Scholar] [CrossRef]
- Baranski, W.; Podhalicz-Dziegielewska, M.; Zdunczyk, S.; Janowski, T. The Diagnosis and Prevalence of Subclinical Endometritis in Cows Evaluated by Different Cytologic Thresholds. Theriogenology 2012, 78, 1939–1947. [Google Scholar] [CrossRef]
- Adnane, M.; Chapwanya, A.; Kaidi, R.; Meade, K.G.; O’Farrelly, C. Profiling Inflammatory Biommarkers in Cervico-Vaginal Mucus (CVM) Postpartum: Potential Early Indicators of Bovine Clinical Endometritis? Theriogenology 2017, 103, 117–122. [Google Scholar] [CrossRef]
- Brewer, A.; Cormican, P.; Lim, J.J.; Chapwanya, A.; O’Farrelly, C.; Meade, K.G. Qualitative and Quantitative Differences in Endometrial Inflammatory Gene Expression Precede the Development of Bovine Uterine Disease. Sci. Rep. 2020, 10, 18275. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of General 16S Ribosomal RNA Gene PCR Primers for Classical and Next-Generation Sequencing-Based Diversity Studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.a.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590-6. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team R: A Language and Environment for Statistical Computing. Available online: http://www.r-project.org/ (accessed on 2 February 2023).
- Lonergan, P.; Fair, T.; Forde, N.; Rizos, D. Embryo development in dairy cattle. Theriogenology 2016, 86, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Lietaer, L.; Pascottini, O.B.; Hernandez-Sanabria, E.; Kerckhof, F.M.; Lacoere, T.; Boon, N.; Vlaminck, L.; Opsomer, G.; van de Wiele, T. Low Microbial Biomass within the Reproductive Tract of Mid-Lactation Dairy Cows: A Study Approach. J. Dairy Sci. 2021, 104, 6159–6174. [Google Scholar] [CrossRef]
- Miranda-CasoLuengo, R.; Lu, J.; Williams, E.J.; Miranda-CasoLuengo, A.A.; Carrington, S.D.; Evans, A.C.O.; Meijer, W.G. Delayed Differentiation of Vaginal and Uterine Microbiomes in Dairy Cows Developing Postpartum Endometritis. PLoS ONE 2019, 14, e0200974. [Google Scholar] [CrossRef] [Green Version]
- Peterson, G.; Allen, C.R.; Holling, C.S. Ecological Resilience, Biodiversity, and Scale. Ecosystems 1998, 1, 6–18. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicalho, M.L.S.; Santin, T.; Rodrigues, M.X.; Marques, C.E.; Lima, S.F.; Bicalho, R.C. Dynamics of the microbiota found in the vaginas of dairy cows during the transition period: Associations with uterine diseases and reproductive outcome. J. Dairy Sci. 2017, 100, 3043–3058. [Google Scholar] [CrossRef]
- Gilbert, R.O.; Santos, N.R. Dynamics of postpartum endometrial cytology and bacteriology and their relationship to fertility in dairy cows. Theriogenology 2016, 85, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Ballas, P.; Reinländer, U.; Schlegl, R.; Ehling-Schulz, M.; Drillich, M.; Wagener, K. Characterization of intrauterine cultivable aerobic microbiota at the time of insemination in dairy cows with and without mild endometritis. Theriogenology 2021, 159, 28–34. [Google Scholar] [CrossRef]
- Jeon, S.J.; Vieira-Neto, A.; Gobikrushanth, M.; Daetz, R.; Mingoti, R.D.; Parize, A.C.B.; de Freitas, S.L.; da Costa, A.N.L.; Bicalho, R.C.; Lima, S.; et al. Uterine Microbiota Progression from Calving until Establishment of Metritis in Dairy Cows. Appl. Environ. Microbiol. 2015, 81, 6324–6332. [Google Scholar] [CrossRef] [Green Version]
- Azawi, O.; Omran, S.; Hadad, J. A Study of Endometritis Causing Repeat Breeding of Cycling Iraqi Buffalo Cows. Reprod. Domest. Anim. 2008, 43, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.R.V.; Karstrup, C.C.; Pedersen, H.G.; Agerholm, J.S.; Jensen, T.K.; Klitgaard, K. Revisiting Bovine Pyometra—New Insights into the Disease Using a Culture-Independent Deep Sequencing Approach. Vet. Microbiol. 2015, 175, 319–324. [Google Scholar] [CrossRef]
- Pascottini, O.B.; Van Schyndel, S.J.; Spricigo, J.F.W.; Rousseau, J.; Weese, J.S.; LeBlanc, S.J. Dynamics of uterine microbiota in postpartum dairy cows with clinical or subclinical endometritis. Sci. Rep. 2020, 10, 12353. [Google Scholar] [CrossRef]
- Moore, S.G.; Ericsson, A.C.; Behura, S.K.; Lamberson, W.R.; Evans, T.J.; McCabe, M.S.; Poock, S.E.; Lucy, M.C. Concurrent and long-term associations between the endometrial microbiota and endometrial transcriptome in postpartum dairy cows. BMC Genom. 2019, 20, 405. [Google Scholar] [CrossRef] [PubMed]
- Bicalho, M.L.S.; Lima, S.; Higgins, C.H.; Machado, V.S.; Lima, F.S.; Bicalho, R.C. Genetic and functional analysis of the bovine uterine microbiota. Part II: Purulent vaginal discharge versus healthy cows. J. Dairy Sci. 2017, 100, 3863–3874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.L.; Liu, M.C.; Xu, J.; An, L.G.; Wang, J.F.; Zhu, Y.H. Uterine Microbiota of Dairy Cows with Clinical and Subclinical Endometritis. Front. Microbiol. 2018, 9, 2691. [Google Scholar] [CrossRef] [PubMed]
- Paiano, R.B.; Moreno, L.Z.; Gomes, V.T.M.; Parra, B.M.; Barbosa, M.R.; Sato, M.I.Z.; Bonilla, J.; Pugliesi, G.; Baruselli, P.S.; Moreno, A.M. Assessment of the main pathogens associated with clinical and subclinical endometritis in cows by culture and MALDI-TOF mass spectrometry identification. J. Dairy Sci. 2022, 105, 3367–3376. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, A.A.M.J.; Munden, S.; McCabe, E.; Hurley, D.; Fanning, S.; Chapwanya, A.; Butaye, P. The Endometrial Microbiota—16S rRNA Gene Sequence Signatures in Healthy, Pregnant and Endometritis Dairy Cows. Vet. Sci. 2023, 10, 215. https://doi.org/10.3390/vetsci10030215
Becker AAMJ, Munden S, McCabe E, Hurley D, Fanning S, Chapwanya A, Butaye P. The Endometrial Microbiota—16S rRNA Gene Sequence Signatures in Healthy, Pregnant and Endometritis Dairy Cows. Veterinary Sciences. 2023; 10(3):215. https://doi.org/10.3390/vetsci10030215
Chicago/Turabian StyleBecker, Anne A. M. J., Stacie Munden, Evonne McCabe, Daniel Hurley, Séamus Fanning, Aspinas Chapwanya, and Patrick Butaye. 2023. "The Endometrial Microbiota—16S rRNA Gene Sequence Signatures in Healthy, Pregnant and Endometritis Dairy Cows" Veterinary Sciences 10, no. 3: 215. https://doi.org/10.3390/vetsci10030215