

Extracellular Vesicles in Sickle Cell Disease: A Promising Tool

Abstract
:1. Introduction
2. Extracellular Vesicles
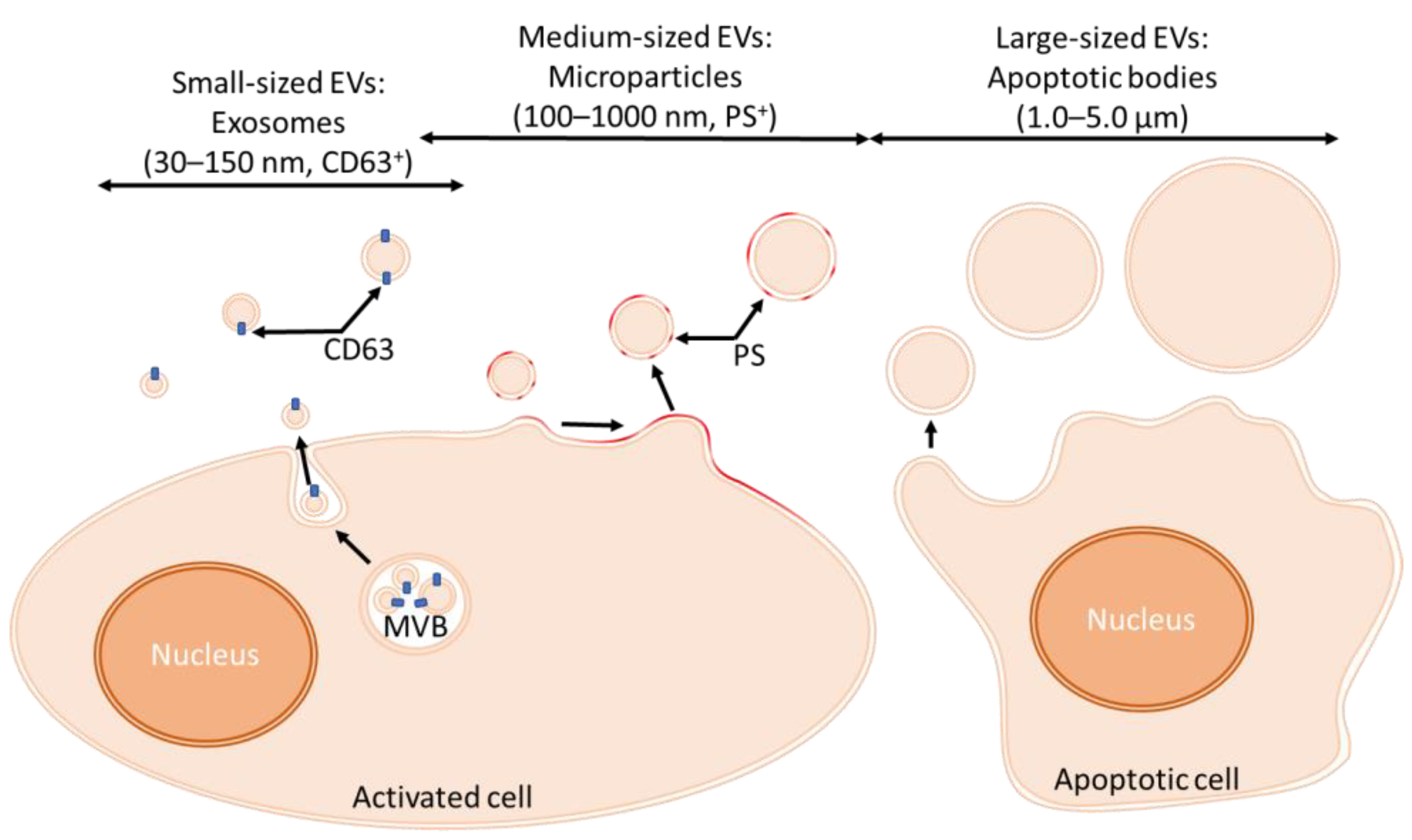
2.1. Classification and Biogenesis of EVs
2.1.1. Classification of EVs
2.1.2. Production of EVs
2.2. Isolation of EVs
2.3. Composition of EVs
3. Pathophysiology of SCD
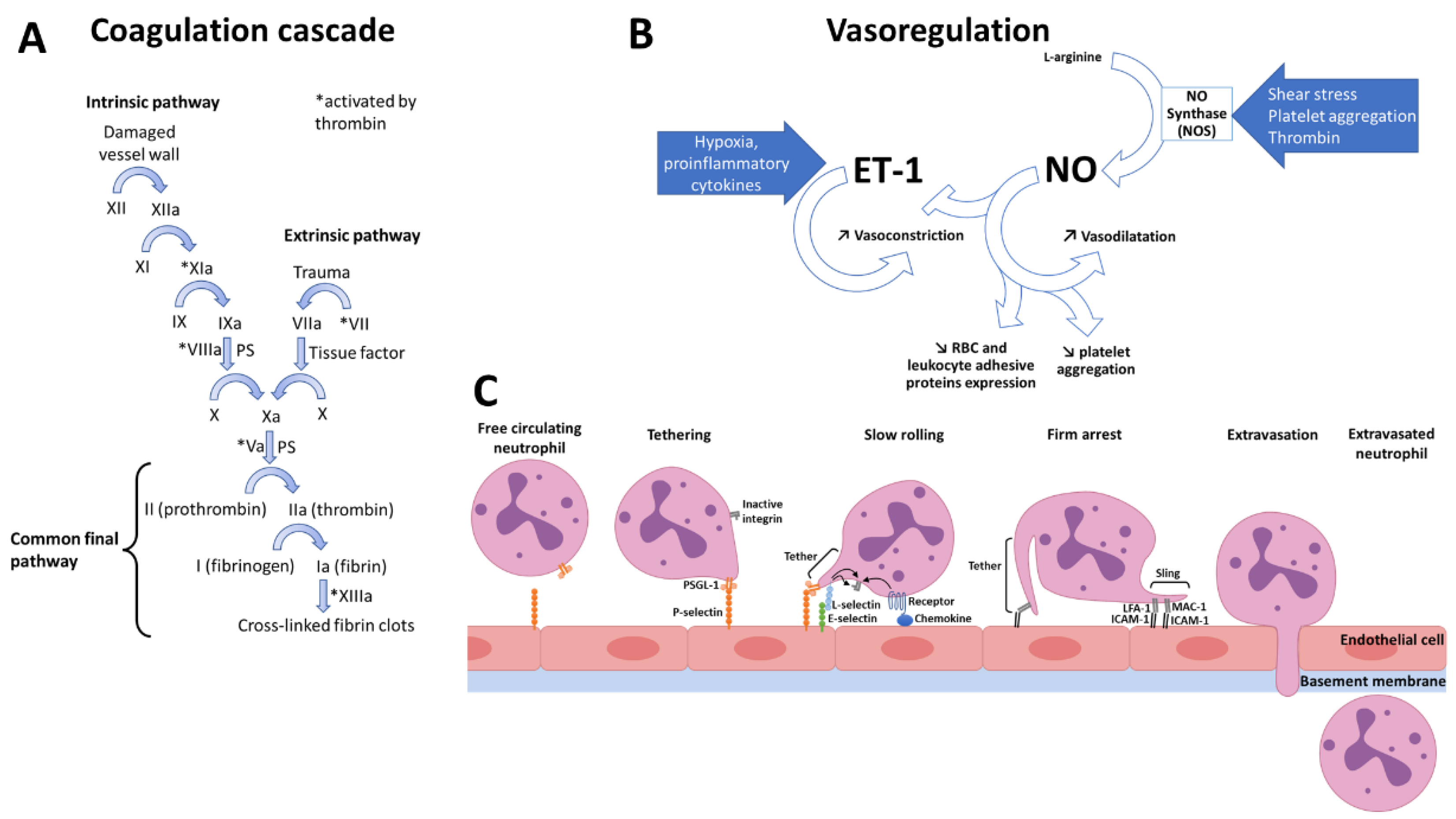
3.1. Physiological Hemostasis and Inflammation
3.1.1. Normal Hemostasis
3.1.2. Normal Inflammation
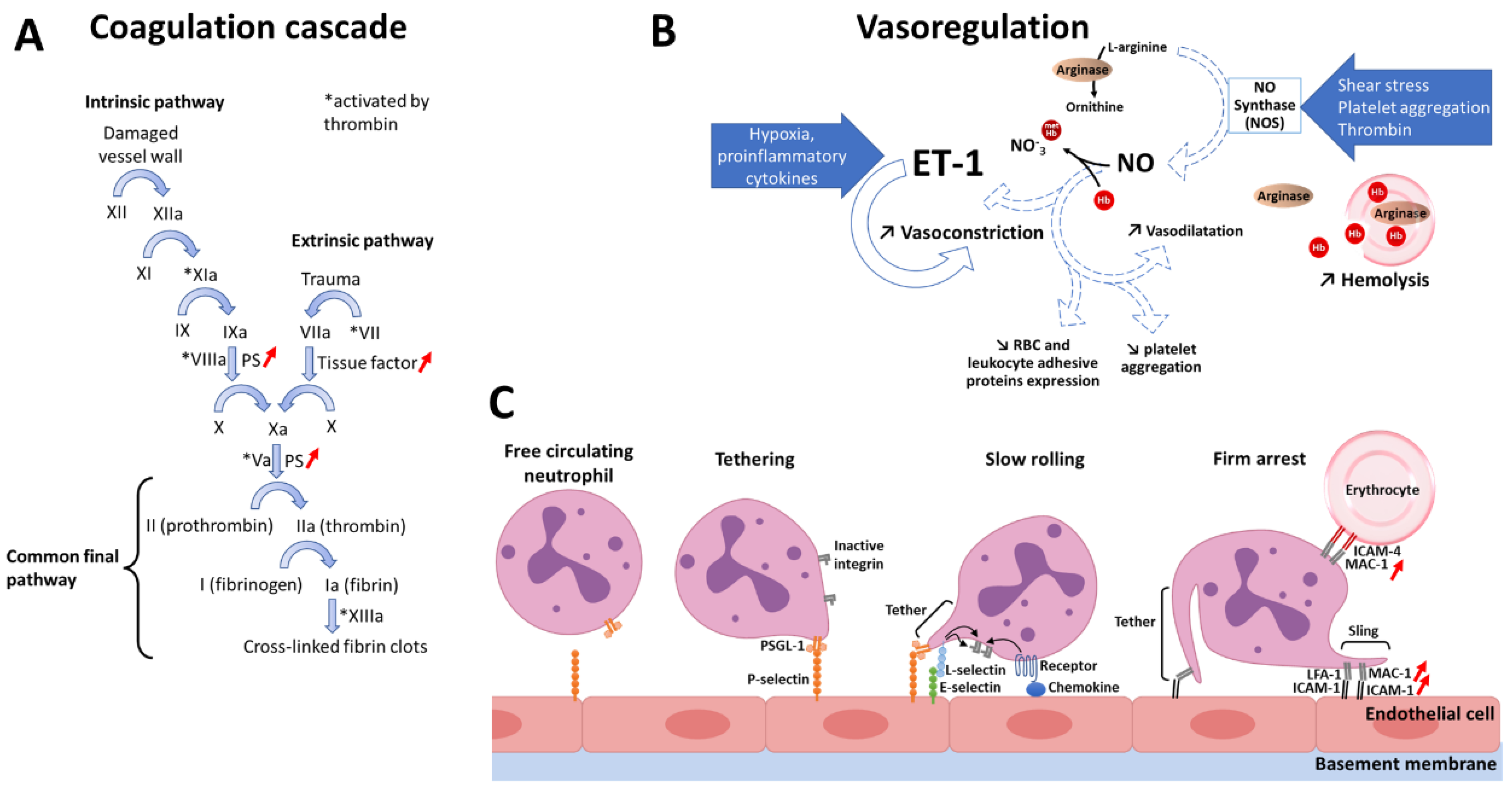
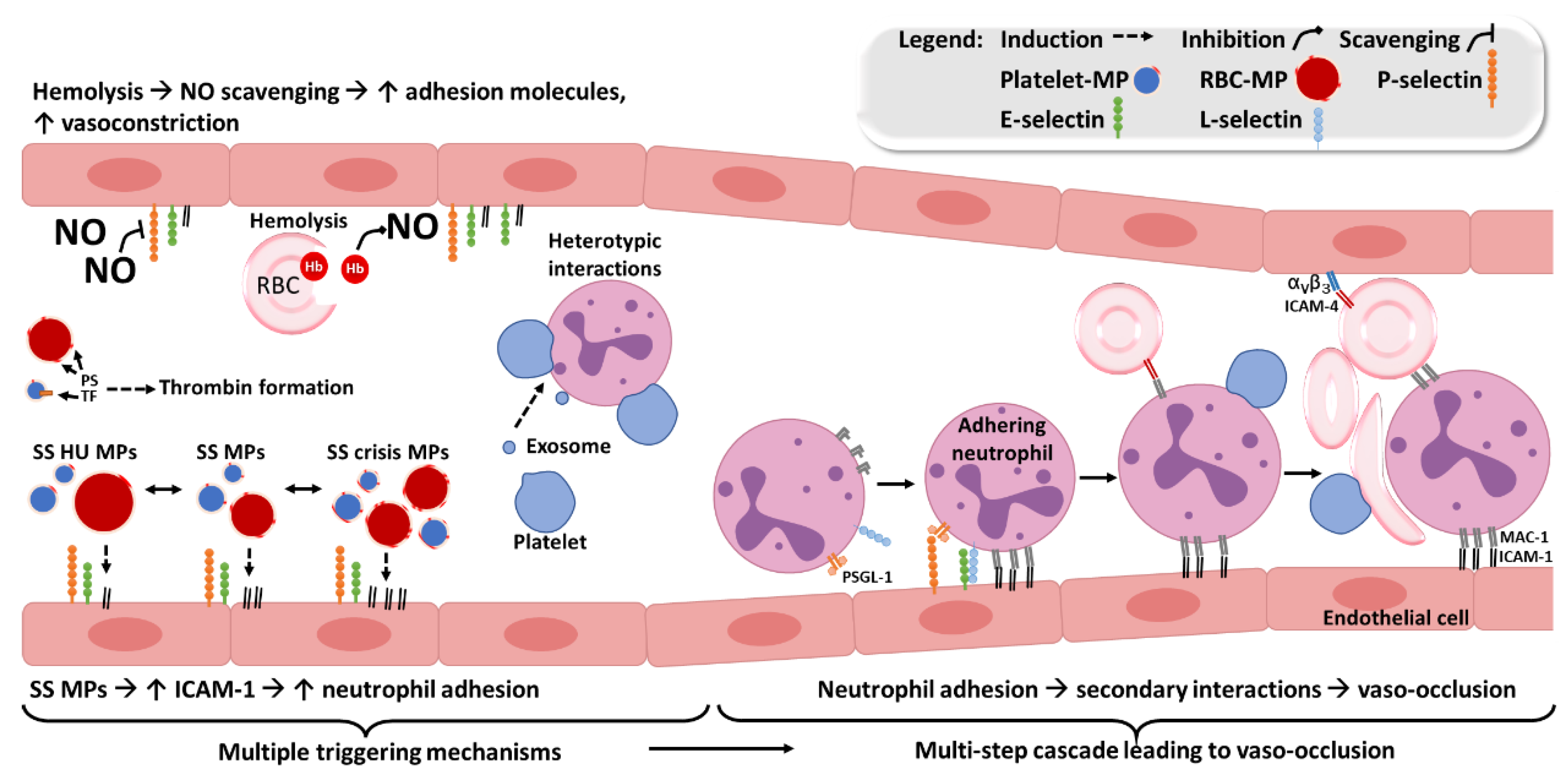
3.2. Dysregulated Mechanisms in SCD
3.2.1. Pro-Coagulant State
3.2.2. Decreased Nitric Oxide Bioavailability
3.2.3. Pro-Inflammatory State
3.3. EVs as Novel Biomarkers in SCD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference (Number of Included Patients) | Method | EV Type | EV Cell Type-of-Origin | EVs Concentration |
|---|---|---|---|---|
| Dembélé et al. [136] (232 SCA patients) | Flow cytometry | MPs | RBCs, platelets, monocytes, endothelial cells, progenitor cells | RBC-MPs/mL: 6678 (SCA), 1533 (Controls); PLT-MPs/µL: 3320 (SCA), 2627 (Controls) |
| Kasar et al. [138] (45 SCD patients) | Flow cytometry | MPs | RBCs, platelets, endothelial cells, monocytes | RBC-MPs (events/µL): 7.59 (SCD), 0.10 (Controls); PLT-MPs (events/µL): 12.58 (SCD), 1.59 (Controls) |
| Shet et al. [137] (16 SCD patients) | Flow cytometry | MPs | RBCs, platelets, monocytes | RBC-MPs/µL: ~650 (SCD), ~30 (Controls); PLT-MPs/µL: ~50 (SCD), ~50 (Controls) |
| Gerotziafas et al. [142] (92 SCA patients) | Flow cytometry | MPs | RBCs, platelets | RBC-MPs/µL: 1370 (SCA), 69 (Controls); PLT-MPs/µL: 1897 (SCA), 752 (Controls) |
| Garnier et al. [143] (33 SCD patients) | Flow cytometry | MPs | RBCs, platelets, monocytes, endothelial cells, leukocytes | RBC-MPs/µL: 631 (SCA), 260 (HbSC); PLT-MPs/µL: 6485 (SCA), 4014 (HbSC) |
| Lappin-carr et al. [144] (33 SCD patients) | Imaging flow cytometry | Exosomes | RBCs, endothelial cells, hematopoietic progenitors, lymphocytes, monocytes, platelets | RBC-Exo/µL: 31,338 (SCD), 9661 (Controls); PLT-Exo/µL: 2702 (SCD), 1116 (Controls) |
| Khalyfa et al. [145] (32 SCA patients) | Imaging flow cytometry, electron microscopy | Exosomes | Endothelial cells, endothelial progenitor cells, monocytes, platelets, RBCs | RBC-Exo/µL: 2,760,753 (SCA), 1,768,125 (Controls); PLT-Exo/µL: 5653 (SCA), 5435 (Controls) |
3.4. Effects of EVs in SCD
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piel, F.B.; Patil, A.P.; Howes, R.E.; Gething, P.W.; Temperley, W.H.; Hay, S.I.; Piel, F.B.; Patil, A.P.; Howes, R.E.; Nyangiri, O.A.; et al. Global Epidemiology of Sickle Haemoglobin in Neonates: A Contemporary Geostatistical Model-Based Map and Population Estimates. Lancet 2013, 381, 142–151. [Google Scholar] [CrossRef]
- Osunkwo, I.; Manwani, D.; Kanter, J. Current and Novel Therapies for the Prevention of Vaso-Occlusive Crisis in Sickle Cell Disease. Ther. Adv. Hematol. 2020, 11, 204062072095500. [Google Scholar] [CrossRef] [PubMed]
- Burger, D.; Schock, S.; Thompson, C.S.; Montezano, A.C.; Hakim, A.M.; Touyz, R.M. Microparticles: Biomarkers and Beyond. Clin. Sci. 2013, 124, 423–441. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Hugel, B.; Ohan, J.; Lesèche, G. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques a role for apoptosis in plaque thrombogenicity. Circulation 1999, 99, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Ridger, V.C.; Boulanger, C.M.; Angelillo-Scherrer, A.; Badimon, L.; Blanc-Brude, O.; Bochaton-Piallat, M.L.; Boilard, E.; Buzas, E.I.; Caporali, A.; Dignat-George, F.; et al. Microvesicles in Vascular Homeostasis and Diseases. Position Paper of the European Society of Cardiology (ESC) Working Group on Atherosclerosis and Vascular Biology. Thromb. Haemost. 2017, 117, 1296–1316. [Google Scholar] [CrossRef]
- Garnier, Y.; Claude, L.; Hermand, P.; Sachou, E.; Claes, A.; Desplan, K.; Chahim, B.; Roger, P.M.; Martino, F.; Colin, Y.; et al. Plasma microparticles of intubated COVID-19 patients cause endothelial cell death, neutrophil adhesion and netosis, in a phosphatidylserine-dependent manner. Br. J. Haematol. 2022, 196, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Amabile, N.; Heiss, C.; Chang, V.; Angeli, F.S.; Damon, L.; Rame, E.J.; McGlothlin, D.; Grossman, W.; De Marco, T.; Yeghiazarians, Y. Increased CD62e+ endothelial microparticle levels predict poor outcome in pulmonary hypertension patients. J. Heart Lung Transplant. 2009, 28, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Amabile, N.; Guérin, A.P.; Tedgui, A.; Boulanger, C.M.; London, G.M. Predictive value of circulating endothelial microparticles for cardiovascular mortality in end-stage renal failure: A pilot study. Nephrol. Dial. Transplant. 2012, 27, 1873–1880. [Google Scholar] [CrossRef]
- Nader, E.; Garnier, Y.; Connes, P.; Romana, M. Extracellular Vesicles in Sickle Cell Disease: Plasma Concentration, Blood Cell Types Origin Distribution and Biological Properties. Front. Med. 2021, 8, 728693. [Google Scholar] [CrossRef]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- György, B.; Szabó, T.G.; Pásztói, M.; Pál, Z.; Misják, P.; Aradi, B.; László, V.; Pállinger, É.; Pap, E.; Kittel, Á.; et al. Membrane Vesicles, Current State-of-the-Art: Emerging Role of Extracellular Vesicles. Cell Mol. Life Sci. 2011, 68, 2667–2688. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- el Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular Vesicles: Biology and Emerging Therapeutic Opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Witwer, K.W.; Soekmadji, C.; Hill, A.F.; Wauben, M.H.; Buzás, E.I.; di Vizio, D.; Falcon-Perez, J.M.; Gardiner, C.; Hochberg, F.; Kurochkin, I.V.; et al. Updating the MISEV Minimal Requirements for Extracellular Vesicle Studies: Building Bridges to Reproducibility. J. Extracell. Vesicles 2017, 6, 1396823. [Google Scholar] [CrossRef]
- Hade, M.D.; Suire, C.N.; Suo, Z. Mesenchymal Stem Cell-Derived Exosomes: Applications in Regenerative Medicine. Cells 2021, 10, 1959. [Google Scholar] [CrossRef]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Zhao, Z.; Wijerathne, H.; Godwin, A.K.; Soper, S.A. Isolation and analysis methods of extracellular vesicles (EVs). Extracell. Vesicles Circ. Nucl. Acids 2021, 2, 80. [Google Scholar] [CrossRef]
- Morel, O.; Toti, F.; Morel, N.; Freyssinet, J.M. Mircroparticles in Endothelial Cell and Vascular Homeostasis: Are They Really Noxious? Haematologica 2009, 94, 313–317. [Google Scholar] [CrossRef]
- Hu, Y.; Sun, Y.; Wan, C.; Dai, X.; Wu, S.; Lo, P.C.; Huang, J.; Lovell, J.F.; Jin, H.; Yang, K. Microparticles: Biogenesis, Characteristics and Intervention Therapy for Cancers in Preclinical and Clinical Research. J. Nanobiotechnol. 2022, 20, 189. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT Functions in Exosome Biogenesis, Composition and Secretion Highlights the Heterogeneity of Extracellular Vesicles. J. Cell Sci. 2013, 126 Pt 24, 5553–5565. [Google Scholar] [CrossRef] [Green Version]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef]
- Andreu, Z.; Yáñez-Mó, M. Tetraspanins in Extracellular Vesicle Formation and Function. Front. Immunol. 2014, 5, 442. [Google Scholar] [CrossRef]
- Heijnen, H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived From Exocytosis of Multivesicular Bodies and α-Granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes and Exosomes: Shedding the Confusion between Extracellular Vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Beaudoin, A.R.; Grondin, G. Shedding of Vesicular Material from the Cell Surface of Eukaryotic Cells: Different Cellular Phenomena. Biochim. Biophys. Acta 1991, 1071, 203–219. [Google Scholar] [CrossRef]
- Freyssinet, J.M.; Toti, F.; Hugel, B.; Gidon-Jeangirard, C.; Kunzelmann, C.; Martínez, M.C.; Meyer, D. Apoptosis in Vascular Disease. Thromb. Haemost. 1999, 82, 727–735. [Google Scholar] [CrossRef]
- Gilbert, G.E.; Sims, P.J.; Wiedmer, T.; Furie, B.; Furie, B.C.; Shattil, S.J. Platelet-Derived Microparticles Express High Affinity Receptors for Factor VIII. J. Biol. Chem. 1991, 266, 17261–17268. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Nickenig, G.; Werner, N. Microparticles—Messengers of Biological Information. J. Cell Mol. Med. 2010, 14, 2250–2256. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular Mechanisms Underlying the Formation of Circulating Microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef]
- Connor, D.E.; Exner, T.; Ma, D.D.F.; Joseph, J.E. The Majority of Circulating Platelet-Derived Microparticles Fail to Bind Annexin V, Lack Phospholipid-Dependent Procoagulant Activity and Demonstrate Greater Expression of Glycoprotein Ib. Thromb. Haemost. 2010, 103, 1044–1052. [Google Scholar] [CrossRef] [Green Version]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Dignat-George, F.; Boulanger, C.M. The Many Faces of Endothelial Microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Suire, C.N.; Hade, M.D. Extracellular Vesicles in Type 1 Diabetes: A Versatile Tool. Bioengineering 2022, 9, 105. [Google Scholar] [CrossRef]
- Lacroix, R.; Judicone, C.; Poncelet, P.; Robert, S.; Arnaud, L.; Sampol, J.; Dignat-George, F. Impact of Pre-Analytical Parameters on the Measurement of Circulating Microparticles: Towards Standardization of Protocol. J. Thromb. Haemost. 2012, 10, 437–446. [Google Scholar] [CrossRef]
- Mullier, F.; Bailly, N.; Chatelain, C.; Chatelain, B.; Dogné, J.M. Pre-Analytical Issues in the Measurement of Circulating Microparticles: Current Recommendations and Pending Questions. J. Thromb. Haemost. 2013, 11, 693–696. [Google Scholar] [CrossRef]
- Lacroix, R.; Judicone, C.; Mooberry, M.; Boucekine, M.; Key, N.S.; Dignat-George, F.; Ambrozic, A.; Bailly, N.; Buffat, C.; Buzas, E.; et al. Standardization of Pre-Analytical Variables in Plasma Microparticle Determination: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative Workshop. J. Thromb. Haemost. 2013, 11, 1190–1193. [Google Scholar] [CrossRef]
- Coumans, F.A.W.; Brisson, A.R.; Buzas, E.I.; Dignat-George, F.; Drees, E.E.E.; El-Andaloussi, S.; Emanueli, C.; Gasecka, A.; Hendrix, A.; Hill, A.F.; et al. Methodological Guidelines to Study Extracellular Vesicles. Circ. Res. 2017, 120, 1632–1648. [Google Scholar] [CrossRef]
- Garnier, Y.; Ferdinand, S.; Garnier, M.; Cita, K.-C.; Hierso, R.; Claes, A.; Connes, P.; Hardy-Dessources, M.-D.; Lapouméroulie, C.; Lemonne, N.; et al. Plasma Microparticles of Sickle Patients during Crisis or Taking Hydroxyurea Modify Endothelium Inflammatory Properties. Blood 2020, 136, 247–256. [Google Scholar] [CrossRef]
- Robert, S.; Poncelet, P.; Lacroix, R.; Raoult, D.; Dignat-George, F. More on: Calibration for the Measurement of Microparticles: Value of Calibrated Polystyrene Beads for Flow Cytometry-Based Sizing of Biological Microparticles. J. Thromb. Haemost. 2011, 9, 1676–1678. [Google Scholar] [CrossRef]
- Erdbrügger, U.; Lannigan, J. Analytical Challenges of Extracellular Vesicle Detection: A Comparison of Different Techniques. Cytometry A 2016, 89, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Szatanek, R.; Baj-Krzyworzeka, M.; Zimoch, J.; Lekka, M.; Siedlar, M.; Baran, J. The Methods of Choice for Extracellular Vesicles (EVs) Characterization. Int. J. Mol. Sci. 2017, 18, 1153. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Pisetsky, D.S. The Role of Microparticles in the Pathogenesis of Rheumatic Diseases. Nat. Rev. Rheumatol. 2010, 6, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 8973. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Kovalchuk, S.I.; Popkov, V.A.; Chernikov, V.P.; Zharikova, A.A.; Khutornenko, A.A.; Zorov, S.D.; Plokhikh, K.S.; Zinovkin, R.A.; Evtushenko, E.A.; et al. Do Extracellular Vesicles Derived from Mesenchymal Stem Cells Contain Functional Mito-chondria? Int. J. Mol. Sci. 2022, 23, 7408. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Willis, C.M.; Manferrari, G.; Rogall, R.; Fernandez-Vizarra, E.; Williamson, J.C.; Braga, A.; van den Bosch, A.; Leonardi, T.; et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLoS Biol. 2021, 19, e3001166. [Google Scholar] [CrossRef]
- Boudreau, L.H.; Duchez, A.C.; Cloutier, N.; Soulet, D.; Martin, N.; Bollinger, J.; Paré, A.; Rousseau, M.; Naika, G.S.; Lévesque, T.; et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 2014, 124, 2173–2183. [Google Scholar] [CrossRef]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borràs, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A Compendium for Extracellular Vesicles with Continuous Community Annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef]
- Kim, D.K.; Kang, B.; Kim, O.Y.; Choi, D.S.; Lee, J.; Kim, S.R.; Go, G.; Yoon, Y.J.; Kim, J.H.; Jang, S.C.; et al. EVpedia: An Integrated Database of High-Throughput Data for Systemic Analyses of Extracellular Vesicles. J. Extracell. Vesicles 2013, 2, 20384. [Google Scholar] [CrossRef]
- Simpson, R.J.; Kalra, H.; Mathivanan, S. ExoCarta as a Resource for Exosomal Research. J. Extracell. Vesicles 2012, 1, 18374. [Google Scholar] [CrossRef] [PubMed]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Mathivanan, S.; Ji, H.; Simpson, R.J. Two Distinct Populations of Exosomes Are Released from LIM1863 Colon Carcinoma Cell-Derived Organoids. Mol. Cell. Proteom. 2013, 12, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Crescitelli, R.; Lässer, C.; Szabó, T.G.; Kittel, A.; Eldh, M.; Dianzani, I.; Buzás, E.I.; Lötvall, J. Distinct RNA Profiles in Subpopulations of Extracellular Vesicles: Apoptotic Bodies, Microvesicles and Exosomes. J. Extracell. Vesicles 2013, 2, 20677. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Ofori-Acquah, S.F. Red Cells, Iron, & Erythropoiesis: Erythroid DAMPs Drive Inflammation in SCD. Blood 2014, 123, 3689. [Google Scholar] [CrossRef]
- Dautov, R.F.; Stafford, I.; Liu, S.; Cullen, H.; Madhani, M.; Chirkov, Y.Y.; Horowitz, J.D. Hypoxic Potentiation of Nitrite Effects in Human Vessels and Platelets. Nitric Oxide 2014, 40, 36–44. [Google Scholar] [CrossRef]
- Unic, A.; Derek, L.; Hodak, N.; Marijancevic, D.; Ceprnja, M.; Serdar, T.; Krhac, M.; Romic, Z. Endothelins—Clinical Perspectives. Biochem. Med. 2011, 3, 231–242. [Google Scholar] [CrossRef]
- Boulanger, C.; Luscher, T.F. Release of Endothelin from the Porcine Aorta. Inhibition of Endothelium-Derived Nitric Oxide. J. Clin. Investig. 1990, 85, 587–590. [Google Scholar] [CrossRef]
- Gryglewski, R.J.; Botting, R.M.; Vane, J.R. Mediators Produced by the Endothelial Cell. Hypertension 1988, 12, 530–548. [Google Scholar] [CrossRef]
- Myers, P.R.; Tanner, M.A. Vascular Endothelial Cell Regulation of Extracellular Matrix Collagen: Role of Nitric Oxide. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 717–722. [Google Scholar] [CrossRef]
- Belhassen, L.; Pelle, G.; Sediame, S.; Bachir, D.; Carville, C.; Bucherer, C.; Lacombe, C.; Galacteros, F.; Adnot, S. Endothelial Dysfunction in Patients with Sickle Cell Disease Is Related to Selective Impairment of Shear Stress–Mediated Vasodi-lation. Blood 2001, 97, 1584–1589. [Google Scholar] [CrossRef]
- Tronc, F.; Wassef, M.; Esposito, B.; Henrion, D.; Glagov, S.; Tedgui, A. Role of NO in Flow-Induced Remodeling of the Rabbit Common Carotid Artery. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1256–1262. [Google Scholar] [CrossRef]
- Langille, B.L.; O’Donnell, F. Reductions in Arterial Diameter Produced by Chronic Decreases in Blood Flow Are Endothelium-Dependent. Science 1986, 231, 405–407. [Google Scholar] [CrossRef]
- Langille, B.L.; Bendeck, M.P.; Keeley, F.W. Adaptations of Carotid Arteries of Young and Mature Rabbits to Reduced Carotid Blood Flow. Am. J. Physiol. Heart Circ. Physiol. 1989, 256, H931–H939. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil Extracellular Traps Mediate a Host Defense Response to Human Immunodeficiency Virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Petri, B.; Phillipson, M.; Kubes, P. The Physiology of Leukocyte Recruitment: An In Vivo Perspective. J. Immunol. 2008, 180, 6439–6446. [Google Scholar] [CrossRef]
- Su, Y.; Lei, X.; Wu, L.; Liu, L.; Liu, L. The Role of Endothelial Cell Adhesion Molecules P-Selectin, E-Selectin and Intercellular Adhesion Molecule-1 in Leucocyte Recruitment Induced by Exogenous Methylglyoxal. Immunology 2012, 137, 65–79. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Vestweber, D. How Leukocytes Cross the Vascular Endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Lefort, C.T.; Rossaint, J.; Moser, M.; Petrich, B.G.; Zarbock, A.; Monkley, S.J.; Critchley, D.R.; Ginsberg, M.H.; Fässler, R.; Ley, K. Distinct Roles for Talin-1 and Kindlin-3 in LFA-1 Extension and Affinity Regulation. Blood 2012, 119, 4275–4282. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Heit, B.; Colarusso, P.; Liu, L.; Ballantyne, C.M.; Kubes, P. Intraluminal Crawling of Neutrophils to Emigration Sites: A Molecularly Distinct Process from Adhesion in the Recruitment Cascade. J. Exp. Med. 2006, 203, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Carvalho-Tavares, J.; Hickey, M.J.; Hutchison, J.; Michaud, J.; Sutcliffe, I.T.; Kubes, P. A Role for Platelets and Endothelial Selectins in Tumor Necrosis Factor-Alpha-Induced Leukocyte Recruitment in the Brain Microvasculature. Circ. Res. 2000, 87, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Foxman, E.F.; Campbell, J.J.; Butcher, E.C. Multistep Navigation and the Combinatorial Control of Leukocyte Chemotaxis. J. Cell Biol. 1997, 139, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Mozzarelli, A.; Hofrichter, J.; Eaton, W.A. Delay Time of Hemoglobin S Polymerization Prevents Most Cells from Sickling in Vivo. Science 1987, 237, 500–506. [Google Scholar] [CrossRef]
- Zhang, D.; Xu, C.; Manwani, D.; Frenette, P.S. Neutrophils, Platelets, and Inflammatory Pathways at the Nexus of Sickle Cell Disease Pathophysiology. Blood 2016, 127, 801–809. [Google Scholar] [CrossRef]
- Ataga, K.I.; Key, N.S. Hypercoagulability in Sickle Cell Disease: New Approaches to an Old Problem. Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Educ. Program 2007, 2, 91–96. [Google Scholar] [CrossRef]
- de Franceschi, L.; Cappellini, M.D.; Olivieri, O. Thrombosis and Sickle Cell Disease. Semin. Thromb. Hemost. 2011, 37, 226–236. [Google Scholar] [CrossRef]
- Ataga, K.I.; Moore, C.G.; Hillery, C.A.; Jones, S.; Whinna, H.C.; Strayhorn, D.; Sohier, C.; Hinderliter, A.; Parise, L.V.; Orringer, E.P. Coagulation Activation and Inflammation in Sickle Cell Disease-Associated Pulmonary Hypertension. Haematologica 2008, 93, 20–26. [Google Scholar] [CrossRef]
- Naik, R.P.; Streiff, M.B.; Haywood, C.; Nelson, J.A.; Lanzkron, S. Venous Thromboembolism in Adults with Sickle Cell Disease: A serious and under-Recognized Complication. Am. J. Med. 2013, 126, 443. [Google Scholar] [CrossRef] [Green Version]
- Noubouossie, D.; Key, N.S.; Ataga, K.I. Coagulation Abnormalities of Sickle Cell Disease: Relationship with Clinical Outcomes and the Effect of Disease Modifying Therapies. Blood Rev. 2016, 30, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Setty, B.N.Y.; Key, N.S.; Rao, A.K.; Gayen-Betal, S.; Krishnan, S.; Dampier, C.D.; Stuart, M.J. Tissue Factor-Positive Monocytes in Children with Sickle Cell Disease: Correlation with Biomarkers of Haemolysis. Br. J. Haematol. 2012, 157, 370–380. [Google Scholar] [CrossRef]
- Solovey, A.; Gui, L.; Key, N.S.; Hebbel, R.P. Tissue Factor Expression by Endothelial Cells in Sickle Cell Anemia. J. Clin. Investig. 1998, 101, 1899–1904. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme Triggers TLR4 Signaling Leading to Endothelial Cell Activation and Vaso-Occlusion in Murine Sickle Cell Disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Setty, B.N.Y.; Betal, S.G. Microvascular Endothelial Cells Express a Phosphatidylserine Receptor: A Functionally Active Receptor for Phosphatidylserine-Positive Erythrocytes. Blood 2008, 111, 905–914. [Google Scholar] [CrossRef]
- Mankelow, T.J.; Griffiths, R.E.; Trompeter, S.; Flatt, J.F.; Cogan, N.M.; Massey, E.J.; Anstee, D.J. Autophagic Vesicles on Mature Human Reticulocytes Explain Phosphatidylserine-Positive Red Cells in Sickle Cell Disease. Blood 2015, 126, 1831–1834. [Google Scholar] [CrossRef]
- Kuypers, F.A.; Jong, K. The Role of Phosphatidylserine in Recognition and Removal of Erythrocytes. Cell. Mol. Biol. 2004, 50, 147–158. [Google Scholar]
- Abed, M.; Feger, M.; Alzoubi, K.; Pakladok, T.; Frauenfeld, L.; Geiger, C.; Towhid, S.T.; Lang, F. Sensitization of Erythrocytes to Suicidal Erythrocyte Death Following Water Deprivation. Kidney Blood Press. Res. 2013, 37, 567–578. [Google Scholar] [CrossRef]
- Joiner, C.H.; Jiang, M.; Franco, R.S. Deoxygenation-Induced Cation Fluxes in Sickle Cells. IV. Modulation by External Calcium. Am. J. Physiol. 1995, 269 Pt 1, C403–C409. [Google Scholar] [CrossRef]
- Yamaja Setty, B.N.; Koneti Rao, A.; Stuart, M.J. Thrombophilia in Sickle Cell Disease: The Red Cell Connection. Blood 2001, 98, 3228–3233. [Google Scholar] [CrossRef]
- Setty, B.N.; Kulkarni, S.; Rao, A.K.; Stuart, M.J. Fetal Hemoglobin in Sickle Cell Disease: Relationship to Erythrocyte Phosphatidylserine Exposure and Coagulation Activation. Blood 2000, 96, 1119–1124. [Google Scholar] [CrossRef]
- Covitz, W.; Espeland, M.; Gallagher, D.; Hellenbrand, W.; Leff, S.; Talner, N. The Heart in Sickle Cell Anemia. The Coop-erative Study of Sickle Cell Disease (CSSCD). Chest 1995, 108, 1214–1219. [Google Scholar] [CrossRef]
- Bucherer, C.; Ladjouzi, J.; Lacombe, C.; Lelièvre, J.C.; Vandewalle, H.; Beuzard, Y.; Galacteros, F. Effect of Deoxygenation on Rheological Behavior of Density Separated Sickle Cell Suspensions. Clin. Hemorheol. Microcirc. 1992, 12, 415–425. [Google Scholar] [CrossRef]
- Chirico, E.N.; Pialoux, V. Role of Oxidative Stress in the Pathogenesis of Sickle Cell Disease. IUBMB Life 2012, 64, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.R.; Kato, G.J.; Poljakovic, M.; Wang, X.; Blackwelder, W.C.; Sachdev, V.; Hazen, S.L.; Vichinsky, E.P.; Morris, S.M.; Gladwin, M.T. Dysregulated Arginine Metabolism, Hemolysis-Associated Pulmonary Hypertension, and Mortality in Sickle Cell Disease. JAMA 2005, 294, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, A.; Gladwin, M.T.; Kim-Shapiro, D.B. Computation of Plasma Hemoglobin Nitric Oxide Scavenging in Hemolytic Anemias. Free Radic. Biol. Med. 2006, 41, 1557–1565. [Google Scholar] [CrossRef]
- Chen, K.; Piknova, B.; Pittman, R.N.; Schechter, A.N.; Popel, A.S. Nitric Oxide from Nitrite Reduction by Hemoglobin in the Plasma and Erythrocytes. Nitric Oxide 2008, 18, 47–60. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary Hyper-tension as a Risk Factor for Death in Patients with Sickle Cell Disease. N. Engl. J. Med. 2004, 3509350, 886–895. [Google Scholar] [CrossRef]
- Kato, G.J.; McGowan, V.; Machado, R.F.; Little, J.A.; Taylor VI, J.; Morris, C.R.; Nichols, J.S.; Wang, X.; Poljakovic, M.; Morris, S.M.; et al. Lactate Dehydrogenase as a Biomarker of Hemolysis-Associated Nitric Oxide Resistance, Priapism, Leg Ulceration, Pulmonary Hypertension, and Death in Patients with Sickle Cell Disease. Blood 2006, 107, 2279–2285. [Google Scholar] [CrossRef]
- Bernaudin, F.; Verlhac, S.; Chevret, S.; Torres, M.; Coic, L.; Arnaud, C.; Kamdem, A.; Hau, I.; Neonato, M.G.; Delacourt, C. G6PD Deficiency, Absence of Alpha-Thalassemia, and Hemolytic Rate at Baseline Are Significant Independent Risk Factors for Abnormally High Cerebral Velocities in Patients with Sickle Cell Anemia. Blood 2008, 112, 4314–4317. [Google Scholar] [CrossRef]
- Baldwin, C.; Nolan, V.G.; Wyszynski, D.F.; Ma, Q.L.; Sebastiani, P.; Embury, S.H.; Bisbee, A.; Farrell, J.; Farrer, L.; Steinberg, M.H. Association of Klotho, Bone Morphogenic Protein 6, and Annexin A2 Polymorphisms with Sickle Cell Osteonecrosis. Blood 2005, 106, 372. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A. Reduced Sickle Erythrocyte Dehydration in Vivo by Endothelin-1 Receptor Antagonists. Am. J. Physiol. Cell Physiol. 2007, 293, C960–C966. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Miner, J.J.; Yago, T.; Yao, L.; Lupu, F.; Xia, L.; McEver, R.P. Differential Regulation of Human and Murine P-Selectin Expression and Function In Vivo. J. Exp. Med. 2010, 207, 2975–2987. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chang, J.; Jang, J.E.; Peired, A.J.; Chiang, E.Y.; Frenette, P.S. Heterotypic Interactions Enabled by Polarized Neutrophil Microdomains Mediate Thromboinflammatory Injury. Nat. Med. 2009, 15, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Aich, A.; Lamarre, Y.; Sacomani, D.P.; Kashima, S.; Covas, D.T.; de la Torre, L.G. Microfluidics in Sickle Cell Disease Research: State of the Art and a Perspective Beyond the Flow Problem. Front. Mol. Biosci. 2021, 7, 558982. [Google Scholar] [CrossRef]
- Morikis, V.A.; Hernandez, A.A.; Magnani, J.L.; Sperandio, M.; Simon, S.I. Targeting Neutrophil Adhesive Events to Address Vaso-Occlusive Crisis in Sickle Cell Patients. Front. Immunol. 2021, 12, 1256. [Google Scholar] [CrossRef]
- Wei, A.; Grigg, A. Granulocyte Colony-Stimulating Factor-Induced Sickle Cell Crisis and Multiorgan Dysfunction in a Patient with Compound Heterozygous Sickle Cell/Beta+ Thalassemia. Blood 2001, 97, 3998–3999. [Google Scholar] [CrossRef]
- Adler, B.K.; Salzman, D.E.; Carabasi, M.H.; Vaughan, W.P.; Reddy, V.V.B.; Prchal, J.T. Fatal Sickle Cell Crisis after Granulocyte Colony-Stimulating Factor Administration. Blood 2001, 97, 3313–3314. [Google Scholar] [CrossRef]
- Abboud, M.; Laver, J.; Blau, C.A. Granulocytosis Causing Sickle-Cell Crisis. Lancet 1998, 351, 959. [Google Scholar] [CrossRef]
- Wali, Y.; Beshlawi, I.; Fawaz, N.; Alkhayat, A.; Zalabany, M.; Elshinawy, M.; Al-Kindi, S.; Al-Rawas, A.H.A.; Klein, C. Coexistence of Sickle Cell Disease and Severe Congenital Neutropenia: First Impressions Can Be Deceiving. Eur. J. Haematol. 2012, 89, 245–249. [Google Scholar] [CrossRef]
- Lard, L.R.; Mul, F.P.J.; de Haas, M.; Roos, D.; Duits, A.J. Neutrophil Activation in Sickle Cell Disease. J. Leukoc. Biol. 1999, 66, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Canalli, A.A.; Franco-Penteado, C.F.; Saad, S.T.O.; Conran, N.; Costa, F.F. Increased Adhesive Properties of Neutrophils in Sickle Cell Disease May Be Reversed by Pharmacological Nitric Oxide Donation. Haematologica 2008, 93, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Lum, A.; Wun, T.; Staunton, D.; Simon, S. Inflammatory Potential of Neutrophils Detected in Sickle Cell Disease. Am. J. Hematol. 2004, 76, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Hermand, P.; Gane, P.; Huet, M.; Jallu, V.; Kaplan, C.; Sonneborn, H.H.; Cartron, J.P.; Bailly, P. Red Cell ICAM-4 Is a Novel Ligand for Platelet-Activated Alpha IIbbeta 3 Integrin. J. Biol. Chem. 2003, 278, 4892–4898. [Google Scholar] [CrossRef] [PubMed]
- Zennadi, R.; Hines, P.C.; de Castro, L.M.; Cartron, J.P.; Parise, L.V.; Telen, M.J. Epinephrine Acts through Erythroid Signaling Pathways to Activate Sickle Cell Adhesion to Endothelium via LW-Alphavbeta3 Interactions. Blood 2004, 104, 3774–3781. [Google Scholar] [CrossRef] [PubMed]
- Zennadi, R.; Whalen, E.J.; Soderblom, E.J.; Alexander, S.C.; Thompson, J.W.; Dubois, L.G.; Moseley, M.A.; Telen, M.J. Erythrocyte Plasma Membrane-Bound ERK1/2 Activation Promotes ICAM-4-Mediated Sickle Red Cell Adhesion to Endothelium. Blood 2012, 119, 1217–1227. [Google Scholar] [CrossRef]
- Wun, T.; Paglieroni, T.; Tablin, F.; Welborn, J.; Nelson, K.; Cheung, A. Platelet Activation and Platelet-Erythrocyte Aggregates in Patients with Sickle Cell Anemia. J. Lab. Clin. Med. 1997, 129, 507–516. [Google Scholar] [CrossRef]
- Brittain, J.E.; Knoll, C.M.; Ataga, K.I.; Orringer, E.P.; Parise, L.V. Fibronectin Bridges Monocytes and Reticulocytes via Integrin Alpha4beta1. Br. J. Haematol. 2008, 141, 872–881. [Google Scholar] [CrossRef]
- Shiu, Y.T.; Udden, M.M.; McIntire, L.V. Perfusion with Sickle Erythrocytes Up-Regulates ICAM-1 and VCAM-1 Gene Expression in Cultured Human Endothelial Cells. Blood 2000, 95, 3232–3241. [Google Scholar] [CrossRef]
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated Autoxidation and Heme Loss Due to Instability of Sickle Hemoglobin. Proc. Natl. Acad. Sci. USA 1988, 85, 237–241. [Google Scholar] [CrossRef]
- Nagababu, E.; Fabry, M.E.; Nagel, R.L.; Rifkind, J.M. Heme Degradation and Oxidative Stress in Murine Models for Hemoglobinopathies: Thalassemia, Sickle Cell Disease and Hemoglobin C Disease. Blood Cells Mol. Dis. 2008, 41, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nader, E.; Conran, N.; Romana, M.; Connes, P. Vasculopathy in Sickle Cell Disease: From Red Blood Cell Sickling to Vascular Dysfunction. Compr. Physiol. 2021, 11, 1785–1803. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; da Silva, M.C.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin Therapy Reverts Heme-Induced Proinflammatory Phenotypic Switching of Macrophages in a Mouse Model of Sickle Cell Disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef]
- Bourne, J.H.; Colicchia, M.; Di, Y.; Martin, E.; Slater, A.; Roumenina, L.T.; Dimitrov, J.D.; Watson, S.P.; Rayes, J. Heme Induces Human and Mouse Platelet Activation through C-Type-Lectin-like Receptor-2. Haematologica 2021, 106, 626–629. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-Induced Neutrophil Extracellular Traps Contribute to the Pathogenesis of Sickle Cell Disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Nur, E.; Biemond, B.J.; van Mierlo, G.J.; Solati, S.; Brandjes, D.P.; Otten, H.M.; Schnog, J.J.; Zeerleder, S. Nucleosomes and Neutrophil Activation in Sickle Cell Disease Painful Crisis. Haematologica 2013, 98, 1797–1803. [Google Scholar] [CrossRef]
- Jansen, F.; Nickenig, G.; Werner, N. Extracellular Vesicles in Cardiovascular Disease: Potential Applications in Diagnosis, Prognosis, and Epidemiology. Circ. Res. 2017, 120, 1649–1657. [Google Scholar] [CrossRef]
- Nébor, D.; Romana, M.; Santiago, R.; Vachiery, N.; Picot, J.; Broquere, C.; Chaar, V.; Doumdo, L.; Odièvre, M.H.; Benkerrou, M.; et al. Fetal Hemoglobin and Hydroxycarbamide Modulate Both Plasma Concentration and Cellular Origin of Circulating Microparticles in Sickle Cell Anemia Children. Haematologica 2013, 98, 862–867. [Google Scholar] [CrossRef]
- Tantawy, A.A.G.; Adly, A.A.M.; Ismail, E.A.R.; Habeeb, N.M.; Farouk, A. Circulating Platelet and Erythrocyte Microparticles in Young Children and Adolescents with Sickle Cell Disease: Relation to Cardiovascular Complications. Platelets 2013, 24, 605–614. [Google Scholar] [CrossRef]
- Westerman, M.; Pizzey, A.; Hirschman, J.; Cerino, M.; Weil-Weiner, Y.; Ramotar, P.; Eze, A.; Lawrie, A.; Purdy, G.; Mackie, I.; et al. Microvesicles in Haemoglobinopathies Offer Insights into Mechanisms of Hypercoagulability, Haemolysis and the Effects of Therapy. Br. J. Haematol. 2008, 142, 126–135. [Google Scholar] [CrossRef]
- Olatunya, O.S.; Lanaro, C.; Longhini, A.L.; Penteado, C.F.F.; Fertrin, K.Y.; Adekile, A.; Saad, S.T.O.; Costa, F.F. Red Blood Cells Microparticles Are Associated with Hemolysis Markers and May Contribute to Clinical Events among Sickle Cell Disease Patients. Ann. Hematol. 2019, 98, 2507–2521. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Murphy, C.; Eakins, E.; Kunde, J.; Corvetta, D.; di Pierro, A.; Negri, G.; Guido, M.; Sainati, L.; Mahon, C.M.; et al. Circulating Microparticles, Protein C, Free Protein S and Endothelial Vascular Markers in Children with Sickle Cell Anaemia. J. Extracell Vesicles 2015, 4, 28414. [Google Scholar] [CrossRef] [PubMed]
- Brunetta, D.M.; de Santis, G.C.; Silva-Pinto, A.C.; Oliveira De Oliveira, L.C.; Covas, D.T. Hydroxyurea Increases Plasma Concentrations of Microparticles and Reduces Coagulation Activation and Fibrinolysis in Patients with Sickle Cell Anemia. Acta Haematol. 2015, 133, 287–294. [Google Scholar] [CrossRef]
- Garnier, Y.; Ferdinand, S.; Connes, P.; Garnier, M.; Etienne-Julan, M.; Lemonne, N.; Romana, M. Decrease of Externalized Phosphatidylserine Density on Red Blood Cell-Derived Microparticles in SCA Patients Treated with Hydroxycarbamide. Br. J. Haematol. 2017, 182, 427–456. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.; Schiffelers, R.; Kuypers, F.; Larkin, S.; Gildengorin, G.; van Solinge, W.; Hoppe, C. Microparticles as Biomarkers of Osteonecrosis of the Hip in Sickle Cell Disease. Br. J. Haematol. 2015, 168, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Dembélé, A.K.; Lapoumeroulie, C.; Diaw, M.; Tessougue, O.; Offredo, L.; Diallo, D.A.; Diop, S.; Elion, J.; Colin-Aronovicz, Y.; Tharaux, P.L.; et al. Cell-Derived Microparticles and Sickle Cell Disease Chronic Vasculopathy in Sub-Saharan Africa: A Multinational Study. Br. J. Haematol. 2021, 192, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Shet, A.S.; Aras, O.; Gupta, K.; Hass, M.J.; Rausch, D.J.; Saba, N.; Koopmeiners, L.; Key, N.S.; Hebbel, R.P. Sickle Blood Contains Tissue Factor-Positive Microparticles Derived from Endothelial Cells and Monocytes. Blood 2003, 102, 2678–2683. [Google Scholar] [CrossRef]
- Kasar, M.; Boğa, C.; Yeral, M.; Asma, S.; Kozanoglu, I.; Ozdogu, H. Clinical Significance of Circulating Blood and Endothelial Cell Microparticles in Sickle-Cell Disease. J. Thromb. Thrombolysis 2014, 38, 167–175. [Google Scholar] [CrossRef]
- van Tits, L.J.; van Heerde, W.L.; Landburg, P.P.; Boderie, M.J.; Muskiet, F.A.J.; Jacobs, N.; Duits, A.J.; Schnog, J.B. Plasma Annexin A5 and Microparticle Phosphatidylserine Levels Are Elevated in Sickle Cell Disease and Increase Further during Painful Crisis. Biochem. Biophys. Res. Commun. 2009, 390, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Hierso, R.; Lemonne, N.; Villaescusa, R.; Lalanne-Mistrih, M.L.; Charlot, K.; Etienne-Julan, M.; Tressières, B.; Lamarre, Y.; Tarer, V.; Garnier, Y.; et al. Exacerbation of Oxidative Stress during Sickle Vaso-Occlusive Crisis Is Associated with Decreased Anti-Band 3 Autoantibodies Rate and Increased Red Blood Cell-Derived Microparticle Level: A Prospective Study. Br. J. Haematol. 2017, 176, 805–813. [Google Scholar] [CrossRef]
- Nebor, D.; Bowers, A.; Connes, P.; Hardy-Dessources, M.D.; Knight-Madden, J.; Cumming, V.; Reid, M.; Romana, M. Plasma Concentration of Platelet-Derived Microparticles Is Related to Painful Vaso-Occlusive Phenotype Severity in Sickle Cell Anemia. PLoS ONE 2014, 9, e87243. [Google Scholar] [CrossRef]
- Gerotziafas, G.T.; van Dreden, P.; Chaari, M.; Galea, V.; Khaterchi, A.; Lionnet, F.; Stankovic-Stojanovic, K.; Blanc-Brude, O.; Woodhams, B.; Maier-Redelsperger, M.; et al. The Acceleration of the Propagation Phase of Thrombin Generation in Patients with Steady-State Sickle Cell Disease Is Associated with Circulating Erythrocyte-Derived Microparticles. Thromb. Haemost. 2012, 107, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Garnier, Y.; Ferdinand, S.; Etienne-Julan, M.; Elana, G.; Petras, M.; Doumdo, L.; Tressières, B.; Lalanne-Mistrih, M.L.; Hardy-Dessources, M.D.; Connes, P.; et al. Differences of Microparticle Patterns between Sickle Cell Anemia and Hemoglobin SC Patients. PLoS ONE 2017, 12, e0177397. [Google Scholar] [CrossRef]
- Lapping-Carr, G.; Khalyfa, A.; Rangel, S.; Darlington, W.; Beyer, E.C.; Peddinti, R.; Cunningham, J.M.; Gozal, D. Exosomes Contribute to Endothelial Integrity and Acute Chest Syndrome Risk: Preliminary Findings. Pediatr. Pulmonol. 2017, 52, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Khalyfa, A.; Khalyfa, A.A.; Akbarpour, M.; Connes, P.; Romana, M.; Lapping-Carr, G.; Zhang, C.; Andrade, J.; Gozal, D. Extracellular Microvesicle MicroRNAs in Children with Sickle Cell Anaemia with Divergent Clinical Phenotypes. Br. J. Haematol. 2016, 174, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Ruf, W.; Rehemtulla, A.; Edgington, T.S. Phospholipid-Independent and -Dependent Interactions Required for Tissue Factor Receptor and Cofactor Function. J. Biol. Chem. 1991, 266, 2158–2166. [Google Scholar] [CrossRef]
- Bach, R.; Gentry, R.; Nemerson, Y. Factor VII Binding to Tissue Factor in Reconstituted Phospholipid Vesicles: Induction of Cooperativity by Phosphatidylserine. Biochemistry 1986, 25, 4007–4020. [Google Scholar] [CrossRef]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating Microparticles: Pathophysiology and Clinical Implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef]
- Castaman, G.; Yu-Feng, L.; Battistin, E.; Rodeghiero, F. Characterization of a Novel Bleeding Disorder with Isolated Prolonged Bleeding Time and Deficiency of Platelet Microvesicle Generation. Br. J. Haematol. 1997, 96, 458–463. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; van Schilfgaarde, M.; van Oerle, R.; Renné, T.; ten Cate, H.; Spronk, H.M.H. Platelet- and Erythrocyte-Derived Microparticles Trigger Thrombin Generation via Factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef]
- Rautou, P.-E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.-C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles from Human Atherosclerotic Plaques Promote Endothelial ICAM-1-Dependent Monocyte Adhesion and Transendothelial Migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial Expression of Autocrine VEGF upon the Uptake of Tumor-Derived Microvesicles Containing Oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, P.K.J.; Holopainen, J.M. Mechanisms of Initiation of Membrane Fusion: Role of Lipids. Biosci. Rep. 2000, 20, 465–482. [Google Scholar] [CrossRef] [PubMed]
- Camus, S.M.; de Moraes, J.A.; Bonnin, P.; Abbyad, P.; le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.C.; Charue, D. Circulating Cell Membrane Microparticles Transfer Heme to Endothelial Cells and Trigger Vasoocclusions in Sickle Cell Disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef]
- Awojoodu, A.O.; Keegan, P.M.; Lane, A.R.; Zhang, Y.; Lynch, K.R.; Platt, M.O.; Botchwey, E.A. Acid Sphingomyelinase Is Activated in Sickle Cell Erythrocytes and Contributes to Inflammatory Microparticle Generation in SCD. Blood 2014, 124, 1941–1950. [Google Scholar] [CrossRef]
- Barry, O.P.; Praticò, D.; Savani, R.C.; FitzGerald, G.A. Modulation of Monocyte-Endothelial Cell Interactions by Platelet Microparticles. J. Clin. Investig. 1998, 102, 136–144. [Google Scholar] [CrossRef]
- Wang, J.G.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.Y.; Mackman, N. Monocytic Microparticles Activate Endothelial Cells in an IL-1beta-Dependent Manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef]
- Nader, E.; Romana, M.; Guillot, N.; Fort, R.; Stauffer, E.; Lemonne, N.; Garnier, Y.; Skinner, S.C.; Etienne-Julan, M.; Robert, M.; et al. Association Between Nitric Oxide, Oxidative Stress, Eryptosis, Red Blood Cell Microparticles, and Vascular Function in Sickle Cell Anemia. Front. Immunol. 2020, 2885, 11. [Google Scholar] [CrossRef]
- Silachev, D.N.; Goryunov, K.V.; Shpilyuk, M.A.; Beznoschenko, O.S.; Morozova, N.Y.; Kraevaya, E.E.; Popkov, V.A.; Pevzner, I.B.; Zorova, L.D.; Evtushenko, E.A.; et al. Effect of MSCs and MSC-Derived Extracellular Vesicles on Human Blood Coagulation. Cells 2019, 8, 258. [Google Scholar] [CrossRef]
- Vats, R.; Brzoska, T.; Bennewitz, M.F.; Jimenez, M.A.; Pradhan-Sundd, T.; Tutuncuoglu, E.; Jonassaint, J.; Gutierrez, E.; Watkins, S.C.; Shiva, S.; et al. Platelet Extracellular Vesicles Drive Inflammasome-IL1β-Dependent Lung Injury in Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2020, 201, 33–46. [Google Scholar] [CrossRef]
- Lapping-Carr, G.; Gemel, J.; Mao, Y.; Beyer, E.C. Circulating Extracellular Vesicles and Endothelial Damage in Sickle Cell Disease. Front. Physiol. 2020, 11, 1063. [Google Scholar] [CrossRef] [PubMed]
- Gemel, J.; Zhang, J.; Mao, Y.; Lapping-carr, G.; Beyer, E.C. Circulating Small Extracellular Vesicles May Contribute to Vaso-Occlusive Crises in Sickle Cell Disease. J. Clin. Med. 2022, 11, 816. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.J.; Jy, W.; Mauro, L.M.; Soderland, C.; Horstman, L.L.; Ahn, Y.S. Endothelial Cells Release Phenotypically and Quantitatively Distinct Microparticles in Activation and Apoptosis. Thromb. Res. 2003, 109, 175–180. [Google Scholar] [CrossRef]
- Prudent, M.; Crettaz, D.; Delobel, J.; Seghatchian, J.; Tissot, J.D.; Lion, N. Differences between Calcium-Stimulated and Storage-Induced Erythrocyte-Derived Microvesicles. Transfus. Apher. Sci. 2015, 53, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O. Progressive Multifocal Leukoencephalopathy in Patients on Immunomodulatory Therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef] [Green Version]




| Exosome | Microparticle | Apoptotic Bodies | |
|---|---|---|---|
| Size (nm) | 30–150 | 100–1000 | 1000–5000 |
| Density (g/cm3) | 1.13–1.19 | 1.04–1.07 | 1.16–1.28 |
| Origin | Multivesicular body | Plasma membrane | Plasma membrane |
| Formation mechanism | Exocytosis of MVB | Budding from PM | Budding from PM |
| Production pathway | ESCRT-dependent * | Ca2+-dependent | Apoptosis-related pathways |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamarre, Y.; Nader, E.; Connes, P.; Romana, M.; Garnier, Y. Extracellular Vesicles in Sickle Cell Disease: A Promising Tool. Bioengineering 2022, 9, 439. https://doi.org/10.3390/bioengineering9090439
Lamarre Y, Nader E, Connes P, Romana M, Garnier Y. Extracellular Vesicles in Sickle Cell Disease: A Promising Tool. Bioengineering. 2022; 9(9):439. https://doi.org/10.3390/bioengineering9090439
Chicago/Turabian StyleLamarre, Yann, Elie Nader, Philippe Connes, Marc Romana, and Yohann Garnier. 2022. "Extracellular Vesicles in Sickle Cell Disease: A Promising Tool" Bioengineering 9, no. 9: 439. https://doi.org/10.3390/bioengineering9090439