
Emerging Promise of Computational Techniques in Anti-Cancer Research: At a Glance

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Anticancer Drug Target Prediction
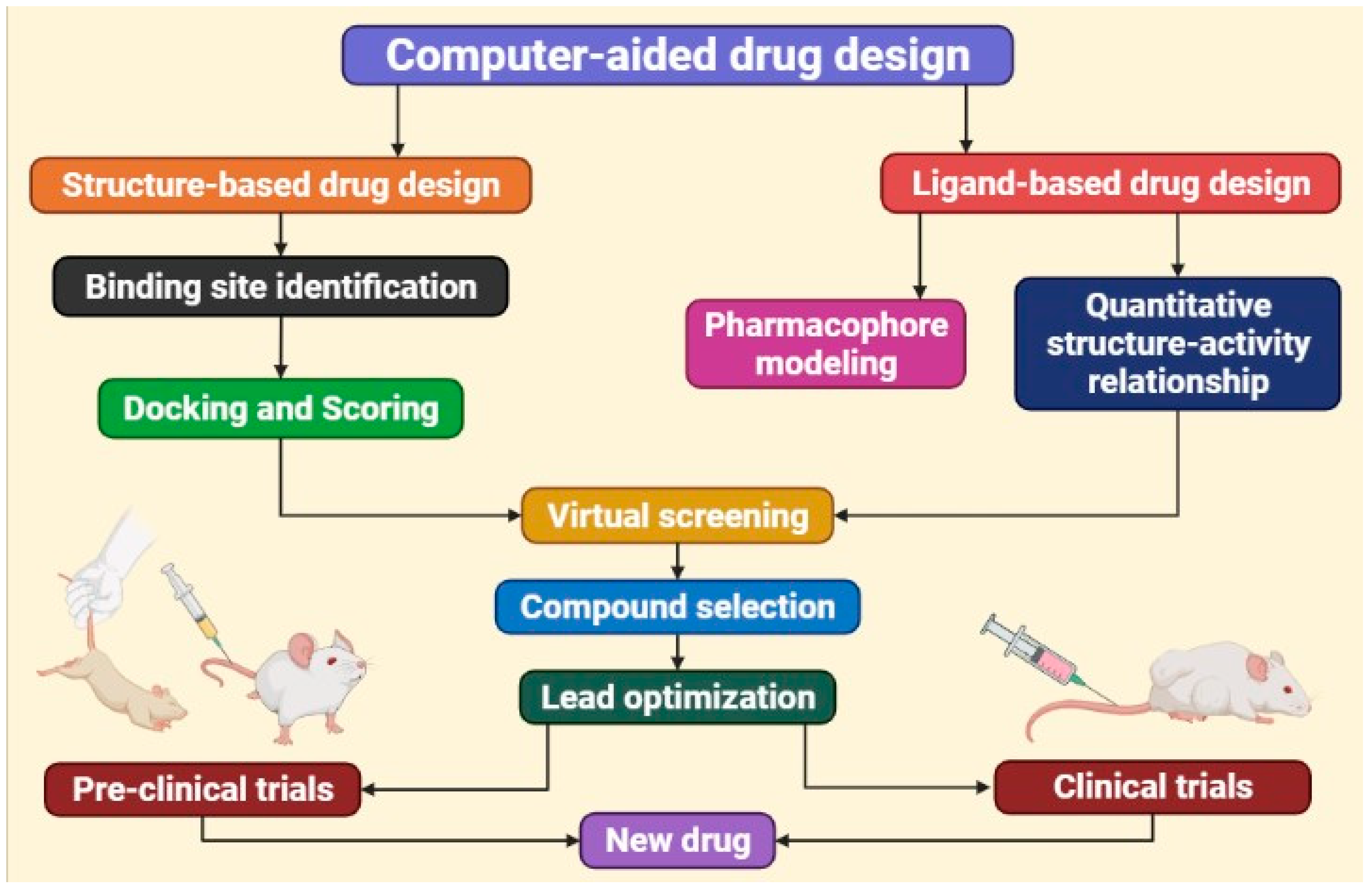
3. Computer-Aided Drug Discovery and Design
3.1. Computer-Aided Drug Design Based on Ligands
3.2. Drug Design Using Structure-Based Computer Assistance

{kind=link}
{kind=link}
{kind=link}
| Compound | Function | Therapeutic Area | Approval Time | References |
|---|---|---|---|---|
| Captopril | ACE inhibitor | Diabetic nephropathy, hypertension, congestive heart failure, myocardial infarction | 1975 | [60,61] |
| Cimetidine | H2 receptor antagonist | Heartburn and peptic ulcer therapy | 1978 | [62] |
| Dorzolamide | Inhibitor of carbonic anhydrase | Antiglaucoma agent | 1989 | [63,64] |
| Saquinavir | Inhibitor of HIV-1 protease | Antiretroviral medication to treat HIV or AIDS | 1995 | [65,66] |
| Zanamivir | Inhibitor of neuraminidase | Antiviral (influenza A and influenza B) | 1999 | [67,68,69,70] |
| Nelfinavir | Inhibitor of HIV protease | Antiretroviral medication to treat HIV or AIDS | 1999 | [71] |
| Lopinavir | HIV protease inhibitor with peptidomimetic properties | Antiretroviral medication used to treat HIV/AIDS in patients who have developed resistance to other protease inhibitors. | 2000 | [72] |
| Darunavir | Inhibitor of nonpeptic HIV protease | Antiretroviral for HIV/AIDS | 2006 | [73,74] |
| Imatinib | Inhibitor of tyrosine kinase | Chronic myeloid leukemia | 1990 | [75,76] |
| Gefitinib | Epidermal growth factor receptor (EGFR) kinase inhibitor | Non-small-cell lung cancer (NSCLC) | 2003 | [77,78] |
| Erlotinib | EGFR kinase inhibitor | Pancreatic cancer, NSCLC | 2005 | [79] |
| Sorafenib | Vascular endothelial growth factor receptor (VEGFR) kinase inhibitor | Thyroid cancer, renal cancer, liver cancer | 2005 | [80,81] |
| Lapatinib | Erb-B2 receptor tyrosine kinase 2 (ERBB2)/EGFR inhibitor | Breast cancer | 2007 | [82,83] |
| Abiraterone | Inhibitor of androgen synthesis | Hormone refractory prostate cancer or metastatic castration-resistant prostate cancer | 2011 | [84,85] |
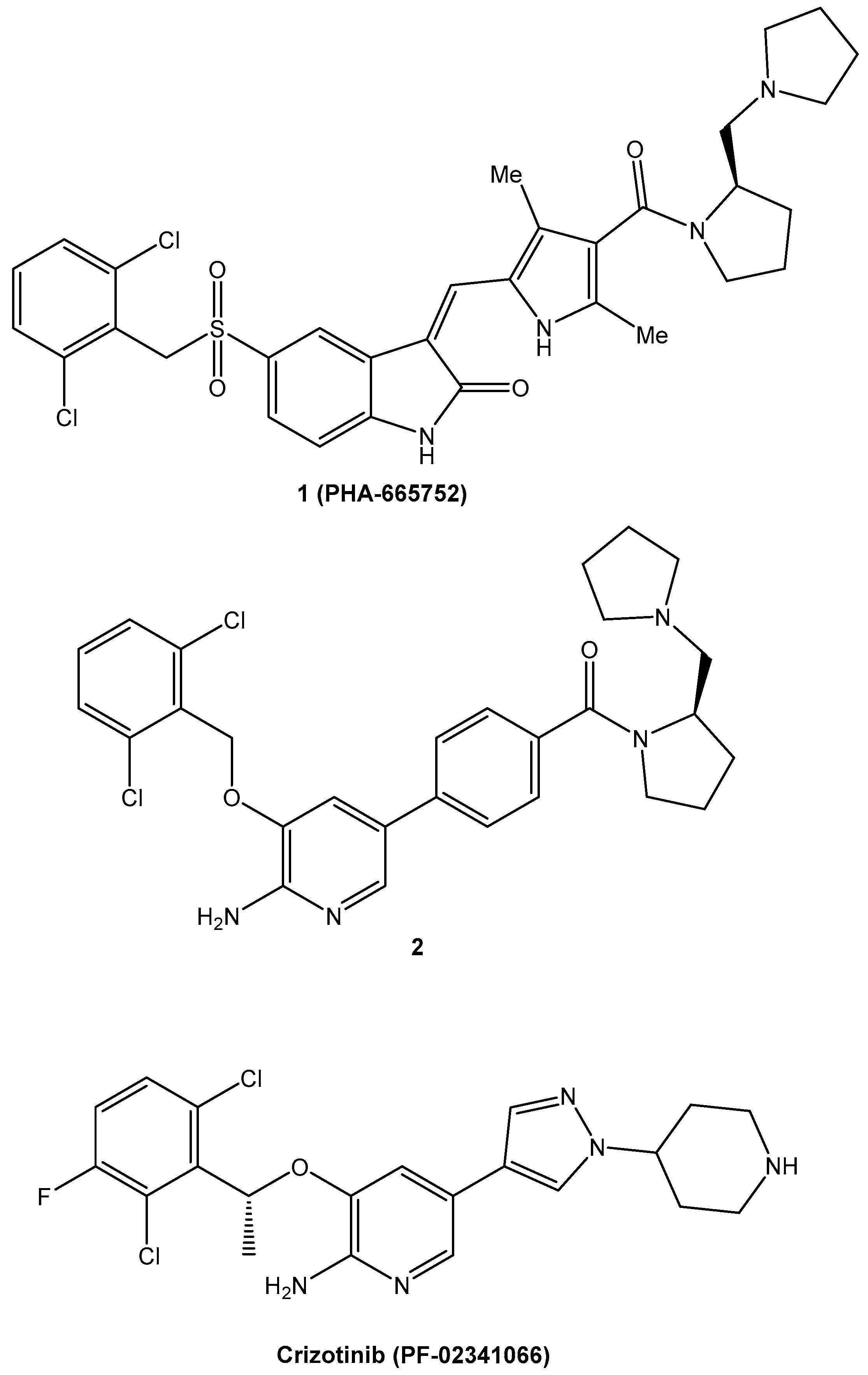
| Crizotinib | Anaplastic lymphoma kinase (ALK) inhibitor | NSCLC | 2011 | [86,87,88] |
4. Anticancer Small Organic Molecules Design via a Computational Approach
4.1. Anticancer Small Molecule Design
4.2. Computational Method for Anticancer Peptide Design
5. Structure-Based Approach
5.1. Docking of Molecules
5.2. Pharmacophore Mapping Based on Structure
6. Drug Development Based on Ligands
6.1. Searching for Similarities
6.2. Ligand-Based Pharmacophore Mapping
6.3. Modeling with QSAR
7. Artificial Intelligence Aids in the Discovery of Anticancer Drugs
8. Discovering New Drug Binding Sites through the Use of MD Simulation
9. Integration of Structure- and Ligand-Based Approaches
9.1. Pseudoreceptor Modeling
9.2. Proteochemometric Modeling
10. Current Trends in Computational Approaches for Anticancer Drug Delivery Systems
11. Successful Stories in Computational Drug Discovery
12. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mak, L.; Liggi, S.; Tan, L.; Kusonmano, K.; Rollinger, J.; Koutsoukas, A.; Glen, R.C.; Kirchmair, J. Anti-cancer Drug Development: Computational Strategies to Identify and Target Proteins Involved in Cancer Metabolism. Curr. Pharm. Des. 2013, 19, 532–577. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Islam, M.R.; Akash, S.; Harun-Or-Rashid, M.; Ray, T.K.; Rahaman, M.S.; Islam, M.; Anika, F.; Hosain, M.K.; Aovi, F.I.; et al. Recent advancements of nanoparticles application in cancer and neurodegenerative disorders: At a glance. Biomed. Pharmacother. 2022, 153, 113305. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Behl, T.; Islam, R.; Alam, N.; Islam, M.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Bungau, S.G. Emerging Management Approach for the Adverse Events of Immunotherapy of Cancer. Molecules 2022, 27, 3798. [Google Scholar] [CrossRef]
- Rahman, M.M.; Bibi, S.; Rahaman, M.S.; Rahman, F.; Islam, F.; Khan, M.S.; Hasan, M.M.; Parvez, A.; Hossain, M.A.; Maeesa, S.K.; et al. Natural therapeutics and nutraceuticals for lung diseases: Traditional significance, phytochemistry, and pharmacology. Biomed. Pharmacother. 2022, 150, 113041. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, M.R.; Shohag, S.; Ahasan, M.T.; Sarkar, N.; Khan, H.; Hasan, A.M.; Cavalu, S.; Rauf, A. Microbiome in cancer: Role in carcinogenesis and impact in therapeutic strategies. Biomed. Pharmacother. 2022, 149, 112898. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Abu-Izneid, T.; Khalil, A.A.; Imran, M.; Shah, Z.A.; Bin Emran, T.; Mitra, S.; Khan, Z.; Alhumaydhi, F.A.; Aljohani, A.S.M.; et al. Berberine as a Potential Anticancer Agent: A Comprehensive Review. Molecules 2021, 26, 7368. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Islam, R.; Shohag, S.; Hossain, E.; Rahaman, S.; Islam, F.; Ahmed, M.; Mitra, S.; Khandaker, M.U.; Idris, A.M.; et al. The Multifunctional Role of Herbal Products in the Management of Diabetes and Obesity: A Comprehensive Review. Molecules 2022, 27, 1713. [Google Scholar] [CrossRef]
- Bernard, W.S.; Christopher, P.W. World Cancer Report 2014; World Health Organization: Geneva, Switzerland, 2014.
- Das Swadesh, K.; Bhutia, S.K.; Kegelman, T.P.; Peachy, L.; Oyesanya, R.A.; Dasgupta, S.; Sokhi, U.K.; Azab, B.; Dash, R.; Quinn, B.A.; et al. MDA-9/syntenin: A positive gatekeeper of melanoma metastasis. Front. Biosci. 2012, 17, 1–15. [Google Scholar] [CrossRef]
- Shay, L.A.; Carpentier, M.Y.; Vernon, S.W. Prevalence and correlates of fear of recurrence among adolescent and young adult versus older adult post-treatment cancer survivors. Support. Care Cancer 2016, 24, 4689–4696. [Google Scholar] [CrossRef]
- Basith, S.; Cui, M.; Macalino, S.J.Y.; Choi, S. Expediting the Design, Discovery and Development of Anticancer Drugs using Computational Approaches. Curr. Med. Chem. 2017, 24, 4753–4778. [Google Scholar] [CrossRef]
- Workman, P.; Collins, I. Modern cancer drug discovery: Integrating targets, technologies and treatments for personalized medicine. In Cancer Drug Design and Discovery, 2nd ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 3–53. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Heiden, M.G.V. Altered metabolite levels in cancer: Implications for tumour biology and cancer therapy. Nat. Rev. Cancer 2016, 16, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Khotskaya, Y.B.; Mills, G.B.; Shaw, K.R.M. Next-Generation Sequencing and Result Interpretation in Clinical Oncology: Challenges of Personalized Cancer Therapy. Annu. Rev. Med. 2017, 68, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waitkus, M.S.; Diplas, B.H.; Yan, H. Biological Role and Therapeutic Potential of IDH Mutations in Cancer. Cancer Cell 2018, 34, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Saeed, K.; Rahkama, V.; Eldfors, S.; Bychkov, D.; Mpindi, J.P.; Yadav, B.; Paavolainen, L.; Aittokallio, T.; Heckman, C.; Wennerberg, K.; et al. Comprehensive Drug Testing of Patient-derived Conditionally Reprogrammed Cells from Castration-resistant Prostate Cancer. Eur. Urol. 2017, 71, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Senkowski, W.; Jarvius, M.; Rubin, J.; Lengqvist, J.; Gustafsson, M.G.; Nygren, P.; Kultima, K.; Larsson, R.; Fryknäs, M. Large-Scale Gene Expression Profiling Platform for Identification of Context-Dependent Drug Responses in Multicellular Tumor Spheroids. Cell Chem. Biol. 2016, 23, 1428–1438. [Google Scholar] [CrossRef] [Green Version]
- Raynal, N.J.-M.; Da Costa, E.M.; Lee, J.T.; Gharibyan, V.; Ahmed, S.; Zhang, H.; Sato, T.; Malouf, G.G.; Issa, J.-P.J. Repositioning FDA-Approved Drugs in Combination with Epigenetic Drugs to Reprogram Colon Cancer Epigenome. Mol. Cancer Ther. 2017, 16, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Park, S.I.; Kim, S.J.; McCauley, L.K.; Gallick, G.E. Preclinical Mouse Models of Human Prostate Cancer and Their Utility in Drug Discovery. Curr. Protoc. Pharmacol. 2010, 51, 14.15.1–14.15.27. [Google Scholar] [CrossRef] [Green Version]
- Nishiguchi, A.; Matsusaki, M.; Kano, M.R.; Nishihara, H.; Okano, D.; Easano, Y.; Shimoda, H.; Kishimoto, S.; Iwai, S.; Akashi, M. In vitro 3D blood/lymph-vascularized human stromal tissues for preclinical assays of cancer metastasis. Biomaterials 2018, 179, 144–155. [Google Scholar] [CrossRef]
- Cheng, F.; Liang, H.; Butte, A.J.; Eng, C.; Nussinov, R. Personal Mutanomes Meet Modern Oncology Drug Discovery and Precision Health. Pharmacol. Rev. 2019, 71, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuruvilla, F.G.; Shamji, A.F.; Sternson, S.; Hergenrother, P.J.; Schreiber, S.L. Dissecting glucose signalling with diversity-oriented synthesis and small-molecule microarrays. Nature 2002, 416, 653–657. [Google Scholar] [CrossRef]
- Sun, X.; Vilar, S.; Tatonetti, N.P. High-throughput methods for combinatorial drug discovery. Sci. Transl. Med. 2013, 5, 205rv1. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhu, F.; Ma, X.; Cao, Z.W.; Li, Y.X.; Chen, Y.Z. Mechanisms of drug combinations: Interaction and network perspectives. Nat. Rev. Drug Discov. 2009, 8, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Omlin, A.; De Bono, J.S. Development of Therapeutic Combinations Targeting Major Cancer Signaling Pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Du, Y.; Li, L.; Wei, D.-Q. Bioinformatics Approaches for Anti-cancer Drug Discovery. Curr. Drug Targets 2019, 21, 3–17. [Google Scholar] [CrossRef]
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yan, C.C.; Zhang, X.; Zhang, X.; Dai, F.; Yin, J.; Zhang, Y. Drug–target interaction prediction: Databases, web servers and computational models. Brief. Bioinform. 2015, 17, 696–712. [Google Scholar] [CrossRef] [Green Version]
- Lazo, J.S.; Sharlow, E.R. Drugging Undruggable Molecular Cancer Targets. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 23–40. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, T.; e Soares, G.A.B.; Chopra, H.; Rahman, M.; Hasan, Z.; Swain, S.S.; Cavalu, S. Applications of Phyto-Nanotechnology for the Treatment of Neurodegenerative Disorders. Materials 2022, 15, 804. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Islam, F.; Parvez, A.; Azad, M.A.K.; Ashraf, G.M.; Ullah, M.F.; Ahmed, M. Citrus limon L. (lemon) seed extract shows neuro-modulatory activity in an in vivo thiopental-sodium sleep model by reducing the sleep onset and enhancing the sleep duration. J. Integr. Neurosci. 2022, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Islam, F.; Afsana Mim, S.; Khan, M.S.; Islam, M.R.; Haque, M.A.; Mitra, S.; Emran, T.B.; Rauf, A. Multifunctional Therapeutic Approach of Nanomedicines against Inflammation in Cancer and Aging. J. Nanomater. 2022, 2022, 4217529. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- Rahman, M.M.; Dhar, P.S.; Sumaia; Anika, F.; Ahmed, L.; Islam, M.R.; Sultana, N.A.; Cavalu, S.; Pop, O.; Rauf, A. Exploring the plant-derived bioactive substances as antidiabetic agent: An extensive review. Biomed. Pharmacother. 2022, 152, 113217. [Google Scholar] [CrossRef]
- Takarabe, M.; Kotera, M.; Nishimura, Y.; Goto, S.; Yamanishi, Y. Drug target prediction using adverse event report systems: A pharmacogenomic approach. Bioinformatics 2012, 28, i611–i618. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Ferdous, K.S.; Ahmed, M.; Islam, M.T.; Khan, R.; Perveen, A.; Ashraf, G.; Uddin, S. Hutchinson-Gilford Progeria Syndrome: An Overview of the Molecular Mechanism, Pathophysiology and Therapeutic Approach. Curr. Gene Ther. 2021, 21, 216–229. [Google Scholar] [CrossRef]
- Rahman, M.M.; Ahmed, M.; Islam, M.T.; Khan, M.R.; Sultana, S.; Maeesa, S.K.; Hasan, S.; Hossain, M.A.; Ferdous, K.S.; Mathew, B.; et al. Nanotechnology-based Approaches and Investigational Therapeutics against COVID-19. Curr. Pharm. Des. 2021, 28, 948–968. [Google Scholar] [CrossRef]
- Rahman, M.; Rahaman, S.; Islam, R.; Hossain, E.; Mithi, F.M.; Ahmed, M.; Saldías, M.; Akkol, E.K.; Sobarzo-Sánchez, E. Multifunctional Therapeutic Potential of Phytocomplexes and Natural Extracts for Antimicrobial Properties. Antibiotics 2021, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Bibi, S.; Meem, A.F.K.; Islam, M.; Rahaman, S.; Bepary, S.; Rahman, M.; Rahman, M.; Elzaki, A.; Kajoak, S.; et al. Natural Bioactive Molecules: An Alternative Approach to the Treatment and Control of COVID-19. Int. J. Mol. Sci. 2021, 22, 12638. [Google Scholar] [CrossRef] [PubMed]
- Mominur Rahman, M.; Islam, F.; Saidur Rahaman, M.; Sultana, N.A.; Fahim, N.F.; Ahmed, M. Studies on the prevalence of HIV/AIDS in Bangladesh including other developing countries. Adv. Tradit. Med. 2021, 21, 1–12. [Google Scholar] [CrossRef]
- Rahman, M.; Rahaman, S.; Islam, R.; Rahman, F.; Mithi, F.M.; Alqahtani, T.; Almikhlafi, M.A.; Alghamdi, S.Q.; Alruwaili, A.S.; Hossain, S.; et al. Role of Phenolic Compounds in Human Disease: Current Knowledge and Future Prospects. Molecules 2021, 27, 233. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. Drug–target interaction prediction by random walk on the heterogeneous network. Mol. BioSyst. 2012, 8, 1970–1978. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.-C.; Jensen, L.J.; Bork, P. Drug Target Identification Using Side-Effect Similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Rosenheim, O.; King, H. The Ring-system of sterols and bile acids. Part II. J. Soc. Chem. Ind. 1932, 51, 954–957. [Google Scholar] [CrossRef]
- Kaldor, S.W.; Kalish, V.J.; Davies, J.F.; Shetty, B.V.; Fritz, J.E.; Appelt, K.; Burgess, J.A.; Campanale, K.M.; Chirgadze, N.Y.; Clawson, D.K.; et al. Viracept (Nelfinavir Mesylate, AG1343): A Potent, Orally Bioavailable Inhibitor of HIV-1 Protease. J. Med. Chem. 1997, 40, 3979–3985. [Google Scholar] [CrossRef]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Brief. Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L. The many roles of computation in drug discovery. Science 2004, 303, 1813–1818. [Google Scholar] [CrossRef]
- Prada-Gracia, D.; Huerta-Yépez, S.; Moreno-Vargas, L.M. Application of computational methods for anticancer drug discovery, design, and optimization. Boletín Médico Hosp. Infant. México 2016, 73, 411–423. [Google Scholar] [CrossRef]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sana, F.; Ayesha, J.; Talath, F.; Sharequa, H. Computational Drug Designing of Anticancer Drugs. Int. J. Pharm. Res. Sch. 2018, 7, 58–70. [Google Scholar] [CrossRef]
- de Sousa, N.F.; Scotti, L.; de Moura, É.P.; dos Santos Maia, M.; Rodrigues, G.C.S.; de Medeiros, H.I.R.; Lopes, S.M.; Scotti, M.T. Computer Aided Drug Design Methodologies with Natural Products in the Drug Research Against Alzheimer’s Disease. Curr. Neuropharmacol. 2021, 20, 857–885. [Google Scholar] [CrossRef]
- Kulkarni, A.M.; Kumar, V.; Parate, S.; Lee, G.; Yoon, S.; Lee, K.W. Identification of New KRAS G12D Inhibitors through Computer-Aided Drug Discovery Methods. Int. J. Mol. Sci. 2022, 23, 1309. [Google Scholar] [CrossRef] [PubMed]
- Tariq, A.; Rehman, H.M.; Mateen, R.M.; Ali, M.; Mutahir, Z.; Afzal, M.S.; Sajjad, M.; Gul, R.; Saleem, M. A computer aided drug discovery based discovery of lead-like compounds against KDM5A for cancers using pharmacophore modeling and high-throughput virtual screening. Proteins Struct. Funct. Bioinform. 2021, 90, 645–657. [Google Scholar] [CrossRef]
- Magwenyane, A.M.; Ugbaja, S.C.; Amoako, D.G.; Somboro, A.M.; Khan, R.B.; Kumalo, H.M. Heat Shock Protein 90 (HSP90) Inhibitors as Anticancer Medicines: A Review on the Computer-Aided Drug Discovery Approaches over the Past Five Years. Comput. Math. Methods Med. 2022, 2022, 2147763. [Google Scholar] [CrossRef]
- Cushman, D.W.; Ondetti, M.A. Design of angiotensin converting enzyme inhibitors. Nat. Med. 1999, 5, 1110–1112. [Google Scholar] [CrossRef]
- Ondetti, M.A.; Rubin, B.; Cushman, D.W. Design of specific inhibitors of angiotensin-converting enzyme: New class of orally active antihypertensive agents. Science 1977, 196, 441–444. [Google Scholar] [CrossRef]
- Brimblecombe, R.; Duncan, W.; Durant, G.; Ganellin, C.; Parsons, M.; Black, J. The pharmacology of cimetidine, a new histamine H2-receptor antagonist. J. Cereb. Blood Flow Metab. 2010, 160, S52–S53. [Google Scholar] [CrossRef] [Green Version]
- Blankenstein, M. Some Observations on the Epidemiology of Barrett’s Oesophagus and Adenocarcinoma of the Oesophagus; Erasmus Universiteit Rotterdam (EUR): Rotterdam, The Netherlands, 2006; p. 114. [Google Scholar]
- Angeli, A.; Kartsev, V.; Petrou, A.; Pinteala, M.; Vydzhak, R.M.; Panchishin, S.Y.; Brovarets, V.; De Luca, V.; Capasso, C.; Geronikaki, A.; et al. New Sulfanilamide Derivatives Incorporating Heterocyclic Carboxamide Moieties as Carbonic Anhydrase Inhibitors. Pharmaceuticals 2021, 14, 828. [Google Scholar] [CrossRef] [PubMed]
- Aubier, M.; Barnes, P.J. Theophylline and phosphodiesterase inhibitors. Eur. Respir. J. 1995, 8, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Clinical Thyroidology for the Public. American Thyroid Association. 2017, Volume 10. Available online: https://www.thyroid.org/patient-thyroid-information/ct-for-patients/may-2017/ (accessed on 21 April 2022).
- Raugi, D.N.; Smith, R.A.; Gottlieb, G.S. Four Amino Acid Changes in HIV-2 Protease Confer Class-Wide Sensitivity to Protease Inhibitors. J. Virol. 2016, 90, 1062–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, K.; Dyason, J.C.; Maggioni, A.; von Itzstein, M.; Downard, K.M. Binding of a natural anthocyanin inhibitor to influenza neuraminidase by mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 6563–6572. [Google Scholar] [CrossRef]
- Gupta, D.; Gupta, S.V.; Dahan, A.; Tsume, Y.; Hilfinger, J.; Lee, K.-D.; Amidon, G.L. Increasing Oral Absorption of Polar Neuraminidase Inhibitors: A Prodrug Transporter Approach Applied to Oseltamivir Analogue. Mol. Pharm. 2013, 10, 512–522. [Google Scholar] [CrossRef]
- Peng, A.W.; Hussey, E.K.; Rosolowski, B.; Blumer, J.L. Pharmacokinetics and tolerability of a single inhaled dose of zanamivir in children. Curr. Ther. Res. 2000, 61, 36–46. [Google Scholar] [CrossRef]
- Serafim, M.S.M.; Júnior, V.S.D.S.; Gertrudes, J.C.; Maltarollo, V.G.; Honorio, K.M. Machine learning techniques applied to the drug design and discovery of new antivirals: A brief look over the past decade. Expert Opin. Drug Discov. 2021, 16, 961–975. [Google Scholar] [CrossRef]
- Barragan, P.; Podzamczer, D. Lopinavir/ritonavir: A protease inhibitor for HIV-1 treatment. Expert Opin. Pharmacother. 2008, 9, 2363–2375. [Google Scholar] [CrossRef]
- Amano, M.; Koh, Y.; Das, D.; Li, J.; Leschenko, S.; Wang, Y.-F.; Boross, P.I.; Weber, I.T.; Ghosh, A.K.; Mitsuya, H. A Novel Bis-Tetrahydrofuranylurethane-Containing Nonpeptidic Protease Inhibitor (PI), GRL-98065, Is Potent against Multiple-PI-Resistant Human Immunodeficiency Virus In Vitro. Antimicrob. Agents Chemother. 2007, 51, 2143–2155. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-C.E.; Yu, X.; Zhang, Y.; Tie, Y.; Wang, Y.-F.; Yashchuk, S.; Ghosh, A.K.; Harrison, R.W.; Weber, I.T. Potent Antiviral HIV-1 Protease Inhibitor GRL-02031 Adapts to the Structures of Drug Resistant Mutants with Its P1′-Pyrrolidinone Ring. J. Med. Chem. 2012, 55, 3387–3397. [Google Scholar] [CrossRef] [Green Version]
- Petrola, M.J.; De Castro, A.J.M.; Pitombeira, M.H.D.S.; Barbosa, M.C.; Quixadá, A.T.D.S.; Duarte, F.B.; Gonçalves, R.P. Serum concentrations of nitrite and malondialdehyde as markers of oxidative stress in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Rev. Bras. De Hematol. E Hemoter. 2012, 34, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Liu, M.; Xu, S.; Wang, Y.; Li, Y.; Li, Y.; Zhang, H.; Liu, H.; Chen, J. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, sensitizes lung cancer cells to treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Oncotarget 2016, 7, 84951. [Google Scholar] [CrossRef] [Green Version]
- Masago, K.; Togashi, Y.; Fukudo, M.; Terada, T.; Irisa, K.; Sakamori, Y.; Fujita, S.; Kim, Y.H.; Mio, T.; Inui, K.-I.; et al. Good Clinical Response to Erlotinib in a Non-Small Cell Lung Cancer Patient Harboring Multiple Brain Metastases and a Double Active Somatic Epidermal Growth Factor Gene Mutation. Case Rep. Oncol. 2010, 3, 98–105. [Google Scholar] [CrossRef]
- Negrier, S.; Jäger, E.; Porta, C.; McDermott, D.; Moore, M.; Bellmunt, J.; Anderson, S.; Cihon, F.; Lewis, J.; Escudier, B.; et al. Efficacy and safety of sorafenib in patients with advanced renal cell carcinoma with and without prior cytokine therapy, a subanalysis of TARGET. Med. Oncol. 2009, 27, 899–906. [Google Scholar] [CrossRef]
- Chang, Y.S.; Adnane, J.; Trail, P.A.; Levy, J.; Henderson, A.; Xue, D.; Bortolon, E.; Ichetovkin, M.; Chen, C.; McNabola, A.; et al. Sorafenib (BAY 43-9006) inhibits tumor growth and vascularization and induces tumor apoptosis and hypoxia in RCC xenograft models. Cancer Chemother. Pharmacol. 2006, 59, 561–574. [Google Scholar] [CrossRef]
- Xia, W.; Liu, Z.; Zong, R.; Liu, L.; Zhao, S.; Bacus, S.S.; Mao, Y.; He, J.; Wulfkuhle, J.D.; Petricoin, E.F.; et al. Truncated ErbB2 Expressed in Tumor Cell Nuclei Contributes to Acquired Therapeutic Resistance to ErbB2 Kinase Inhibitors. Mol. Cancer Ther. 2011, 10, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Mi, L.-Z.; Schürpf, T.; Walz, T.; Springer, T.A. Mechanisms for Kinase-mediated Dimerization of the Epidermal Growth Factor Receptor. J. Biol. Chem. 2012, 287, 38244–38253. [Google Scholar] [CrossRef] [Green Version]
- Asmane, I.; Céraline, J.; Duclos, B.; Rob, L.; Litique, V.; Barthélémy, P.; Bergerat, J.P.; Dufour, P.; Kurtz, J.E. New Strategies for Medical Management of Castration-Resistant Prostate Cancer. Oncology 2011, 80, 1–11. [Google Scholar] [CrossRef]
- Jagusch, C.; Negri, M.; Hille, U.E.; Hu, Q.; Bartels, M.; Jahn-Hoffmann, K.; Mendieta, M.A.P.-B.; Rodenwaldt, B.; Müller-Vieira, U.; Schmidt, D. Synthesis, biological evaluation and molecular modelling studies of methyleneimidazole substituted biaryls as inhibitors of human 17α-hydroxylase-17,20-lyase (CYP17). Part I: Heterocyclic modifications of the core structure. Bioorg. Med. Chem. 2008, 16, 1992–2010. [Google Scholar] [CrossRef] [PubMed]
- Chavez, C.; Hoffman, M.A. Complete remission of ALK-negative plasma cell granuloma (inflammatory myofibroblastic tumor) of the lung induced by celecoxib: A case report and review of the literature. Oncol. Lett. 2013, 5, 1672–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodig, S.J.; Shapiro, G.I. Crizotinib, a small-molecule dual inhibitor of the c-Met and ALK receptor tyrosine kinases. Curr. Opin. Investig. Drugs 2010, 11, 1477–1490. [Google Scholar] [PubMed]
- Cui, W.; Aouidate, A.; Wang, S.; Yu, Q.; Li, Y.; Yuan, S. Discovering Anti-Cancer Drugs via Computational Methods. Front. Pharmacol. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2018, 93, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- González, M.G.; Janssen, A.P.; Ijzerman, A.P.; Heitman, L.H.; van Westen, G.J. Oncological drug discovery: AI meets structure-based computational research. Drug Discov. Today 2022, 27, 1661–1670. [Google Scholar] [CrossRef]
- Rosenthal, J.; Carelli, R.; Omar, M.; Brundage, D.; Halbert, E.; Nyman, J.; Hari, S.N.; Van Allen, E.M.; Marchionni, L.; Umeton, R.; et al. Building Tools for Machine Learning and Artificial Intelligence in Cancer Research: Best Practices and a Case Study with the PathML Toolkit for Computational Pathology. Mol. Cancer Res. 2022, 20, 202–206. [Google Scholar] [CrossRef]
- Luthra, A.; Mastrogiacomo, B.; Smith, S.A.; Chakravarty, D.; Schultz, N.; Sanchez-Vega, F. Computational methods and translational applications for targeted next-generation sequencing platforms. Genes Chromosom. Cancer 2022, 61, 322–331. [Google Scholar] [CrossRef]
- Arafat, Y.; Reyes-Aldasoro, C.C. Computational Image Analysis Techniques, Programming Languages and Software Platforms Used in Cancer Research: A Scoping Review. medRxiv 2022. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Gulhan, D.C.; Lee, J.J.-K.; Melloni, G.E.M.; Park, P.J. Computational analysis of cancer genome sequencing data. Nat. Rev. Genet. 2021, 23, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Kolmar, L.; Autour, A.; Ma, X.; Vergier, B.; Eduati, F.; Merten, C.A. Technological and computational advances driving high-throughput oncology. Trends Cell Biol. 2022; in press. [Google Scholar] [CrossRef]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In silico Methods for Design of Kinase Inhibitors as Anticancer Drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Krishna, S.; Siddiqi, M.I. Virtual screening strategies: Recent advances in the identification and design of anti-cancer agents. Methods 2015, 71, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Magistrato, A.; Sgrignani, J.; Krause, R.; Cavalli, A. Single or Multiple Access Channels to the CYP450s Active Site? An Answer from Free Energy Simulations of the Human Aromatase Enzyme. J. Phys. Chem. Lett. 2017, 8, 2036–2042. [Google Scholar] [CrossRef]
- Sgrignani, J.; Magistrato, A. Influence of the Membrane Lipophilic Environment on the Structure and on the Substrate Access/Egress Routes of the Human Aromatase Enzyme. A Computational Study. J. Chem. Inf. Model. 2012, 52, 1595–1606. [Google Scholar] [CrossRef]
- Ritacco, I.; Saltalamacchia, A.; Spinello, A.; Ippoliti, E.; Magistrato, A. All-Atom Simulations Disclose How Cytochrome Reductase Reshapes the Substrate Access/Egress Routes of Its Partner CYP450s. J. Phys. Chem. Lett. 2020, 11, 1189–1193. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Salmaso, V.; Sturlese, M.; Cuzzolin, A.; Moro, S. Exploring Protein-Peptide Recognition Pathways Using a Supervised Molecular Dynamics Approach. Structure 2017, 25, 655–662.e2. [Google Scholar] [CrossRef] [Green Version]
- Ciemny, M.; Kurcinski, M.; Kamel, K.; Kolinski, A.; Alam, N.; Schueler-Furman, O.; Kmiecik, S. Protein–peptide docking: Opportunities and challenges. Drug Discov. Today 2018, 23, 1530–1537. [Google Scholar] [CrossRef]
- Lammi, C.; Sgrignani, J.; Arnoldi, A.; Grazioso, G. Biological Characterization of Computationally Designed Analogs of peptide TVFTSWEEYLDWV (Pep2-8) with Increased PCSK9 Antagonistic Activity. Sci. Rep. 2019, 9, 2343. [Google Scholar] [CrossRef] [Green Version]
- Lammi, C.; Sgrignani, J.; Roda, G.; Arnoldi, A.; Grazioso, G. Inhibition of PCSK9D374Y/LDLR Protein–Protein Interaction by Computationally Designed T9 Lupin Peptide. ACS Med. Chem. Lett. 2018, 10, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Spodzieja, M.; Lach, S.; Iwaszkiewicz, J.; Cesson, V.; Kalejta, K.; Olive, D.; Michielin, O.; Speiser, D.E.; Zoete, V.; Derré, L.; et al. Design of short peptides to block BTLA/HVEM interactions for promoting anticancer T-cell responses. PLoS ONE 2017, 12, e0179201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Dias, R. Molecular Docking Algorithms. Curr. Drug Targets 2008, 9, 1040–1047. [Google Scholar] [CrossRef] [PubMed]
- Halperin, I.; Ma, B.; Wolfson, H.; Nussinov, R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins Struct. Funct. Bioinform. 2002, 47, 409–443. [Google Scholar] [CrossRef]
- Salmaso, V.; Moro, S. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 2018, 9, 923. [Google Scholar] [CrossRef] [Green Version]
- Kong, W.; Midena, G.; Chen, Y.; Athanasiadis, P.; Wang, T.; Rousu, J.; He, L.; Aittokallio, T. Systematic review of computational methods for drug combination prediction. Comput. Struct. Biotechnol. J. 2022, 20, 2807–2814. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef]
- Lu, P.; Bevan, D.R.; Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Bassaganya-Riera, J. Computer-Aided Drug Discovery. In Accelerated Path to Cures; Springer: Cham, Switzerland, 2018; pp. 7–24. [Google Scholar] [CrossRef]
- Pirhadi, S.; Shiri, F.; Ghasemi, J.B. Methods and Applications of Structure Based Pharmacophores in Drug Discovery. Curr. Top. Med. Chem. 2013, 13, 1036–1047. [Google Scholar] [CrossRef]
- Wolber, G.; Dornhofer, A.A.; Langer, T. Efficient overlay of small organic molecules using 3D pharmacophores. J. Comput. Aided. Mol. Des. 2006, 20, 773–788. [Google Scholar] [CrossRef]
- Chen, J.; Lai, L. Pocket v.2: Further Developments on Receptor-Based Pharmacophore Modeling. J. Chem. Inf. Model. 2006, 46, 2684–2691. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Costa, D.B.; Oxnard, G.R.; Huberman, M.; Gainor, J.F.; Lennes, I.T.; Muzikansky, A.; Shaw, A.T.; Azzoli, C.G.; Heist, R.S.; et al. Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann. Oncol. 2018, 29, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Yang, H.; Chen, Y.; Li, Q.; He, S.; Jiang, X.; Feng, F.; Qu, W.; Sun, H. The Development of Pharmacophore Modeling: Generation and Recent Applications in Drug Discovery. Curr. Pharm. Des. 2018, 24, 3424–3439. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012-2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef]
- Buckle, D.R.; Erhardt, P.W.; Ganellin, C.R.; Kobayashi, T.; Perun, T.J.; Proudfoot, J.; Senn-Bilfinger, J. Glossary of terms used in medicinal chemistry. Part II (IUPAC recommendations 2013). Pure Appl. Chem. 2013, 85, 1725–1758. [Google Scholar] [CrossRef]
- Chao, W.R.; Yean, D.; Amin, K.; Green, C.; Jong, L. Computer-aided rational drug design: A novel agent (SR13668) designed to mimic the unique anticancer mechanisms of dietary indole-3-carbinol to block Akt signaling. J. Med. Chem. 2007, 50, 3412–3415. [Google Scholar] [CrossRef]
- Mendenhall, J.; Meiler, J. Improving quantitative structure-activity relationship models using Artificial Neural Networks trained with dropout. J. Comput. Aided. Mol. Des. 2016, 30, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Muhsin, M.; Graham, J.; Kirkpatrick, P. Fresh from the pipeline: Gefitinib. Nat. Rev. Cancer 2003, 3, 556–557. [Google Scholar] [CrossRef]
- Grünwald, V.; Hidalgo, M. Development of the epidermal growth factor receptor inhibitor TarcevaTM (OSI-774). Adv. Exp. Med. Biol. 2003, 532, 235–246. [Google Scholar] [CrossRef]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A Unique Structure for Epidermal Growth Factor Receptor Bound to GW572016 (Lapatinib). Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarman, M.; Elaine Barrie, S.; Liera, J.M. The 16,17-Double Bond Is Needed for Irreversible Inhibition of Human Cytochrome P45017α by Abiraterone (17-(3-Pyridyl)androsta-5,16-dien-3β-ol) and Related Steroidal Inhibitors. J. Med. Chem. 1998, 41, 5375–5381. [Google Scholar] [CrossRef]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK -Rearranged Inflammatory Myofibroblastic Tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reker, D.; Rodrigues, T.; Schneider, P.; Schneider, G. Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl. Acad. Sci. USA 2014, 111, 4067–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, T.; Werner, M.; Roth, J.; Da Cruz, E.H.G.; Marques, M.C.; Akkapeddi, P.; Lobo, S.A.; Koeberle, A.; Corzana, F.; Da Silva Júnior, E.N.; et al. Machine intelligence decrypts β-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem. Sci. 2018, 9, 6899–6903. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Born, J.; Manica, M.; Oskooei, A.; Cadow, J.; Rodríguez Martínez, M. PaccmannRL: Designing Anticancer Drugs From Transcriptomic Data via Reinforcement Learning. In Research in Computational Molecular Biology; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hua, L.; Zhai, W.; Zhao, Y. Novel Computational Approaches and Applications in Cancer Research. BioMed Res. Int. 2017, 2017, 9509280. [Google Scholar] [CrossRef]
- Jeon, M.; Kim, S.; Park, S.; Lee, H.; Kang, J. In silico drug combination discovery for personalized cancer therapy. BMC Syst Biol. 2018, 12 (Suppl. 2), 16. [Google Scholar] [CrossRef]
- Kinney, G.G.; O’Brien, J.A.; Lemaire, W.; Burno, M.; Bickel, D.J.; Clements, M.K.; Chen, T.B.; Wisnoski, D.D.; Lindsley, C.W.; Tiller, P.R.; et al. A Novel Selective Positive Allosteric Modulator of Metabotropic Glutamate Receptor Subtype 5 Has in Vivo Activity and Antipsychotic-Like Effects in Rat Behavioral Models. J. Pharmacol. Exp. Ther. 2005, 313, 199–206. [Google Scholar] [CrossRef]
- Tautermann, C.S. GPCR structures in drug design, emerging opportunities with new structures. Bioorg. Med. Chem. Lett. 2014, 24, 4073–4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flock, T.; Ravarani, C.N.J.; Sun, D.; Venkatakrishnan, A.J.; Kayikci, M.; Tate, C.G.; Veprintsev, D.; Babu, M.M. Universal allosteric mechanism for Gα activation by GPCRs. Nature 2015, 524, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVree, B.T.; Mahoney, J.P.; Vélez-Ruiz, G.A.; Rasmussen, S.G.F.; Kuszak, A.J.; Edwald, E.; Fung, J.-J.; Manglik, A.; Masureel, M.; Du, Y.; et al. Allosteric coupling from G protein to the agonist-binding pocket in GPCRs. Nature 2016, 535, 182–186. [Google Scholar] [CrossRef] [Green Version]
- Sabbadin, D.; Moro, S. Supervised Molecular Dynamics (SuMD) as a Helpful Tool To Depict GPCR–Ligand Recognition Pathway in a Nanosecond Time Scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Deganutti, G.; Cuzzolin, A.; Ciancetta, A.; Moro, S. Understanding allosteric interactions in G protein-coupled receptors using Supervised Molecular Dynamics: A prototype study analysing the human A3 adenosine receptor positive allosteric modulator LUF6000. Bioorg. Med. Chem. 2015, 23, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, A.; Sturlese, M.; Deganutti, G.; Salmaso, V.; Sabbadin, D.; Ciancetta, A.; Moro, S. Deciphering the Complexity of Ligand-Protein Recognition Pathways Using Supervised Molecular Dynamics (SuMD) Simulations. J. Chem. Inf. Model. 2016, 56, 687–705. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.C.S.; Xu, Y.; Tan, L.; Vogel, H.; Cheng, J.; Wu, D.; Yuan, S. Enhancing the Signaling of GPCRs via Orthosteric Ions. ACS Cent. Sci. 2020, 6, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Seeliger, M.A. Targeting Conformational Plasticity of Protein Kinases. ACS Chem. Biol. 2014, 10, 190–200. [Google Scholar] [CrossRef]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Foloppe, N.; Fisher, L.M.; Howes, R.; Potter, A.; Robertson, A.G.S.; Surgenor, A.E. Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening. Bioorg. Med. Chem. 2006, 14, 4792–4802. [Google Scholar] [CrossRef] [PubMed]
- Song, C.H.; Yang, S.H.; Park, E.; Cho, S.H.; Gong, E.Y.; Khadka, D.B.; Cho, W.J.; Lee, K. Structure-based virtual screening and identification of a novel androgen receptor antagonist. J. Biol. Chem. 2012, 287, 30769–30780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.H.; Song, H.Y.; Zhang, J.X.; Han, B.C.; Wei, X.N.; Ma, X.H.; Cui, W.K.; Chen, Y.Z. Identifying novel type ZBGs and nonhydroxamate HDAC inhibitors through a SVM based virtual screening approach. Mol. Inform. 2010, 29, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Luan, F.; Cordeiro, M.N.D.S. Rational drug design for anti-cancer chemotherapy: Multi-target QSAR models for the in silico discovery of anti-colorectal cancer agents. Bioorg. Med. Chem. 2012, 20, 4848–4855. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, L.; Yu, M.; Xu, L.-H.; Cheng, B.; Lin, Y.-S.; Gu, Q.; He, X.-H.; Xu, J. Discovering new mTOR inhibitors for cancer treatment through virtual screening methods and in vitro assays. Sci. Rep. 2016, 6, 18987. [Google Scholar] [CrossRef] [Green Version]
- Allen, B.K.; Mehta, S.; Ember, S.W.J.; Schönbrunn, E.; Ayad, N.; Schürer, S.C. Large-Scale Computational Screening Identifies First in Class Multitarget Inhibitor of EGFR Kinase and BRD4. Sci. Rep. 2015, 5, 16924. [Google Scholar] [CrossRef] [Green Version]
- Svensson, F.; Karlén, A.; Sköld, C. Virtual Screening Data Fusion Using Both Structure- and Ligand-Based Methods. J. Chem. Inf. Model. 2011, 52, 225–232. [Google Scholar] [CrossRef]
- Tanrikulu, Y.; Schneider, G. Pseudoreceptor models in drug design: Bridging ligand- and receptor-based virtual screening. Nat. Rev. Drug Discov. 2008, 7, 667–677. [Google Scholar] [CrossRef]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef]
- Pei, J.; Zhou, J.; Xie, G.; Chen, H.; He, X. PARM: A practical utility for drug design. J. Mol. Graph. Model. 2001, 19, 448–454. [Google Scholar] [CrossRef]
- Peng, T.; Pei, J.; Zhou, J. 3D-QSAR and Receptor Modeling of Tyrosine Kinase Inhibitors with Flexible Atom Receptor Model (FLARM). J. Chem. Inf. Comput. Sci. 2002, 43, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.J.; Coffman, V.C.; Wu, J.Q. Contractile-ring assembly in fission yeast cytokinesis: Recent advances and new perspectives. Cytoskeleton 2012, 69, 751–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rödl, C.B.; Tanrikulu, Y.; Wisniewska, J.M.; Proschak, E.; Schneider, G.; Steinhilber, D.; Hofmann, B. Potent inhibitors of 5-lipoxygenase identified using pseudoreceptors. ChemMedChem 2011, 6, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Lapinsh, M.; Prusis, P.; Gutcaits, A.; Lundstedt, T.; Wikberg, J.E. Development of proteo-chemometrics: A novel technology for the analysis of drug-receptor interactions. Biochim. Et Biophys. Acta Gen. Subj. 2001, 1525, 180–190. [Google Scholar] [CrossRef]
- Van Westen, G.J.P.; Wegner, J.K.; Geluykens, P.; Kwanten, L.; Vereycken, I.; Peeters, A.; Ijzerman, A.P.; Van Vlijmen, H.W.T.; Bender, A. Which Compound to Select in Lead Optimization? Prospectively Validated Proteochemometric Models Guide Preclinical Development. PLoS ONE 2011, 6, e27518. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Huang, Q.; Zhang, Y.; Zhang, Q.; Liu, Q.; Gao, J.; Cao, Z.; Zhu, R. Screening of selective histone deacetylase inhibitors by proteochemometric modeling. BMC Bioinform. 2012, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 Inhibition Potentiates Antiangiogenic Cancer Therapy and Prevents Metastasis in Preclinical Models. Sci. Transl. Med. 2014, 6, 242ra84. [Google Scholar] [CrossRef]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, A.; Nueno, V.I.P.; Lentini, G.; Ritchie, D.W. Recent Trends and Future Prospects in Computational GPCR Drug Discovery: From Virtual Screening to Polypharmacology. Curr. Top. Med. Chem. 2013, 13, 1069–1097. [Google Scholar] [CrossRef] [PubMed]
- Anighoro, A.; Stumpfe, D.; Heikamp, K.; Beebe, K.; Neckers, L.M.; Bajorath, J.; Rastelli, G. Computational polypharmacology analysis of the heat shock protein 90 interactome. J. Chem. Inf. Model. 2015, 55, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Barrera, D.; Santos, N.; Avila, E.; Ordaz-Rosado, D.; Rivas-Suarez, M.; Halhali, A.; Rodriguez, P.; Gamboa-Dominguez, A.; et al. Astemizole Synergizes Calcitriol Antiproliferative Activity by Inhibiting CYP24A1 and Upregulating VDR: A Novel Approach for Breast Cancer Therapy. PLoS ONE 2012, 7, e45063. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kang, S.; Kim, W. Drug Repositioning for Cancer Therapy Based on Large-Scale Drug-Induced Transcriptional Signatures. PLoS ONE 2016, 11, e0150460. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Chang, P.M.-H.; Hsu, C.-W.; Huang, C.-Y.F.; Ng, K.-L. Drug repositioning for non-small cell lung cancer by using machine learning algorithms and topological graph theory. BMC Bioinform. 2016, 17, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Dider, S.; Han, W.; Xu, H.; Zhao, Z.; Xie, L. Toward Repurposing Metformin as a Precision Anti-Cancer Therapy Using Structural Systems Pharmacology. Sci. Rep. 2016, 6, 20441. [Google Scholar] [CrossRef]
- Kung, P.P.; Jones, R.A.; Richardson, P. Crizotinib (Xalkori): The First-in-Class ALK/ROS Inhibitor for Non-small Cell Lung Cancer. In Innovative Drug Synthesis; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 119–156. [Google Scholar] [CrossRef]
- Cui, J.J.; McTigue, M.; Kania, R.; Edwards, M. Case History: XalkoriTM (Crizotinib), a Potent and Selective Dual Inhibitor of Mesenchymal Epithelial Transition (MET) and Anaplastic Lymphoma Kinase (ALK) for Cancer Treatment. Annu. Rep. Med. Chem. 2013, 48, 421–434. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Christensen, J.G.; Burrows, J.; Salgia, R. c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET Amplification Identifies a Small and Aggressive Subgroup of Esophagogastric Adenocarcinoma With Evidence of Responsiveness to Crizotinib. J. Clin. Oncol. 2011, 29, 4803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, R.; Petak, I.; Kollar, M.; Pinter, F.; Varkondi, E.; Kohanka, A.; Barti-Juhasz, H.; Schönleber, J.; Brauswetter, D.; Kopper, L.; et al. Major partial response to crizotinib, a dual MET/ALK inhibitor, in a squamous cell lung (SCC) carcinoma patient with de novo c-MET amplification in the absence of ALK rearrangement. Lung Cancer 2014, 83, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Bryson, H.M.; Sorkin, E.M. Cladribine. Drugs 1993, 46, 872–894. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Duggan, S. Darolutamide: First Approval. Drugs 2019, 79, 1813–1818. [Google Scholar] [CrossRef]
- Markham, A. Erdafitinib: First Global Approval. Drugs 2019, 79, 1017–1021. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.; Diamond, S.; Boer, J.; Harris, J.J.; Li, Y.; Rupar, M.; Behshad, E.; Gardiner, C.; Collier, P.; et al. The Janus Kinase 2 Inhibitor Fedratinib Inhibits Thiamine Uptake: A Putative Mechanism for the Onset of Wernicke’s Encephalopathy. Drug Metab. Dispos. 2014, 42, 1656–1662. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y. Selinexor: First Global Approval. Drugs 2019, 79, 1485–1494. [Google Scholar] [CrossRef]
- Syed, Y.Y. Zanubrutinib: First Approval. Drugs 2020, 80, 91–97. [Google Scholar] [CrossRef]
- Kim, E.S. Abemaciclib: First Global Approval. Drugs 2017, 77, 2063–2070. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Apalutamide: A Review in Non-Metastatic Castration-Resistant Prostate Cancer. Drugs 2019, 79, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Encorafenib and Binimetinib: First Global Approvals. Drugs 2018, 78, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Sidaway, P. Cemiplimab effective in cutaneous SCC. Nat. Rev. Clin. Oncol. 2018, 15, 472. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.A. Duvelisib: First Global Approval. Drugs 2018, 78, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Gilteritinib: First Global Approval. Drugs 2019, 79, 331–339. [Google Scholar] [CrossRef] [PubMed]



| Database | Simple Explanation | Reference |
|---|---|---|
| Gene Expression Omnibus (GEO) | GEO is a free, open-access repository for functional genomics data that accepts submissions of MIAME-compliant data. | Gene expression omnibus. Available online: http://www.ncbi.nlm.nih.gov/geo (accessed on 23 January 2022) |
| The Cancer Genome Atlas (TCGA) | Genomic statistics from >10,000 patient tissue samples from >30 prevalent cancers, such as exome, SNP, methylation, mRNA, miRNA, and clinical. | The Cancer Genome Atlas Program. Available online: http://cancergenome.nih.gov (accessed on 23 January 2022) |
| Genetic Association Database (GAD) | A database of information on genetic associations with serious illnesses and disorders. | Gender and Development Program. GAD Activities Sex Disaggregated Data. Available online: http://www.tapi.dost.gov.ph/resources/gad-databases (accessed on 23 January 2022) |
| Catalogue Of Somatic Mutations In Cancer (COSMIC) | A thorough resource for learning about somatic mutations’ effects on human cancer. | Catalogue Of Somatic Mutations In Cancer. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 23 January 2022) |
| Online Mendelian Inheritance in Man (OMIM) | Relationship between genetic traits, especially diseases, and genes. | An Online Catalog of Human Genes and Genetic Disorders. Available online: http://www.omim.org (accessed on 23 January 2022) |
| Database | Simple Explanation | Reference |
|---|---|---|
| Therapeutic Target (TTD) | A database that offers details on the diseases targeted, the investigated and undiscovered therapeutic protein and nucleic acid targets, the relevant methods, and the medications that are specific to each target. | Therapeutic Target Database. Available online: https://openebench.bsc.es/tool/ttd (accessed on 23 January 2022) |
| Genomics of Drug Sensitivity in Cancer (GDSC) | A database of 138 identified anticancer compounds (on average 525 cell lines studied for each drug) representing more than 1000 distinct cancer cell lines. | Genomics of Drug Sensitivity in Cancer. Available online: http://www.cancerrxgene.org/download (accessed on 23 January 2022) |
| DrugBank | Complete drug target data, including information on sequencing, structure, and route, together with detailed drug (i.e., chemical, pharmacological, and pharmaceutical) data. | Drug bank online. Available online: http://www.drugbank.ca/ (accessed on 23 January 2022) |
| PharmGKB | A freely accessible online knowledge repository that collects, organizes, synthesizes, and disseminates information about the influence of genetic variation on pharmacological response. | Online Knowledge Repository. Available online: https://www.pharmgkb.org/ (accessed on 23 January 2022) |
| Cancer Cell Line Encyclopedia (CCLE) | Genomic data, including information on DNA copy number, mRNA expression, and mutations, from more than 1000 cancer cell lines. | Cancer Cell Line Encyclopedia. Available online: https://portals.broadinstitute.org/ccle (accessed on 23 January 2022) |
| Name | Molecular Formula | ATC Code | Therapeutic Area | Target and Function | Year of Approval |
|---|---|---|---|---|---|
| Alpelisib | C19H22F3N5O2S | L01EM03 | Breast cancer | PI3K inhibitor | 2019 [181] |
| Cladribine | C10H12ClN5O3 | L04AA40 | Hairy cell leukemia | Adenosine deaminase inhibitor | 2019 [182] |
| Darolutamide | C19H19ClN6O2 | L02BB06 | Prostate cancer | Androgen receptor inhibitor | 2019 [183] |
| Entrectinib | C31H34F2N6O2 | L01EX14 | Non-small-cell lung cancer and solid tumors | Tyrosine kinase inhibitor | 2019 [88] |
| Erdafitinib | C25H30N6O2 | L01EN01 | Urothelial carcinoma | FGFR tyrosine inhibitor | 2019 [184] |
| Fedratinib Hydrochloride | C27H36N6O3S | L01EJ02 | Myelofibrosis | Tyrosine kinase inhibitor | 2019 [185] |
| Selinexor | C17H11F6N7O | L01XX66 | Multiple myeloma | Nuclear export inhibitor | 2019 [186] |
| Zanubrutinib | C27H29N5O3 | L01EL03 | Mantle cell lymphoma | Bruton′s tyrosine kinase inhibitor | 2019 [187] |
| Abemaciclib | C27H32F2N8 | L01EF03 | Breast cancer | Cyclin-dependent kinase inhibitor | 2018 [188] |
| Apalutamide | C21H15F4N5O2S | L02BB05 | Prostate cancer | Androgen receptor inhibitor | 2018 [189] |
| Binimetinib | C17H15BrF2N4O3 | L01EE03 | Melanoma | MEk1 and MEK2 inhibitor | 2018 [190] |
| Dacomitinib | C24H27ClFN5O3 | L01EB07 | Non-small-cell lung cancer | Oral kinase inhibitor | 2018 [191] |
| Duvelisib | C22H17ClN6O | L01EM04 | Chronic lymphocytic leukemia (CLL) and follicular lymphoma (FL) | PI3K kinase inhibitor | 2018 [192] |
| Encorafenib | C22H27Cl1F1N7O4S1 | L01EC03 | Colorectal cancer and melanoma | BRAF kinase inhibitor | 2018 [190] |
| Gilteritinib Fumarate | C62H92N16O10 | L01EX13 | Acute myeloid leukemia | Tyrosine kinase inhibitor | 2018 [193] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; Islam, M.R.; Rahman, F.; Rahaman, M.S.; Khan, M.S.; Abrar, S.; Ray, T.K.; Uddin, M.B.; Kali, M.S.K.; Dua, K.; et al. Emerging Promise of Computational Techniques in Anti-Cancer Research: At a Glance. Bioengineering 2022, 9, 335. https://doi.org/10.3390/bioengineering9080335
Rahman MM, Islam MR, Rahman F, Rahaman MS, Khan MS, Abrar S, Ray TK, Uddin MB, Kali MSK, Dua K, et al. Emerging Promise of Computational Techniques in Anti-Cancer Research: At a Glance. Bioengineering. 2022; 9(8):335. https://doi.org/10.3390/bioengineering9080335
Chicago/Turabian StyleRahman, Md. Mominur, Md. Rezaul Islam, Firoza Rahman, Md. Saidur Rahaman, Md. Shajib Khan, Sayedul Abrar, Tanmay Kumar Ray, Mohammad Borhan Uddin, Most. Sumaiya Khatun Kali, Kamal Dua, and et al. 2022. "Emerging Promise of Computational Techniques in Anti-Cancer Research: At a Glance" Bioengineering 9, no. 8: 335. https://doi.org/10.3390/bioengineering9080335