
A Bioinformatics View on Acute Myeloid Leukemia Surface Molecules by Combined Bayesian and ABC Analysis
 , ,
, ,
Abstract
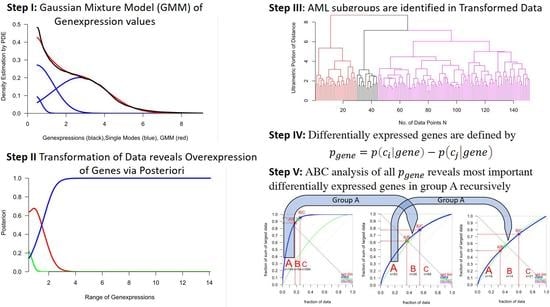
:
1. Introduction
2. Materials and Methods
2.1. Gene Expression Datasets
2.2. Calculation of Bayesian Decision Borders for Expressed and Unexpressed Genes
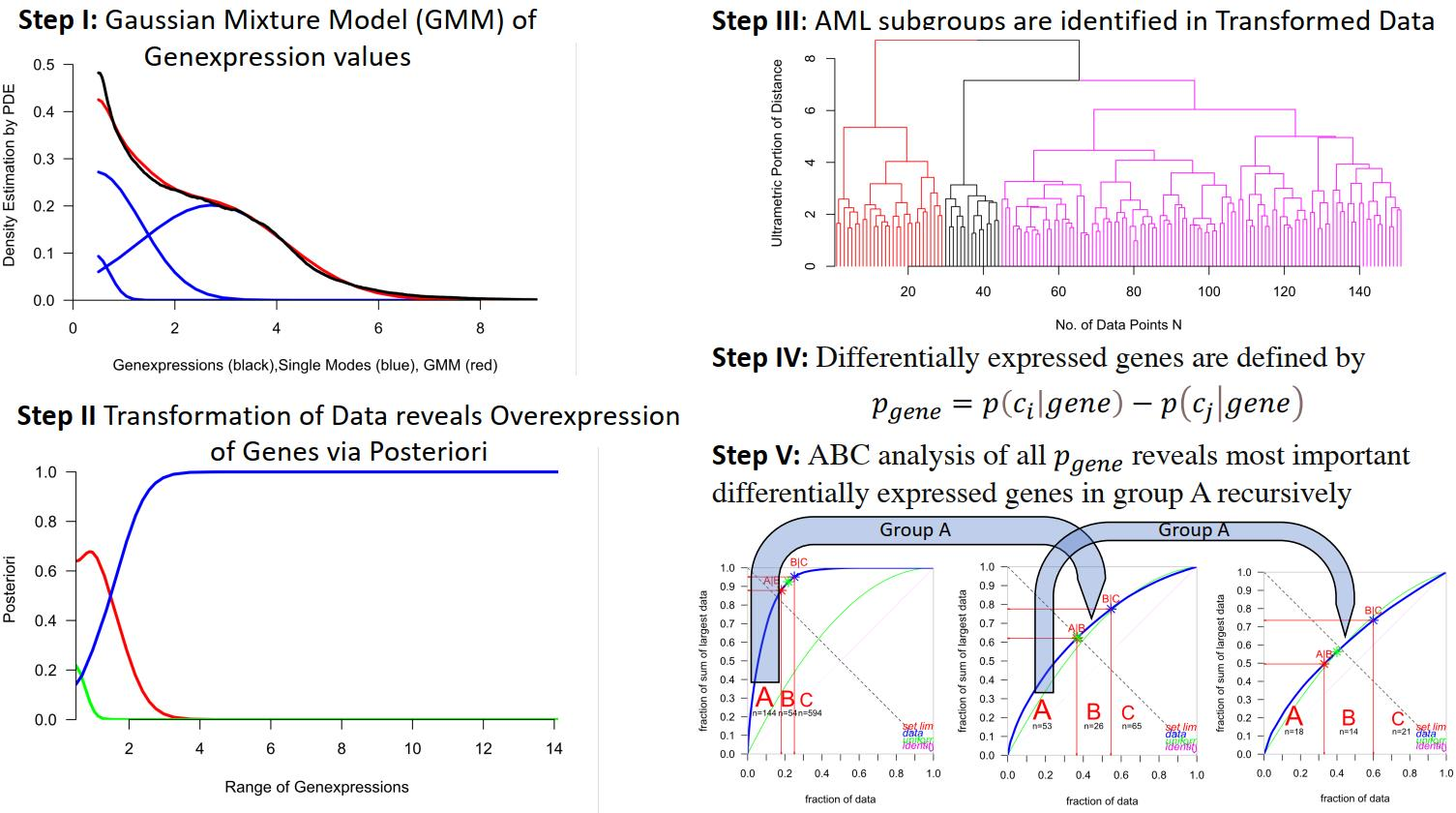
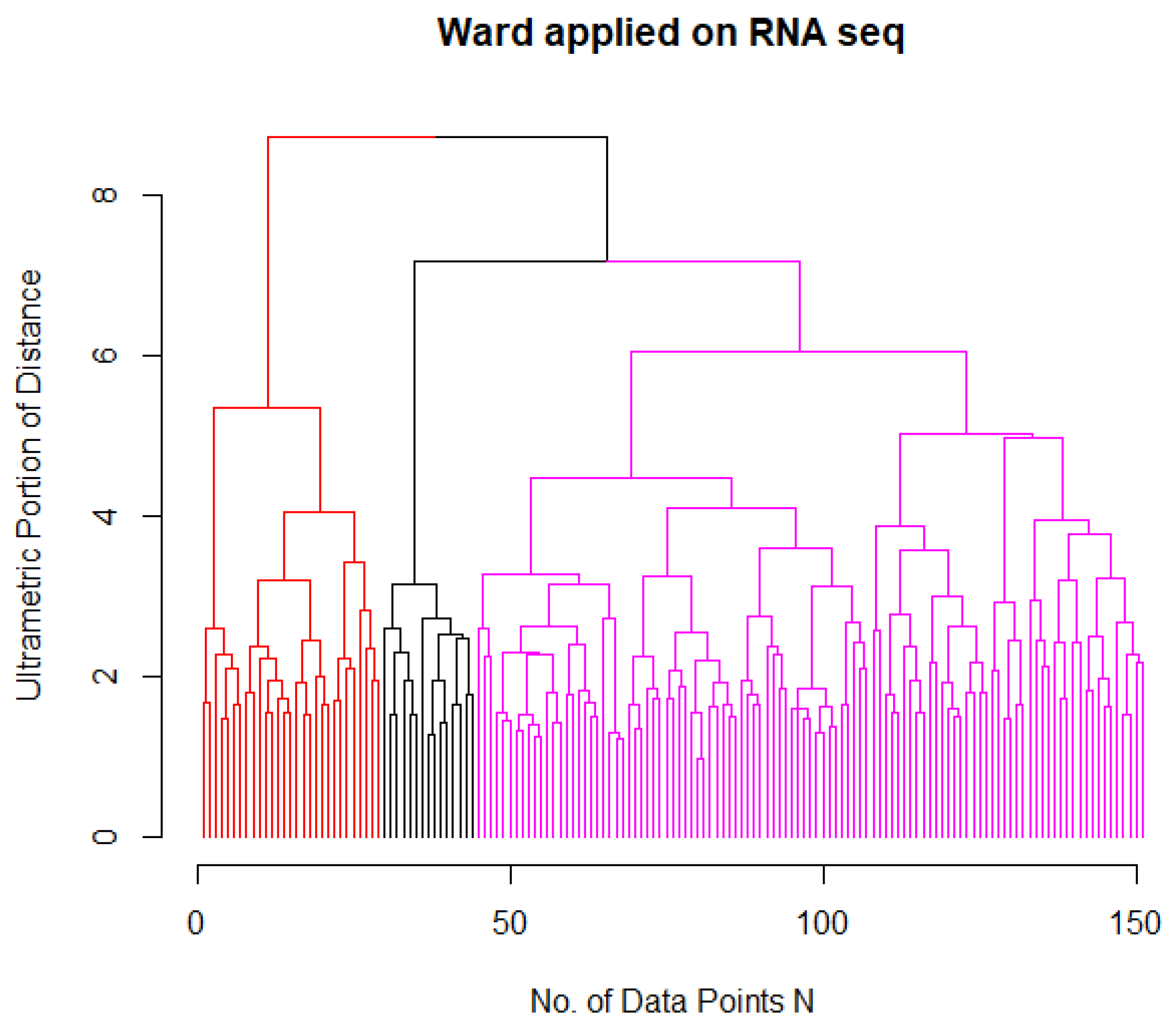
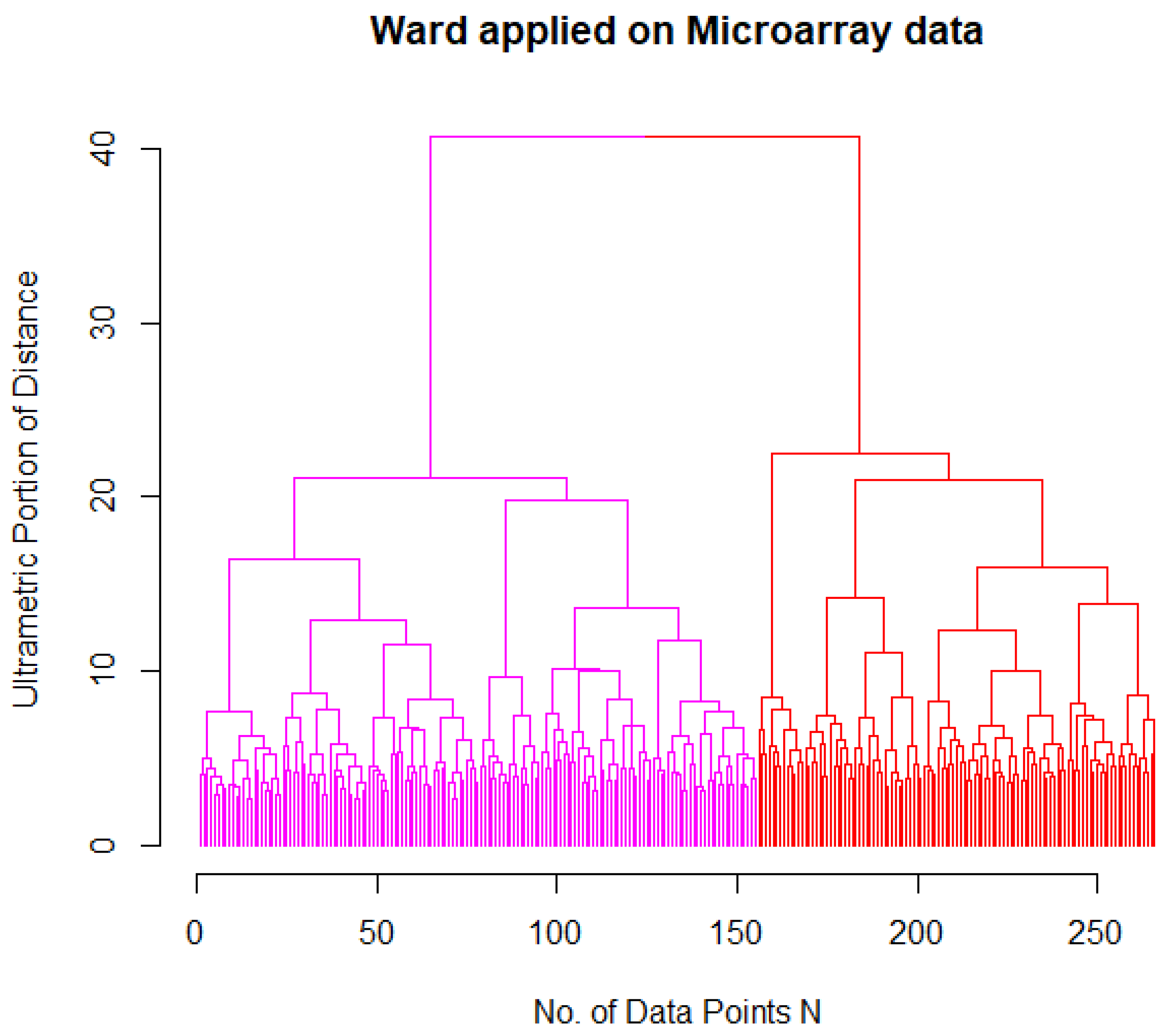
2.3. Identification of AML Subgroups
2.4. Selection of Differentially Expressed Genes (Deg)
3. Results
3.1. Subgroups of AML Based on the Expression of CD Genes
3.2. Differential Expression of CD Genes in AML Subtypes
3.3. Differential Expression of CD Genes in AML Compared to Normal Samples
3.4. Validation of Target Genes by Literature Screening

{kind=link}
{kind=link}
{kind=link}
| Index in Table 2/Table 3 | Gene | Evidence |
|---|---|---|
| 21,14 | CD339 | [27,28] |
| 1 | CD3D | [40] |
| 2 | CD9 | [41,42,43,44] |
| 3 | CD11b | [41,42,43,44] |
| 4 | CD14 | [19,43] |
| 5 | CD15 | [45,46] |
| 6 | CD18 | [47] |
| 7 | CD24 | [48] |
| 8 | CD37 | [49] |
| 9 | CD50 | [50] |
| 10 | CD52 | [51] |
| 11 | CD55 | [52] |
| 12 | CD62L | [53] |
| 13 | CD66b | [54] |
| 14 | CD66c | [54] |
| 15 | CD83 | [55,56] |
| 16 | CD87 | [57] |
| 17 | CD88 | [30] |
| 18 | CD98 | [58,59] |
| 19 | CD210A | [60,61] |
| 20 | CD282 | [57] |
| 23 | CD369 | [62] |
| 1 | CD1D | [63] |
| 2 | CD7 | [64,65] |
| 3 | CD13 | [45,46] |
| 4 | CD44 | [60,66] |
| 5 | CD79A | [67,68] |
| 6 | CD84 | [29,69] |
| 7 | CD88 | [30] |
| 9 | CD148 | [31] |
| 11 | CD227 | [24,32,33] |
| 12 | CD230 | [70] |
| 13 | CD261 | x |
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garraway, L.A. Genomics-Driven Oncology: Framework for an Emerging Paradigm. J. Clin. Oncol. 2013, 31, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.-J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013, 32, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Sanz, M.A.; Fenaux, P.; Tallman, M.S.; Estey, E.H.; Löwenberg, B.; Naoe, T.; Lengfelder, E.; Döhner, H.; Burnett, A.K.; Chen, S.-J.; et al. Management of acute promyelocytic leukemia: Updated recommendations from an expert panel of the European LeukemiaNet. Blood 2019, 133, 1630–1643. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [Green Version]
- Ultsch, A.; Lotsch, J. Computed ABC Analysis for Rational Selection of Most Informative Variables in Multivariate Data. PLoS ONE 2015, 10, e0129767. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Béné, M.-C.; De Vos, J.; Hernández, J.M.; Hofmann, W.-K.; Mills, K.I.; et al. Clinical Utility of Microarray-Based Gene Expression Profiling in the Diagnosis and Subclassification of Leukemia: Report From the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Thrun, M.C.; Gehlert, T.; Ultsch, A. Analyzing the fine structure of distributions. PLoS ONE 2020, 15, e0238835. [Google Scholar] [CrossRef] [PubMed]
- Ultsch, A.; Thrun, M.C.; Hansen-Goos, O.; Lötsch, J. Identification of Molecular Fingerprints in Human Heat Pain Thresholds by Use of an Interactive Mixture Model R Toolbox (AdaptGauss). Int. J. Mol. Sci. 2015, 16, 25897–25911. [Google Scholar] [CrossRef]
- Fraley, C.; E Raftery, A. Model-Based Clustering, Discriminant Analysis, and Density Estimation. J. Am. Stat. Assoc. 2002, 97, 611–631. [Google Scholar] [CrossRef]
- Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 1974, 19, 716–723. [Google Scholar] [CrossRef]
- Smirnov, N. Table for Estimating the Goodness of Fit of Empirical Distributions. Ann. Math. Stat. 1948, 19, 279–281. [Google Scholar] [CrossRef]
- Thrun, M.C.; Stier, Q. Fundamental clustering algorithms suite. SoftwareX 2021, 13, 100642. [Google Scholar] [CrossRef]
- Thrun, M.C. Projection Based Clustering through Self-Organization and Swarm Intelligence; Springer: Heidelberg, Germany, 2018. [Google Scholar]
- Hedges, L.V.; Olkin, I. Statistical Methods for Meta-Analysis; Academic Press: New York, NY, USA, 1985. [Google Scholar]
- Bennett, J.M.; Catovsky, D.; Daniel, M.-T.; Flandrin, G.; Galton, D.A.G.; Gralnick, H.R.; Sultan, C. Proposals for the Classification of the Acute Leukaemias French-American-British (FAB) Co-operative Group. Br. J. Haematol. 1976, 33, 451–458. [Google Scholar] [CrossRef]
- Haferlach, T.; Kohlmann, A.; Schnittger, S.; Dugas, M.; Hiddemann, W.; Kern, W.; Schoch, C. AML M3 and AML M3 variant each have a distinct gene expression signature but also share patterns different from other genetically defined AML subtypes. Genes Chromosom. Cancer 2005, 43, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.; Kohlmann, A.; Schnittger, S.; Brors, B.; Dugas, M.; Mergenthaler, S.; Kern, W.; Hiddemann, W.; Eils, R.; Haferlach, T. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc. Natl. Acad. Sci. USA 2002, 99, 10008–10013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.S.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like Leukemia Stem Cells in Acute Myeloid Leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matutes, E.; Morilla, R.; Farahat, N.; Carbonell, F.; Swansbury, J.; Dyer, M.; Catovsky, D. Definition of acute biphenotypic leukemia. Haematologica 1997, 82, 64–66. [Google Scholar] [PubMed]
- Grieselhuber, N.; Klco, J.; Verdoni, A.M.; Lamprecht, T.; Sarkaria, S.; Wartman, L.D.; Ley, T.J. Notch signaling in acute promyelocytic leukemia. Leukemia 2013, 27, 1548–1557. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, E.; Itoh, M.; Tojo, N.; Koyama, T.; Nara, N.; Tohda, S. Flow cytometric analysis of Notch1 and Jagged1 expression in normal blood cells and leukemia cells. Exp. Ther. Med. 2012, 4, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, M.; Hirtreiter, C.; Rehli, M.; Rehm, A.; A Kunz-Schughart, L.; Andreesen, R.; Hennemann, B. CD84 expression on human hematopoietic progenitor cells. Exp. Hematol. 2003, 31, 798–805. [Google Scholar] [CrossRef]
- Ismail, A.A.; Borkowska-Rzeszotek, S.; Kubiś, E.; Bujko, K.; Brzeźniakiewicz-Janus, K.; Bolkun, L.; Kloczko, J.; Moniuszko, M.; Basak, G.W.; Wiktor-Jedrzejczak, W.; et al. Activation of the complement cascade enhances motility of leukemic cells by downregulating expression of HO-1. Leukemia 2016, 31, 446–458. [Google Scholar] [CrossRef]
- Kresinsky, A.; Bauer, R.; Schnöder, T.M.; Berg, T.; Meyer, D.; Ast, V.; König, R.; Serve, H.; Heidel, F.H.; Böhmer, F.-D.; et al. Loss of DEP-1 (Ptprj) promotes myeloproliferative disease in FLT3-ITD acute myeloid leukemia. Haematologica 2018, 103, e505–e509. [Google Scholar] [CrossRef]
- Brossart, P.; Schneider, A.; Dill, P.; Schammann, T.; Grünebach, F.; Wirths, S.; Kanz, L.; Bühring, H.J.; Brugger, W. The epithelial tumor antigen MUC1 is expressed in hematological malignancies and is recognized by MUC1-specific cytotoxic T-lymphocytes. Cancer Res. 2001, 61, 6846–6850. [Google Scholar]
- Fatrai, S.; Schepers, H.; Tadema, H.; Vellenga, E.; Daenen, S.M.; Schuringa, J.J. Mucin1 expression is enriched in the human stem cell fraction of cord blood and is upregulated in majority of the AML cases. Exp. Hematol. 2008, 36, 1254–1265. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, T.; Liu, X.; Haubner, S.; Bücklein, V.; Hänel, G.; Krupka, C.; Solis-Mezarino, V.; Herzog, F.; Subklewe, M. Integrated multiomic approach for identification of novel immunotherapeutic targets in AML. Biomark. Res. 2022, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.A.; Loken, M.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Aplenc, R.; Bernstein, I.D.; Gamis, A.S.; Alonzo, T.A.; Meshinchi, S. CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2016, 34, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wang, M.; Hu, Y.; Xing, H.; Chen, X.; Zhang, Y.; Zhu, P. Significance of CD71 expression by flow cytometry in diagnosis of acute leukemia. Leuk. Lymphoma 2013, 55, 892–898. [Google Scholar] [CrossRef]
- Boyer, T.; Guihard, S.; Roumier, C.; Peyrouze, P.; Gonzales, F.; Berthon, C.; Quesnel, B.; Preudhomme, C.; Behal, H.; Duhamel, A.; et al. Tetraspanin CD81 is an adverse prognostic marker in acute myeloid leukemia. Oncotarget 2016, 7, 62377–62385. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.C.; Dunphy, C.H. Immunoreactivity of MIC2 (CD99) and Terminal Deoxynucleotidyl Transferase in Bone Marrow Clot and Core Specimens of Acute Myeloid Leukemias and Myelodysplastic Syndromes. Arch. Pathol. Lab. Med. 2006, 130, 153–157. [Google Scholar] [CrossRef]
- Pereira, M.S.; Silva, M.L.M.; Valente, A.N.; De Souza, M.H.O.; Abdelhay, E. Translocation of CD3D gene in an acute myeloid leukemia (M5) with t(11;17)(q23;21). Cancer Genet. Cytogenet. 1993, 71, 173–175. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, N.; Xu, Z.; Xu, J.; Zhang, Y.; Chen, X.; Tan, Y.; Chang, J.; Wang, H. TheCD9+CD11b−HLA-DR−immunophenotype can be used to diagnose acute promyelocytic leukemia. Int. J. Lab. Hematol. 2018, 41, 168–175. [Google Scholar] [CrossRef]
- Dong, H.Y.; Kung, J.X.; Bhardwaj, V.; McGill, J. Flow Cytometry Rapidly Identifies All Acute Promyelocytic Leukemias With High Specificity Independent of Underlying Cytogenetic Abnormalities. Am. J. Clin. Pathol. 2011, 135, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, Y.; Tong, Y.; Gao, Q.; Mao, X.; Zhang, W.; Xia, Z.; Fu, C. Stepwise discriminant function analysis for rapid identification of acute promyelocytic leukemia from acute myeloid leukemia with multiparameter flow cytometry. Int. J. Hematol. 2016, 103, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, D.; Carlesso, N.; Gallo, E.; Pregno, P.; de Fabritiis, P.; Petti, M.C.; Mandelli, F. CD9 antigen on acute non-lymphoid leukemia cells: Preferential expression by promyelocytic (M3) subtype. Leuk. Res. 1991, 15, 457–461. [Google Scholar] [CrossRef]
- Kárai, B.; Habók, M.; Reményi, G.; Rejtő, L.; Ujfalusi, A.; Kappelmayer, J.; Hevessy, Z. A novel flow cytometric method for enhancing acute promyelocytic leukemia screening by multidimensional dot-plots. Ann. Hematol. 2019, 98, 1413–1420. [Google Scholar] [CrossRef] [Green Version]
- Orfao, A.; Chillón, M.C.; Bortoluci, A.M.; López-Berges, M.C.; García-Sanz, R.; Gonzalez, M.; Tabernero, M.D.; García-Marcos, M.A.; Rasillo, A.I.; Hernández-Rivas, J. The flow cytometric pattern of CD34, CD15 and CD13 expression in acute myeloblastic leukemia is highly characteristic of the presence of PML-RARalpha gene rearrangements. Haematologica 1999, 84, 405–412. [Google Scholar] [PubMed]
- Zhou, Y.; Jorgensen, J.L.; Wang, S.A.; Ravandi, F.; Cortes, J.; Kantarjian, H.M.; Medeiros, L.J.; Konoplev, S. Usefulness of CD11a and CD18 in Flow Cytometric Immunophenotypic Analysis for Diagnosis of Acute Promyelocytic Leukemia. Am. J. Clin. Pathol. 2012, 138, 744–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raife, T.J.; Lager, D.J.; Kemp, J.D.; Dick, F.R. Expression of CD24 (BA-1) Predicts Monocytic Lineage in Acute Myeloid Leukemia. Am. J. Clin. Pathol. 1994, 101, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Kamashev, D.; Vitoux, D.; De Thé, H. PML–RARA-RXR Oligomers Mediate Retinoid and Rexinoid/cAMP Cross-Talk in Acute Promyelocytic Leukemia Cell Differentiation. J. Exp. Med. 2004, 199, 1163–1174. [Google Scholar] [CrossRef]
- Khaznadar, Z.; Boissel, N.; Agaugué, S.; Henry, G.; Cheok, M.; Vignon, M.; Geromin, D.; Cayuela, J.-M.; Castaigne, S.; Pautas, C.; et al. Defective NK Cells in Acute Myeloid Leukemia Patients at Diagnosis Are Associated with Blast Transcriptional Signatures of Immune Evasion. J. Immunol. 2015, 195, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-W.; Tang, D.; Ahrens, K.P.; She, J.-X.; Braylan, R.C.; Yang, L. All-trans-retinoic acid induces CD52 expression in acute promyelocytic leukemia. Blood 2003, 101, 1977–1980. [Google Scholar] [CrossRef] [Green Version]
- Guc, D.; Canpınar, H.; Kucukaksu, C.; Kansu, E. Expression of complement regulatory proteins CR1, DAF, MCP and CD59in haematological malignancies. Eur. J. Haematol. 2000, 64, 3–9. [Google Scholar] [CrossRef]
- Jambrovics, K.; Uray, I.P.; Keresztessy, Z.; Keillor, J.W.; Fésüs, L.; Balajthy, Z. Transglutaminase 2 programs differentiating acute promyelocytic leukemia cells in all-trans retinoic acid treatment to inflammatory stage through NF-κB activation. Haematologica 2019, 104, 505–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, M.; Zaghloul, A.; Abdulateef, N.; Morsi, H. Membranous Expression of pan CD66, CD66a, CD66b, and CD66c and their Clinical Impact in Acute Leukemia: Cross Sectional Longitudinal Cohort Study in Saudi Arabia. J. Leuk. 2017, 5, 230–239. [Google Scholar] [CrossRef]
- Shrestha, B.; Walton, K.; Reff, J.; Sagatys, E.M.; Tu, N.; Boucher, J.C.; Ghafoor, B.T.; Pidala, J.; Blazar, B.R.; Anasetti, C.; et al. Human CD83 Targeted Chimeric Antigen Receptor T Cell for the Prevention of Graft Versus Host Disease and Treatment of Myeloid Leukemia. Blood 2019, 134, 196. [Google Scholar] [CrossRef]
- Rigolin, G.M.; Della Porta, M.; Bigoni, R.; Tieghi, A.; Cuneo, A.; Castoldi, G. Dendritic cells in acute promyelocytic leukaemia. Br. J. Haematol. 2001, 114, 830–833. [Google Scholar] [CrossRef]
- Atfy, M.; Eissa, M.; Salah, H.E.; El Shabrawy, D.A. Role of urokinase plasminogen activator receptor (CD87) as a prognostic marker in acute myeloid leukemia. Med. Oncol. 2011, 29, 2063–2069. [Google Scholar] [CrossRef]
- Bajaj, J.; Konuma, T.; Lytle, N.K.; Kwon, H.Y.; Ablack, J.N.; Cantor, J.M.; Rizzieri, D.; Chuah, C.; Oehler, V.G.; Broome, E.H.; et al. CD98-Mediated Adhesive Signaling Enables the Establishment and Propagation of Acute Myelogenous Leukemia. Cancer Cell 2016, 30, 792–805. [Google Scholar] [CrossRef] [Green Version]
- Bixby, D.; Wieduwilt, M.J.; Akard, L.P.; Khoury, H.J.; Becker, P.S.; Van Der Horst, E.H.; Ho, W.; Cortes, J.E. A Phase I Study of IGN523, a Novel Anti-CD98 Monoclonal Antibody in Patients with Relapsed or Refractory Acute Myeloid Leukemia (AML). Blood 2015, 126, 3809. [Google Scholar] [CrossRef]
- Coustan-Smith, E.; Song, G.; Shurtleff, S.; Yeoh, A.E.-J.; Chng, W.J.; Chen, S.P.; Rubnitz, J.E.; Pui, C.-H.; Downing, J.R.; Campana, D. Universal monitoring of minimal residual disease in acute myeloid leukemia. JCI Insight 2018, 3, 98561. [Google Scholar] [CrossRef]
- Chen, N.; Xu, Y.; Mou, J.; Rao, Q.; Xing, H.; Tian, Z.; Tang, K.; Wang, M.; Wang, J. Targeting of IL-10R on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood Cancer J. 2021, 11, 144. [Google Scholar] [CrossRef]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef]
- Metelitsa, L.S.; I Weinberg, K.; Emanuel, P.; Seeger, R.C. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia 2003, 17, 1068–1077. [Google Scholar] [CrossRef] [PubMed]
- Sobas, M.; Montesinos, P.; Boluda, B.; Bernal, T.; Vellenga, E.; Nomdedeu, J.; González-Campos, J.; Chillón, M.; Holowiecka, A.; Esteve, J.; et al. An analysis of the impact of CD56 expression in de novo acute promyelocytic leukemia patients treated with upfront all-trans retinoic acid and anthracycline-based regimens. Leuk. Lymphoma 2018, 60, 1030–1035. [Google Scholar] [CrossRef]
- Del Poeta, G.; Stasi, R.; Venditti, A.; Cox, C.; Aronica, G.; Masi, M.; Bruno, A.; Simone, M.D.; Buccisano, F.; Papa, G.; et al. CD7 Expression in Acute Myeloid Leukemia. Leuk. Lymphoma 1995, 17, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Legras, S.; Günthert, U.; Stauder, R.; Curt, F.; Oliferenko, S.; Kluin-Nelemans, H.; Marie, J.; Proctor, S.; Jasmin, C.; Smadja-Joffe, F. A Strong Expression of CD44-6v Correlates With Shorter Survival of Patients With Acute Myeloid Leukemia. Blood 1998, 91, 3401–3413. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Kallakury, B.V.; Ross, J.S.; Azumi, N.; Bagg, A. CD79a Is Heterogeneously Expressed in Neoplastic and Normal Myeloid Precursors and Megakaryocytes in an Antibody Clone–Dependent Manner. Am. J. Clin. Pathol. 2007, 128, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; A Jenkins, K.; Slovak, M.L. CD79 alpha expression in acute myeloid leukemia. High frequency of expression in acute promyelocytic leukemia. Am. J. Pathol. 1996, 149, 1105–1110. [Google Scholar]
- Çelik, H.; Lindblad, K.E.; Popescu, B.; Gui, G.; Goswami, M.; Valdez, J.; DeStefano, C.; Lai, C.; Thompson, J.; Ghannam, J.Y.; et al. Highly multiplexed proteomic assessment of human bone marrow in acute myeloid leukemia. Blood Adv. 2020, 4, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Leung, K.K.; Nguyen, A.; Shi, T.; Tang, L.; Ni, X.; Escoubet, L.; MacBeth, K.J.; DiMartino, J.; Wells, J.A. Multiomics of azacitidine-treated AML cells reveals variable and convergent targets that remodel the cell-surface proteome. Proc. Natl. Acad. Sci. USA 2019, 116, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Gallazzi, M.; Ucciero, M.A.M.; Faraci, D.G.; Mahmoud, A.M.; Al Essa, W.; Gaidano, G.; Mouhssine, S.; Crisà, E. New Frontiers in Monoclonal Antibodies for the Targeted Therapy of Acute Myeloid Leukemia and Myelodysplastic Syndromes. Int. J. Mol. Sci. 2022, 23, 7542. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; Fazi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic Acid and Arsenic Trioxide for Acute Promyelocytic Leukemia. N. Engl. J. Med. 2013, 369, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Zhong, S.; Liu, X.; Mai, H.; Mai, G.; Xu, C.; Zhou, F. CD3D and PRKCQ work together to discriminate between B-cell and T-cell acute lymphoblastic leukemia. Comput. Biol. Med. 2016, 77, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sheng, Q.; Li, J.; Ye, F.; Samuels, D.C.; Shyr, Y. Large Scale Comparison of Gene Expression Levels by Microarrays and RNAseq Using TCGA Data. PLoS ONE 2013, 8, e71462. [Google Scholar] [CrossRef] [PubMed]
- Thrun, M.C. Cluster Analysis of Per Capita Gross Domestic Products. Entrep. Bus. Econ. Rev. 2018, 7, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Thrun, M.; Ultsch, A. Models of Income Distributions for Knowledge Discovery. In European Conference on Data Analysis; University of Essex: Colchester, UK, 2015; pp. 136–137. [Google Scholar]
- Gbadamosi, M.O.; Shastri, V.M.; Elsayed, A.H.; Ries, R.; Olabige, O.; Nguyen, N.H.K.; De Jesus, A.; Wang, Y.-C.; Dang, A.; Hirsch, B.A.; et al. A ten-gene DNA-damage response pathway gene expression signature predicts gemtuzumab ozogamicin response in pediatric AML patients treated on COGAAML0531 and AAML03P1 trials. Leukemia 2022, 36, 2022–2031. [Google Scholar] [CrossRef] [PubMed]


| Ward Cluster | M0 | M1 | M2 | M3 | M4 | M5 | M6 | M7 |
|---|---|---|---|---|---|---|---|---|
| 1 (magenta) | 15 | 34 | 36 | 0 | 17 | 2 | 2 | 1 |
| 2 (red) | 0 | 3 | 1 | 0 | 12 | 13 | 0 | 0 |
| 3 (black) | 0 | 0 | 0 | 15 | 0 | 0 | 0 | 0 |
| Gene | APL vs. AML1 | APL vs. AML2 | AML1 vs. AML2 | |
|---|---|---|---|---|
| 1 | CD3D | 0.9 | 0.92 | 0 |
| 2 | CD9 | 0.89 | 0 | 0 |
| 3 | CD11b | 0 | −1.17 | −0.94 |
| 4 | CD14 | 0 | −0.99 | 0 |
| 5 | CD15 | 0 | −0.77 | 0 |
| 6 | CD18 | −0.97 | −1.69 | −0.72 |
| 7 | CD24 | 0 | −1.29 | −0.79 |
| 8 | CD37 | 0 | −1.09 | 0 |
| 9 | CD50 | 0 | −0.98 | 0 |
| 10 | CD52 | −0.96 | −1.25 | 0 |
| 11 | CD55 | 0.78 | 0 | 0 |
| 12 | CD62L | −1.04 | −1.44 | 0 |
| 13 | CD66b | 0 | −1.49 | −0.82 |
| 14 | CD66c | 0 | −1.06 | 0 |
| 15 | CD83 | −0.7 | −1.4 | 0 |
| 16 | CD87 | 1.08 | 0 | 0 |
| 17 | CD88 | 0 | −1.1 | −1.09 |
| 18 | CD98 | 0.93 | 1.05 | 0 |
| 19 | CD210A | 0 | −1 | −0.79 |
| 20 | CD282 | 0 | −0.81 | −0.75 |
| 21 | CD339 | 0.9 | 0.75 | 0 |
| 22 | CD354 | 0 | 0 | −0.75 |
| 23 | CD369 | 0 | −1.04 | −0.94 |
| Gene | APL vs. AML1 | APL vs. AML2 | AML1 vs. AML2 | |
|---|---|---|---|---|
| 1 | CD1D | 0 | 0.42 | 0 |
| 2 | CD7 | 0 | 0.52 | 0 |
| 3 | CD13 | 0 | 0.39 | 0 |
| 4 | CD44 | 0 | 0.53 | 0 |
| 5 | CD79A | −0.39 | 0 | 0 |
| 6 | CD84 | 0.32 | 0.32 | 0 |
| 7 | CD88 | 0 | 0.34 | 0 |
| 8 | CD91 | 0 | 0 | −0.32 |
| 9 | CD148 | 0.88 | 0.89 | 0 |
| 10 | CD197 | 0 | 0 | 0.32 |
| 11 | CD227 | 0 | −0.36 | 0 |
| 12 | CD230 | 0 | 0.32 | 0 |
| 13 | CD261 | 0 | 0.55 | 0.52 |
| 14 | CD339 | 0.45 | 0.42 | 0 |
| Gene | APL vs. Normal | AML1 vs. Normal | AML2 vs. Normal | |
|---|---|---|---|---|
| 1 | CD1D | 0 | 0 | 0.39 |
| 2 | CD3D | 0.72 | 0 | 0 |
| 3 | CD9 | 1 | 0 | 0.31 |
| 4 | CD11b | −1.24 | −1 | 0 |
| 5 | CD13 | 0 | 0 | 0.33 |
| 6 | CD14 | 0 | 0 | 0.49 |
| 7 | CD18 | −1.18 | 0 | 0.52 |
| 8 | CD24 | −1.83 | −1.33 | −0.54 |
| 9 | CD32 | 0 | 0 | 0.34 |
| 10 | CD33 | 0 | 0 | 0.46 |
| 11 | CD37 | 0 | 0 | 0.97 |
| 12 | CD44 | 0 | 0 | 0.45 |
| 13 | CD50 | −0.73 | 0 | 0 |
| 14 | CD52 | −0.86 | 0 | 0.39 |
| 15 | CD62L | −0.98 | 0 | 0.46 |
| 16 | CD66b | −1.91 | −1.24 | −0.42 |
| 17 | CD66c | −1.8 | −1.43 | −0.74 |
| 18 | CD71 | 0 | 0 | −0.44 |
| 19 | CD81 | 0.82 | 0 | 0.53 |
| 20 | CD83 | −0.84 | 0 | 0.56 |
| 21 | CD87 | 0.8 | 0 | 0.4 |
| 22 | CD88 | −0.8 | −0.79 | 0.3 |
| 23 | CD97 | 0 | 0 | 0.73 |
| 24 | CD98 | 1.01 | 0 | 0 |
| 25 | CD99 | 1.44 | 1.14 | 1 |
| 26 | CD107b | 0 | 0 | 0.41 |
| 27 | CD114 | 0 | 0 | 0.55 |
| 28 | CD119 | 0 | 0 | 0.39 |
| 29 | CD120b | 0 | 0 | 0.5 |
| 30 | CD132 | 0.72 | 0 | 0.35 |
| 31 | CD135 | 0.7 | 0.82 | 0.6 |
| 32 | CD157 | 0 | 0 | 0.37 |
| 33 | CD210A | 0 | 0 | 0.68 |
| 34 | CD218b | −0.79 | −0.7 | −0.52 |
| 35 | CD225 | 0 | 0 | −0.39 |
| 36 | CD230 | 0 | 0 | 0.36 |
| 37 | CD233 | −1.29 | −0.83 | −1.09 |
| 38 | CD235a | −0.87 | 0 | −0.81 |
| 39 | CD235b | 0 | 0 | −0.53 |
| 40 | CD236 | 0 | 0 | −0.41 |
| 41 | CD240D | 0 | 0 | −0.44 |
| 42 | CD241 | 0 | 0 | −0.31 |
| 43 | CD256 | 0 | 0 | 0.4 |
| 44 | CD257 | 0 | 0 | 0.53 |
| 45 | CD282 | 0 | 0 | 0.68 |
| 46 | CD339 | 0.92 | 0 | 0 |
| 47 | CD354 | 0 | 0 | 0.75 |
| 48 | CD369 | 0 | 0 | 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thrun, M.C.; Mack, E.K.M.; Neubauer, A.; Haferlach, T.; Frech, M.; Ultsch, A.; Brendel, C. A Bioinformatics View on Acute Myeloid Leukemia Surface Molecules by Combined Bayesian and ABC Analysis. Bioengineering 2022, 9, 642. https://doi.org/10.3390/bioengineering9110642
Thrun MC, Mack EKM, Neubauer A, Haferlach T, Frech M, Ultsch A, Brendel C. A Bioinformatics View on Acute Myeloid Leukemia Surface Molecules by Combined Bayesian and ABC Analysis. Bioengineering. 2022; 9(11):642. https://doi.org/10.3390/bioengineering9110642
Chicago/Turabian StyleThrun, Michael C., Elisabeth K. M. Mack, Andreas Neubauer, Torsten Haferlach, Miriam Frech, Alfred Ultsch, and Cornelia Brendel. 2022. "A Bioinformatics View on Acute Myeloid Leukemia Surface Molecules by Combined Bayesian and ABC Analysis" Bioengineering 9, no. 11: 642. https://doi.org/10.3390/bioengineering9110642