
Verification of the Efficacy of Mexiletine Treatment for the A1656D Mutation on Downgrading Reentrant Tachycardia Using a 3D Cardiac Electrophysiological Model


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
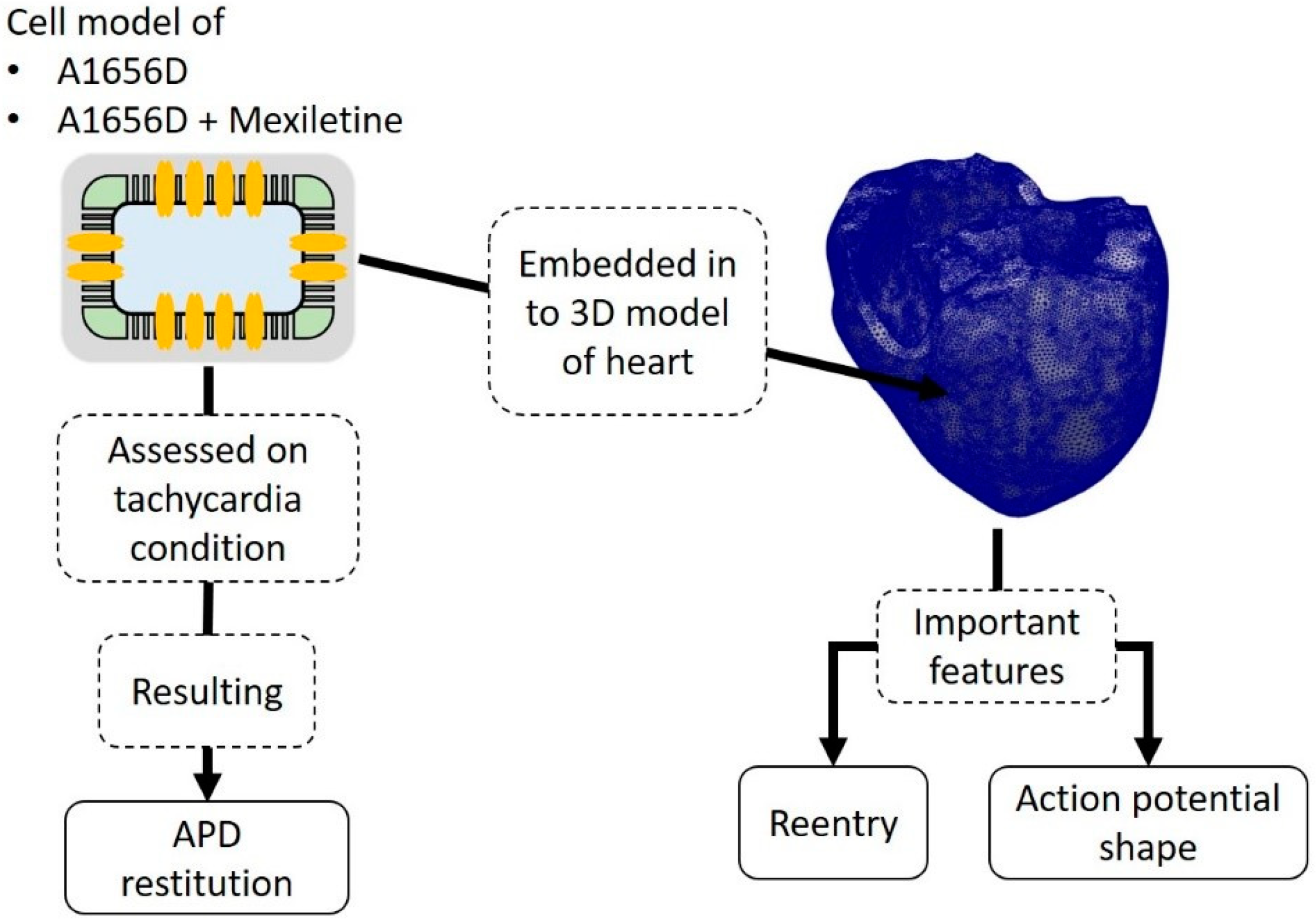
2. Materials and Methods
2.1. Model of Ventricular Cell under A1656D Mutation with Incorporated Drug Effects
2.2. Simulation Protocol
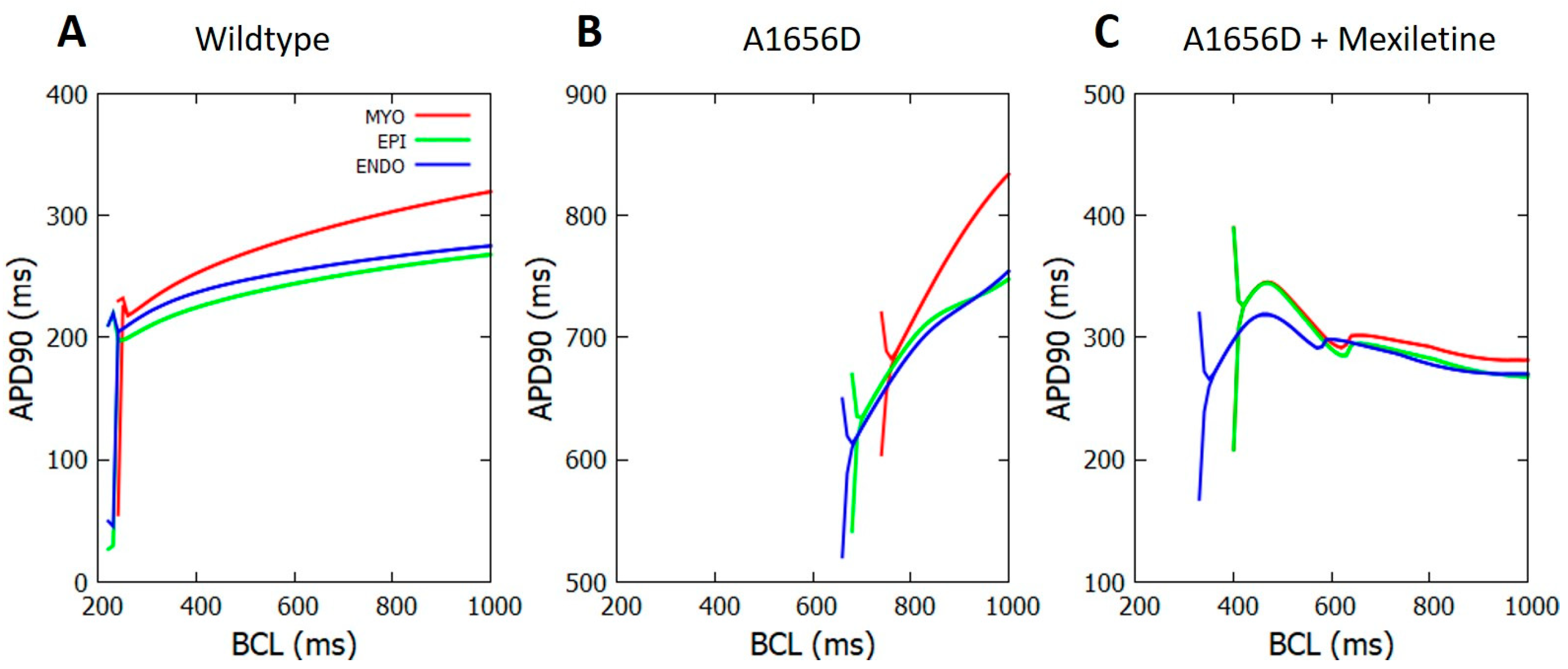
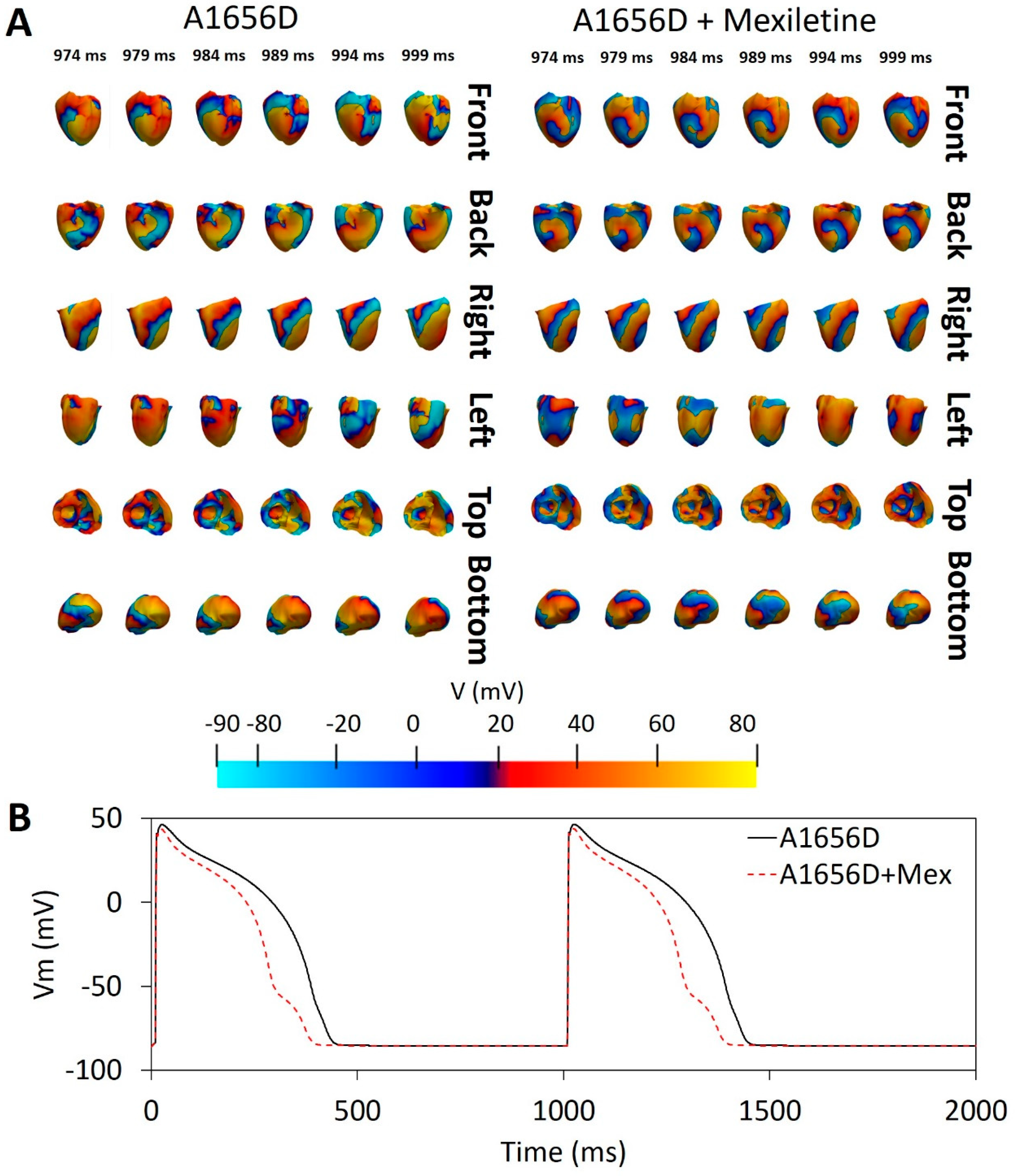
3. Results
4. Discussion
- Mexiletine could reduce APD.
- Mexiletine could shift the alternant occurrence in the cell from a normal to a quicker heart rate, offering extra safety standards during treatment.
- During reentry, mexiletine could reduce the possibility of a spiral wave breakup, which can contribute to ventricular fibrillation.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, E254–E743. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.I.; Chugh, S.S.; Dimarco, J.P.; Albert, C.M.; Anderson, M.E.; Bonow, R.O.; Buxton, A.E.; Chen, P.S.; Estes, M.; Jouven, X.; et al. Sudden Cardiac Death Prediction and Prevention Report From a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation 2010, 122, 2335. [Google Scholar] [CrossRef] [PubMed]
- Irawati, S.; Wasir, R.; Floriaan Schmidt, A.; Islam, A.; Feenstra, T.; Buskens, E.; Wilffert, B.; Hak, E. Long-Term Incidence and Risk Factors of Cardiovascular Events in Asian Populations: Systematic Review and Meta-Analysis of Population–Based Cohort Studies. Curr. Med. Res. Opin. 2018, 35, 291–299. [Google Scholar] [CrossRef]
- Tosaki, A. ArrhythmoGenoPharmacoTherapy. Front. Pharmacol. 2020, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Elias, A.; Benito, B. Ion Channel Disorders and Sudden Cardiac Death. Int. J. Mol. Sci. 2018, 19, 692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A Mutations Associated with an Inherited Cardiac Arrhythmia, Long QT Syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Giudicessi, J.R.; Ackerman, M.J. Genotype- and Phenotype-Guided Management of Congenital Long QT Syndrome. Curr. Probl. Cardiol. 2013, 38, 417–455. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Y.; Liu, N.; Priori, S.G. Sodium Channel Mutations and Arrhythmias. Nat. Rev. Cardiol. 2009, 6, 337–348. [Google Scholar] [CrossRef]
- Bennett, P.B.; Yazawa, K.; Makita, N.; George, A.L. Molecular Mechanism for an Inherited Cardiac Arrhythmia. Nature 1995, 376, 683–685. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Locati, E.H.; Napolitano, C.; Cantù, F.; Towbin, J.A.; Keating, M.T.; Hammoude, H.; Brown, A.M.; Chen, L.-S.K.; et al. Long QT Syndrome Patients With Mutations of the SCN5A and HERG Genes Have Differential Responses to Na+ Channel Blockade and to Increases in Heart Rate. Circulation 1995, 92, 3381–3386. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-Phenotype Correlation in the Long-QT Syndrome. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stramba-Badiale, M.; Priori, S.G.; Napolitano, C.; Locati, E.H.; Viñolas, X.; Haverkamp, W.; Schulze-Bahr, E.; Goulene, K.; Schwartz, P.J. Gene-Specific Differences in the Circadian Variation of Ventricular Repolarization in the Long QT Syndrome: A Key to Sudden Death during Sleep? Ital. Heart J. 2000, 1, 323–328. [Google Scholar] [PubMed]
- Jeyaraj, D.; Haldar, S.M.; Wan, X.; McCauley, M.D.; Ripperger, J.A.; Hu, K.; Lu, Y.; Eapen, B.L.; Sharma, N.; Ficker, E.; et al. Circadian Rhythms Govern Cardiac Repolarization and Arrhythmogenesis. Nature 2012, 483, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Kim, B.G.; Park, J.E.; Ki, C.S.; Huh, J.; Youm, J.B.; Kang, J.S.; Cho, H. Characterization of a Novel LQT3 Variant with a Selective Efficacy of Mexiletine Treatment. Sci. Rep. 2019, 9, 12997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Woltz, R.L.; Wang, C.Y.; Ren, L.; He, P.X.; Yu, S.D.; Liu, X.Q.; Yarov-Yarovoy, V.; Hu, D.; Chiamvimonvat, N.; et al. Gating Properties of Mutant Sodium Channels and Responses to Sodium Current Inhibitors Predict Mexiletine-Sensitive Mutations of Long QT Syndrome 3. Front. Pharmacol. 2020, 11, 1182. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xue, X.; Hu, D.; Liu, W.; Yuan, Y.; Sun, H.; Li, L.; Timothy, K.W.; Zhang, L.; Li, C.; et al. Inhibition of Late Sodium Current by Mexiletine: A Novel Pharmotherapeutical Approach in Timothy Syndrome. Circ. Arrhythm. Electrophysiol. 2013, 6, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Yang, Z.; Robinson, V.M.; Li, J.; Gao, C.; Guo, D.; Kowey, P.R.; Yan, G.X. Heterogeneous Distribution of INa-L Determines Interregional Differences in Rate Adaptation of Repolarization. Heart Rhythm. 2015, 12, 1295–1303. [Google Scholar] [CrossRef]
- Badri, M.; Patel, A.; Patel, C.; Liu, G.; Goldstein, M.; Robinson, V.M.; Xue, X.; Yang, L.; Kowey, P.R.; Yan, G.X. Mexiletine Prevents Recurrent Torsades de Pointes in Acquired Long QT Syndrome Refractory to Conventional Measures. JACC Clin. Electrophysiol. 2015, 1, 315–322. [Google Scholar] [CrossRef]
- Wu, L.; Ma, J.; Li, H.; Wang, C.; Grandi, E.; Zhang, P.; Luo, A.; Bers, D.M.; Shryock, J.C.; Belardinelli, L. Late Sodium Current Contributes to the Reverse Rate-Dependent Effect of IKr Inhibition on Ventricular Repolarization. Circulation 2011, 123, 1713. [Google Scholar] [CrossRef] [Green Version]
- Benditt, D.; Woodrow Benson, D.; Abbott, J.A.; Herre, J.M.; Scheinman, M.M. Permanent Cardiac Pacing in Patients with the Long QT Syndrome. J. Am. Coll. Cardiol. 1987, 10, 600–607. [Google Scholar] [CrossRef]
- Dorostkar, P.C.; Eldar, M.; Belhassen, B.; Scheinman, M.M. Long-Term Follow-Up of Patients With Long-QT Syndrome Treated With β-Blockers and Continuous Pacing. Circulation 1999, 100, 2431–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Y.; Liu, N.; Bloise, R.; Napolitano, C.; Priori, S.G. Gating Properties of SCN5A Mutations and the Response to Mexiletine in Long-QT Syndrome Type 3 Patients. Circulation 2007, 116, 1137–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzanti, A.; Maragna, R.; Faragli, A.; Monteforte, N.; Bloise, R.; Memmi, M.; Novelli, V.; Baiardi, P.; Bagnardi, V.; Etheridge, S.P.; et al. Gene-Specific Therapy With Mexiletine Reduces Arrhythmic Events in Patients With Long QT Syndrome Type 3. J. Am. Coll. Cardiol. 2016, 67, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Liang, J.A.; Franceschi, W.H.; Huang, Q.; Pashakhanloo, F.; Sung, E.; Boyle, P.M.; Trayanova, N.A. Assessment of Arrhythmia Mechanism and Burden of the Infarcted Ventricles Following Remuscularization with Pluripotent Stem Cell-Derived Cardiomyocyte Patches Using Patient-Derived Models. Cardiovasc. Res. 2022, 118, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Paci, M.; Passini, E.; Severi, S.; Hyttinen, J.; Rodriguez, B. Phenotypic Variability in LQT3 Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes and Their Response to Antiarrhythmic Pharmacologic Therapy: An in Silico Approach. Heart Rhythm. 2017, 14, 1704–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, M.; Ruan, Y.; Pandit, S.V.; Shah, K.; Berenfeld, O.; Blaufox, A.; Cerrone, M.; Noujaim, S.F.; Denegri, M.; Jalife, J.; et al. KCNJ2 Mutation in Short QT Syndrome 3 Results in Atrial Fibrillation and Ventricular Proarrhythmia. Proc. Natl. Acad. Sci. USA 2013, 110, 4291–4296. [Google Scholar] [CrossRef] [Green Version]
- Qu, Z.; Garfinkel, A.; Chen, P.S.; Weiss, J.N. Mechanisms of Discordant Alternans and Induction of Reentry in Simulated Cardiac Tissue. Circulation 2000, 102, 1664–1670. [Google Scholar] [CrossRef] [Green Version]
- ten Tusscher, K.H.W.J.; Noble, D.; Noble, P.J.; Panfilov, A.V. A Model for Human Ventricular Tissue. Am. J. Physiol. -Heart Circ. Physiol. 2004, 286, H1573–H1589. [Google Scholar] [CrossRef]
- Hodgkin, A.L.; Huxley, A.F. A Quantitative Description of Membrane Current and Its Application to Conduction and Excitation in Nerve. J. Physiol. 1952, 117, 500. [Google Scholar] [CrossRef]
- Krogh-Madsen, T.; Christini, D.J. Action Potential Duration Dispersion and Alternans in Simulated Heterogeneous Cardiac Tissue with a Structural Barrier. Biophys. J. 2007, 92, 1138–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnemeier, H.; Wiegand, U.K.H.; Brandes, A.; Kluge, N.; Katus, H.A.; Richardt, G.; Potratz, J. Circadian Profile of Cardiac Autonomic Nervous Modulation in Healthy Subjects: J. Cardiovasc. Electrophysiol. 2003, 14, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, L. The Role of Mexiletine in the Management of Long QT Syndrome. J. Electrocardiol. 2018, 51, 1061–1065. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Chen, P.S.; Qu, Z.; Karagueuzian, H.S.; Garfinkel, A. Ventricular Fibrillation. Circ. Res. 2000, 87, 1103–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalife, J. Ventricular Fibrillation: Mechanisms of Initiation and Maintenance. Annu. Rev. Physiol. 2003, 62, 25–50. [Google Scholar] [CrossRef]
- Janusek, D.; Svehlikova, J.; Zelinka, J.; Weigl, W.; Zaczek, R.; Opolski, G.; Tysler, M.; Maniewski, R. The Roles of Mid-Myocardial and Epicardial Cells in T-Wave Alternans Development: A Simulation Study. Biomed. Eng. Online 2018, 17, 57. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, R.J.; Picton, P.; Nanthakumar, K.; Mak, S.; Chauhan, V.S. Endocardial and Epicardial Repolarization Alternans in Human Cardiomyopathy: Evidence for Spatiotemporal Heterogeneity and Correlation With Body Surface T-Wave Alternans. J. Am. Coll. Cardiol. 2007, 49, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Aras, K.; Gams, A.; Faye, N.R.; Brennan, J.; Goldrick, K.; Li, J.; Zhong, Y.; Chiang, C.-H.; Smith, E.H.; Poston, M.D.; et al. Electrophysiology and Arrhythmogenesis in the Human Right Ventricular Outflow Tract. Circ. Arrhythm. Electrophysiol. 2022, 15, e010630. [Google Scholar] [CrossRef]
- Itzhaki, I.; Rapoport, S.; Huber, I.; Mizrahi, I.; Zwi-Dantsis, L.; Arbel, G.; Schiller, J.; Gepstein, L. Calcium Handling in Human Induced Pluripotent Stem Cell Derived Cardiomyocytes. PLoS ONE 2011, 6, e18037. [Google Scholar] [CrossRef]
- Law, M.L.; Cohen, H.; Martin, A.A.; Angulski, A.B.B.; Metzger, J.M. Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies. J. Clin. Med. 2020, 9, 520. [Google Scholar] [CrossRef]
- Zhang, J.; Chou, O.H.-I.; Tse, Y.-L.; Ng, K.-M.; Tse, H.-F. Application of Patient-Specific IPSCs for Modelling and Treatment of X-Linked Cardiomyopathies. Int. J. Mol. Sci. 2021, 22, 8132. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lei, W.; Yang, J.; Ni, X.; Ye, L.; Shen, Z.; Hu, S. The Updated View on Induced Pluripotent Stem Cells for Cardiovascular Precision Medicine. Pflugers Arch. 2021, 473, 1137–1149. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qauli, A.I.; Yoo, Y.; Marcellinus, A.; Lim, K.M. Verification of the Efficacy of Mexiletine Treatment for the A1656D Mutation on Downgrading Reentrant Tachycardia Using a 3D Cardiac Electrophysiological Model. Bioengineering 2022, 9, 531. https://doi.org/10.3390/bioengineering9100531
Qauli AI, Yoo Y, Marcellinus A, Lim KM. Verification of the Efficacy of Mexiletine Treatment for the A1656D Mutation on Downgrading Reentrant Tachycardia Using a 3D Cardiac Electrophysiological Model. Bioengineering. 2022; 9(10):531. https://doi.org/10.3390/bioengineering9100531
Chicago/Turabian StyleQauli, Ali Ikhsanul, Yedam Yoo, Aroli Marcellinus, and Ki Moo Lim. 2022. "Verification of the Efficacy of Mexiletine Treatment for the A1656D Mutation on Downgrading Reentrant Tachycardia Using a 3D Cardiac Electrophysiological Model" Bioengineering 9, no. 10: 531. https://doi.org/10.3390/bioengineering9100531