
Annotation of Potential Vaccine Targets and Designing of mRNA-Based Multi-Epitope Vaccine against Lumpy Skin Disease Virus via Reverse Vaccinology and Agent-Based Modeling
 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
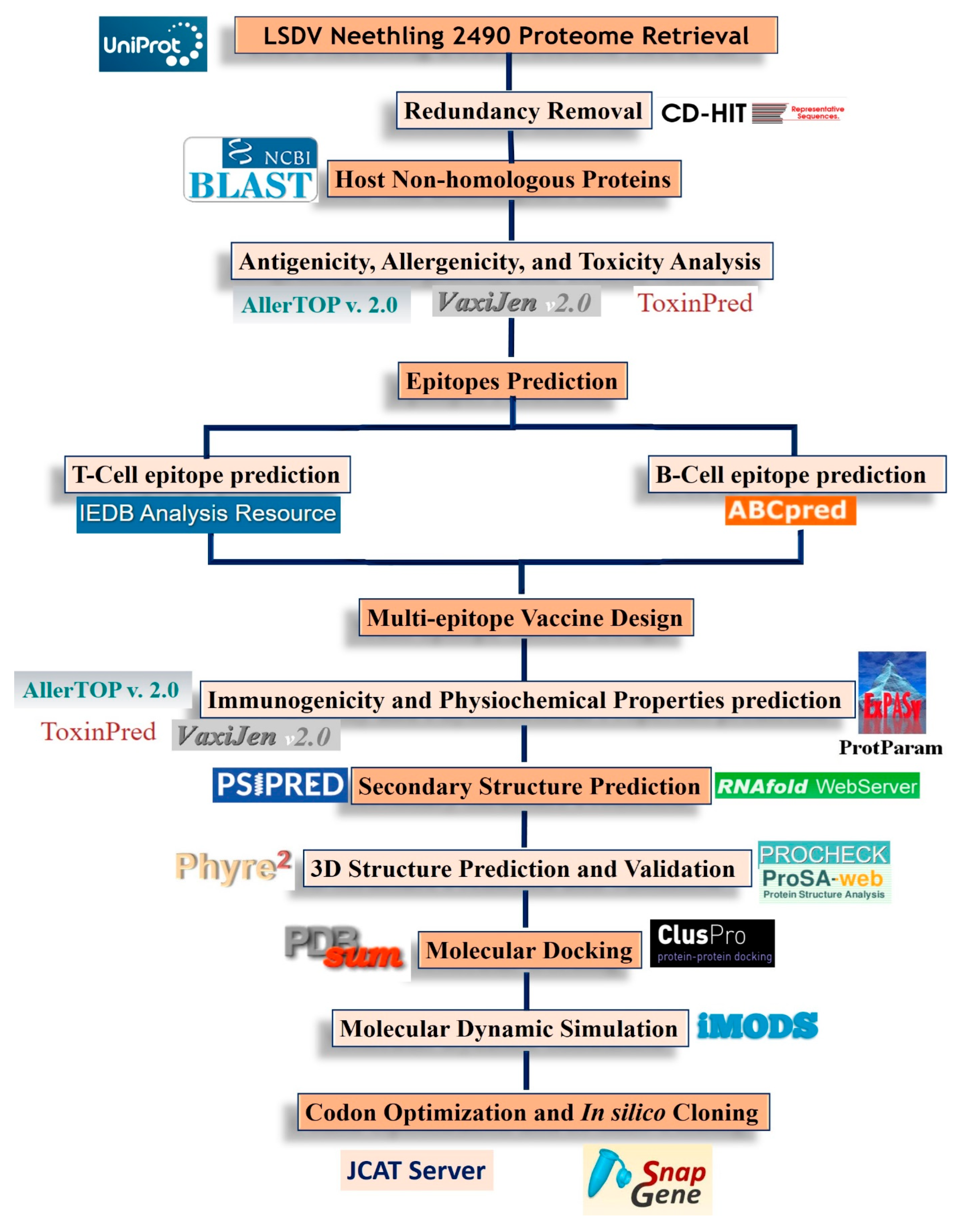
2. Materials and Methods
2.1. Protein Sequence Retrieval
2.2. Epitope Prediction
2.2.1. B-Cell Epitope Prediction
2.2.2. Cytotoxic T-lymphocyte (CTL) Cell Epitope Prediction
2.2.3. Helper T-lymphocyte (HTL) Epitope Prediction
2.3. Evaluation of Predicted Epitopes
2.4. Multi-Epitope Vaccine Construction
2.5. Immunological and Physiochemical Properties Prediction
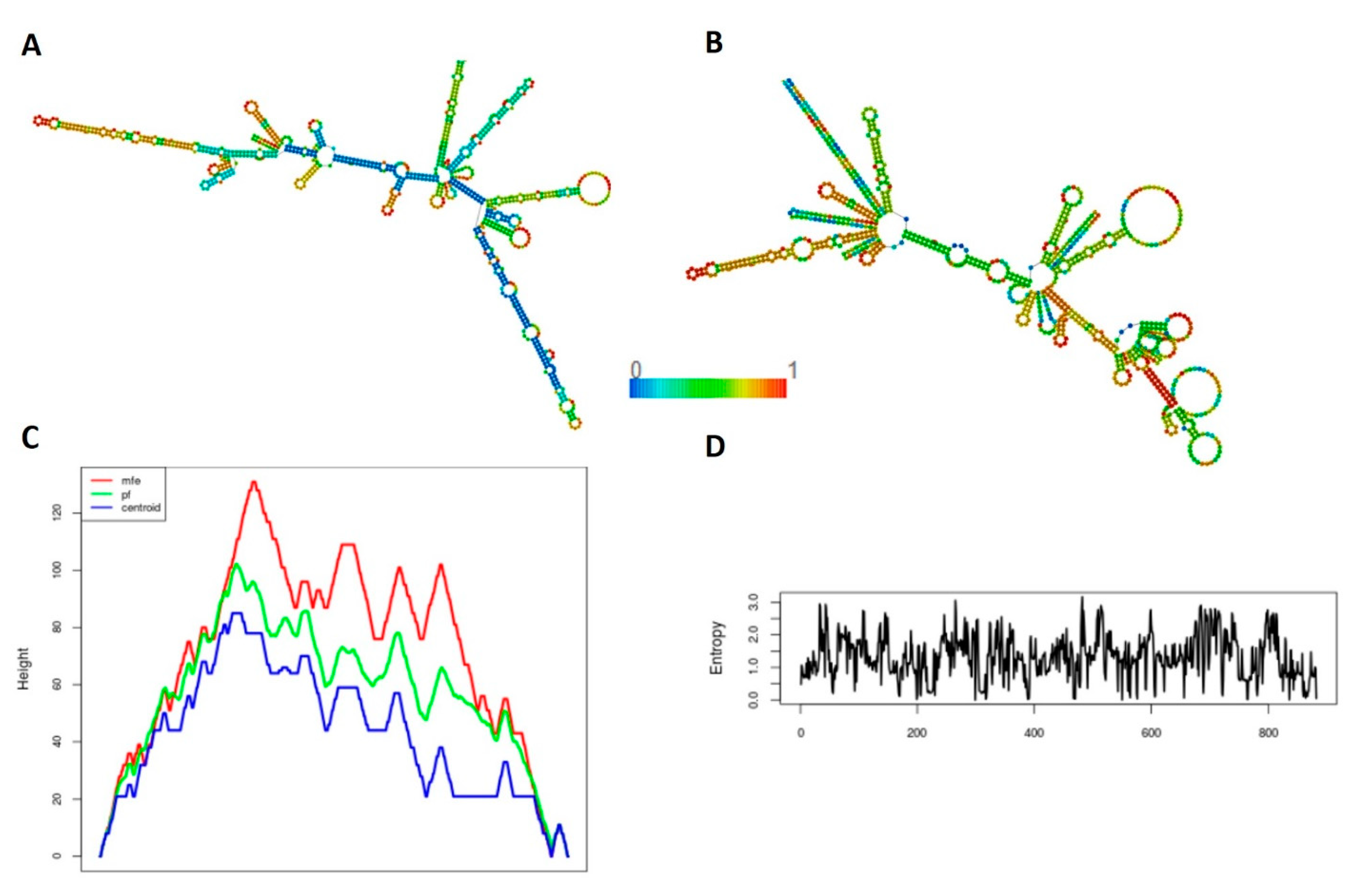
2.6. Secondary Structure Prediction
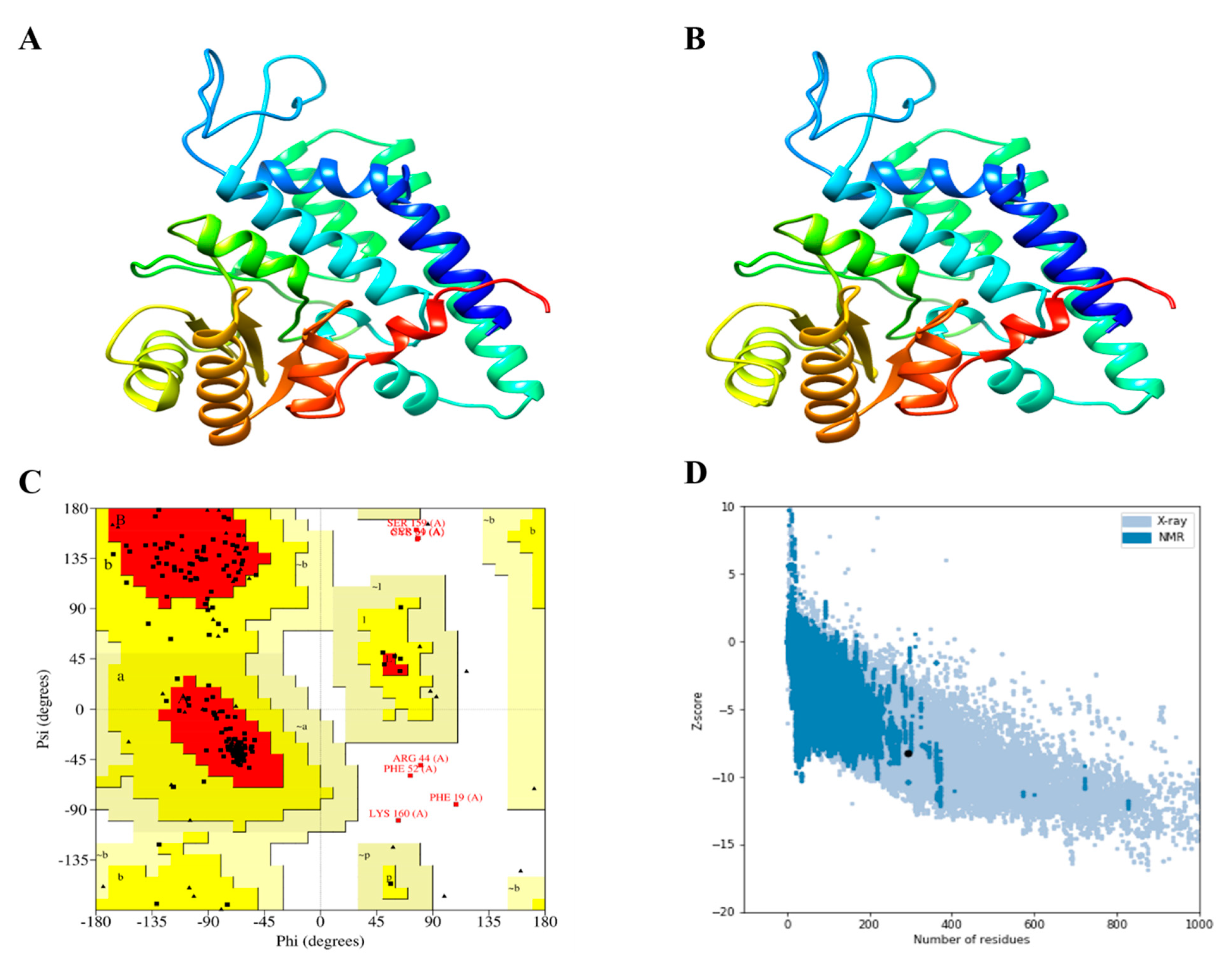
2.7. Tertiary Structure Modeling and Refinement
2.8. Molecular Docking
2.9. Molecular Dynamic Simulation
2.10. Codon Adaptation and In Silico Cloning
2.11. Immune Simulation
3. Results
3.1. LSDV Vaccine Candidate Proteins Prediction
3.2. B-Cell Epitopes Prediction
3.3. Prediction of CTL and HTL Epitopes
3.4. Chimeric Vaccine Construct
3.5. Physiochemical Parameters Prediction of Vaccine
3.6. Secondary Structure Prediction
3.7. D structure Evaluation, Refining, and Validation
3.8. Molecular Docking of Vaccine with TLRs
3.9. MD Simulation
3.10. Codon Adaptation and In Silico Cloning
3.11. Immune Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capstick, P.; Coackley, W. Protection of Cattle Against Lumpy Skin Disease: I.—Trials with a Vaccine Against Neethling Type Infection. Res. Vet. Sci. 1961, 2, 362–368. [Google Scholar] [CrossRef]
- Namazi, F.; Khodakaram Tafti, A. Lumpy skin disease, an emerging transboundary viral disease: A review. Vet. Med. Sci. 2021, 7, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.-J.; Tu, Y.-C.; Wu, C.-H.; Huang, C.-W.; Ting, L.-J.; Huang, Y.-L.; Pan, C.-H.; Chang, C.-Y.; Deng, M.-C.; Lee, F. First detection and phylogenetic analysis of lumpy skin disease virus from Kinmen Island, Taiwan in 2020. J. Vet. Med. Sci. 2022, 84, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, S.; El Idrissi, A.; Mattioli, R.; Tibbo, M.; Njeumi, F.; Raizman, E. Emergence of lumpy skin disease in the Eastern Mediterranean Basin countries. FAO Empres Watch 2013, 29, 1–6. [Google Scholar]
- Hasib, F.M.Y.; Islam, M.S.; Das, T.; Rana, E.A.; Uddin, M.H.; Bayzid, M.; Nath, C.; Hossain, M.A.; Masuduzzaman, M.; Das, S. Lumpy skin disease outbreak in cattle population of Chattogram, Bangladesh. Vet. Med. Sci. 2021, 7, 1616–1624. [Google Scholar] [CrossRef]
- Tuppurainen, E.; Dietze, K.; Wolff, J.; Bergmann, H.; Beltran-Alcrudo, D.; Fahrion, A.; Lamien, C.E.; Busch, F.; Sauter-Louis, C.; Conraths, F.J. Vaccines and Vaccination against Lumpy Skin Disease. Vaccines 2021, 9, 1136. [Google Scholar] [CrossRef]
- Kallerup, R.S.; Foged, C. Classification of vaccines. In Subunit Vaccine Delivery; Springer: Berlin/Heidelberg, Germany, 2015; pp. 15–29. [Google Scholar]
- Kanampalliwar, A.; Rajkumar, S.; Girdhar, A.; Archana, T. Reverse Vaccinology: Basics and Applications. J. Vaccines Vaccin 2013, 4, 194. [Google Scholar] [CrossRef] [Green Version]
- Flower, D.R.; Macdonald, I.K.; Ramakrishnan, K.; Davies, M.N.; Doytchinova, I.A. Computer aided selection of candidate vaccine antigens. Immunome Res. 2010, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, A.; Sharma, G.; Agrawal, P. Computational approaches for vaccine designing. In Bioinformatics; Elsevier: Amsterdam, The Netherlands, 2022; pp. 317–335. [Google Scholar]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CDHTT Suite: A web server for clustering and comparing biological sequences. Biomformahcs 2010, 26, 680–682. [Google Scholar]
- Aiman, S.; Alhamhoom, Y.; Ali, F.; Rahman, N.; Rastrelli, L.; Khan, A.; Ahmed, A.; Khan, A.; Li, C. Multi-epitope chimeric vaccine design against emerging Monkeypox virus via reverse vaccinology techniques-a bioinformatics and immunoinformatics approach. Front. Immunol. 2022, 13, 985450. [Google Scholar] [CrossRef] [PubMed]
- Aiman, S.; Ali, F.; Zia, A.; Aslam, M.; Han, Z.; Shams, S.; Khan, A.; Li, C. Core genome mediated potential vaccine targets prioritization against Clostridium difficile via reverse vaccinology-an immuno-informatics approach. J. Biol. Res. Thessalon. 2022, 29. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Justesen, S.; Lund, O.; Lundegaard, C.; Buus, S. NetMHCIIpan-2.0-Improved pan-specific HLA-DR predictions using a novel concurrent alignment and weight optimization training procedure. Immunome Res. 2010, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P. Peptide toxicity prediction. In Computational Peptidology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 143–157. [Google Scholar]
- Fadaka, A.O.; Sibuyi, N.R.S.; Martin, D.R.; Goboza, M.; Klein, A.; Madiehe, A.M.; Meyer, M. Immunoinformatics design of a novel epitope-based vaccine candidate against dengue virus. Sci. Rep. 2021, 11, 1–22. [Google Scholar] [CrossRef]
- Ahammad, I.; Lira, S.S. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. Int. J. Biol. Macromol. 2020, 162, 820–837. [Google Scholar] [CrossRef]
- Al Tbeishat, H. Novel In Silico mRNA vaccine design exploiting proteins of M. tuberculosis that modulates host immune responses by inducing epigenetic modifications. Sci. Rep. 2022, 12, 4645. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Türeci, O.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gergen, J.; Petsch, B. mRNA-Based Vaccines and Mode of Action. Curr. Top. Microbiol. Immunol. 2021, 440. [Google Scholar] [CrossRef]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP—A server for in silico prediction of allergens. BMC Bioinform. 2013, 14 (Suppl. 6), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [Green Version]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein–Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Berlin/Heidelberg, Germany, 2005; pp. 571–607. [Google Scholar]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neuböck, R.; Hofacker, I.L. The Vienna RNA websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, R.K.; Lim, B.; Kim, D.-Y.; Kim, J.-M. Designing multi-epitope-based vaccine targeting surface immunogenic protein of Streptococcus agalactiae using immunoinformatics to control mastitis in dairy cattle. BMC Vet. Res. 2022, 18, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef] [PubMed]
- López-Blanco, J.R.; Aliaga, J.I.; Quintana-Ortí, E.S.; Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014, 42, W271–W276. [Google Scholar] [CrossRef]
- Grote, A.; Hiller, K.; Scheer, M.; Münch, R.; Nörtemann, B.; Hempel, D.C.; Jahn, D. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005, 33, W526–W531. [Google Scholar] [CrossRef]
- Fu, H.; Liang, Y.; Zhong, X.; Pan, Z.; Huang, L.; Zhang, H.; Xu, Y.; Zhou, W.; Liu, Z. Codon optimization with deep learning to enhance protein expression. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, W.; Guo, J.; Zhao, G.; Sun, S.; Yu, H.; Guo, Y.; Li, J.; Jin, X.; Du, L. In silico design of a DNA-based HIV-1 multi-epitope vaccine for Chinese populations. Hum. Vaccines Immunother. 2015, 11, 795–805. [Google Scholar] [CrossRef] [Green Version]
- Mehmood, A.; Kaushik, A.C.; Wei, D.Q. Prediction and validation of potent peptides against herpes simplex virus type 1 via immunoinformatic and systems biology approach. Chem. Biol. Drug Des. 2019, 94, 1868–1883. [Google Scholar] [CrossRef] [PubMed]
- Khoo, C.; Dahlan, R.; Desa, Z.M.; Syarina, P.; Salim, S.M.; Barker, Z.; Hassan, M.A.; Hassan, R.; Saeid, F.M. Molecular Detection of Lumpy Skin Disease Virus in Malaysia 2021. Int. J. Infect. Dis. 2022, 116, S64. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e1110. [Google Scholar] [CrossRef] [Green Version]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Kar, P.P.; Araveti, P.B.; Kuriakose, A.; Srivastava, A. Design of a multi-epitope protein as a subunit vaccine against lumpy skin disease using an immunoinformatics approach. Sci. Rep. 2022, 12, 19411. [Google Scholar] [CrossRef]
- Shahab, M.; Alzahrani, A.K.; Duan, X.; Aslam, M.; Abida; Imran, M.; Kamal, M.; Alam, M.T.; Zheng, G. An Immunoinformatics Approach to Design Novel and Potent Multi-Epitope-Based Vaccine to Target Lumpy Skin Disease. Biomedicines 2023, 11, 398. [Google Scholar] [CrossRef]
- Li, M.; Zhu, Y.; Niu, C.; Xie, X.; Haimiti, G.; Guo, W.; Yu, M.; Chen, Z.; Ding, J.; Zhang, F. Design of a multi-epitope vaccine candidate against Brucella melitensis. Sci. Rep. 2022, 12, 1–18. [Google Scholar] [CrossRef]
- Li, J.; Qiu, J.; Huang, Z.; Liu, T.; Pan, J.; Zhang, Q.; Liu, Q. Reverse vaccinology approach for the identifications of potential vaccine candidates against Salmonella. Int. J. Med. Microbiol. 2021, 311, 151508. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhu, Y.; Li, Y.; Chen, Z.; Li, Z.; Wang, J.; Li, Z.; Zhang, F.; Ding, J. Design of a recombinant multivalent epitope vaccine based on SARS-CoV-2 and its variants in Immunoinformatics approach. Front. Immunol. 2022, 13, 2116. [Google Scholar]
- Bacchetta, R.; Gregori, S.; Roncarolo, M.-G. CD4+ regulatory T cells: Mechanisms of induction and effector function. Autoimmun. Rev. 2005, 4, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Arpin, C.; Dechanet, J.; Van Kooten, C.; Merville, P.; Grouard, G.; Briere, F.; Banchereau, J.; Liu, Y.-J. Generation of memory B cells and plasma cells in vitro. Science 1995, 268, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
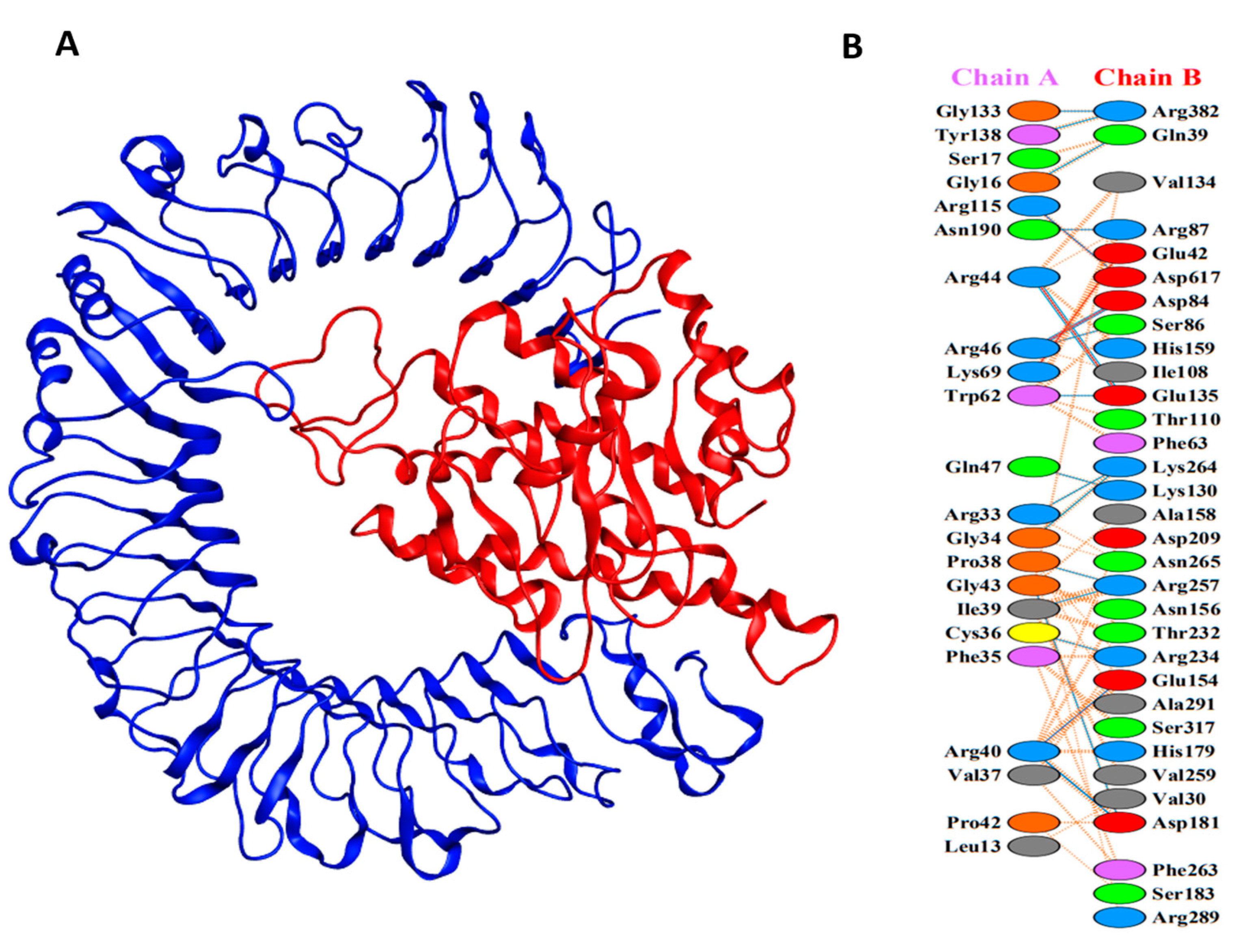{kind=link}
{kind=link}
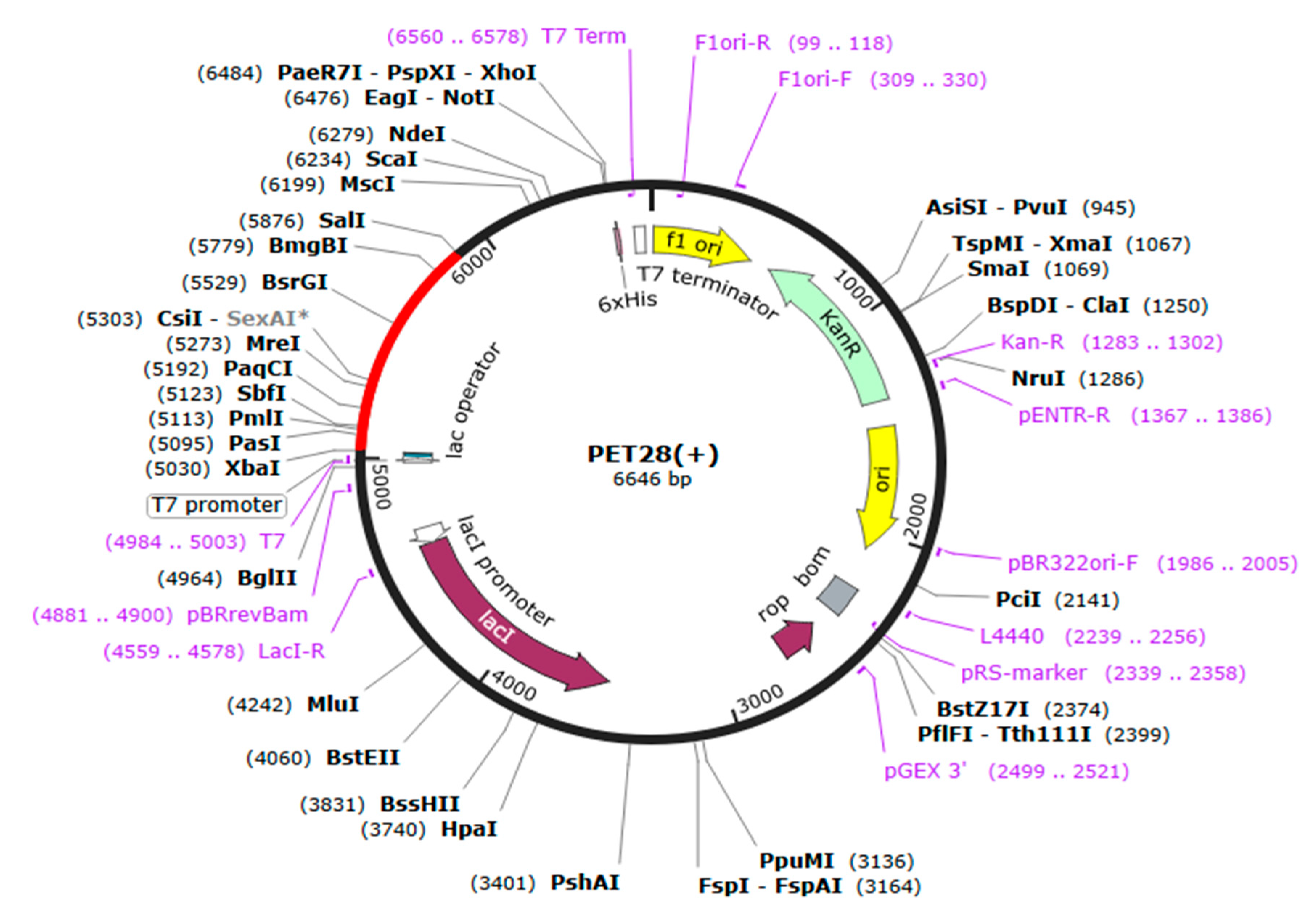{kind=link}
| S/No | Protein GenBank-IDs | Allergenicity AllerTOP2.0 | VaxiJen 2.0 > 0.4 Threshold | ToxinPred |
|---|---|---|---|---|
| 1 | NP_150447.1 | Non-allergen | 0.473 | Non-toxin |
| 2 | NP_150557.1 | Non-allergen | 0.596 | Non-toxin |
| 3 | AAK84969.1 | Non-allergen | 0.473 | Non-toxin |
| Protein IDs | B-Cell Epitopes | ABCPred Score | Antigenicity Score | Allergenicity AllerTop2.0 | ToxinPred |
|---|---|---|---|---|---|
| NP_150447.1 | EGVYLCSITTDTRCNP | 0.93 | 0.6686 | Non-allergen | Non-toxin |
| NSVIGTNYELLCINTK | 0.83 | 1.5331 | Non-allergen | Non-toxin | |
| SITTDTRCNPKNLALK | 0.82 | 1.1784 | Non-allergen | Non-toxin | |
| NP_150557.1 | YGLVKKKNNIWVDVNS | 0.80 | 0.8027 | Non-allergen | Non-toxin |
| LSNIKKSSKGDINACY | 0.70 | 0.5839 | Non-allergen | Non-toxin | |
| SCNYVSYIICVKRLYN | 0.68 | 0.6507 | Non-allergen | Non-toxin | |
| AAK84969.1 | YTTQQYCNVSPFINDN | 0.89 | 0.5361 | Non-allergen | Non-toxin |
| KGCIVEFGSQEKVCVT | 0.82 | 0.5036 | Non-allergen | Non-toxin | |
| SFPKDIKLTSNDFNSN | 0.74 | 0.6487 | Non-allergen | Non-toxin |
| Protein IDs | MHC1-T-Cell Epitopes | IC50 Value | Antigenicity Score | MHCII Epitopes | IC50 Value | Antigenicity Score | Allergenicity AllerTOP2.0 | ToxinPred |
|---|---|---|---|---|---|---|---|---|
| NP_150447 | NVLDYDRSK | 1.097615 | 0.4097 | ALIIKEVKRKYL | 8.85 | 0.5982 | Non-allergen | Non-toxin |
| NSTIALGKN | 2.639064 | 0.6360 | LIIKEVKRKYLS | 9.05 | 0.5862 | Non-allergen | Non-toxin | |
| TVNFLNSTI | 3.753617 | 0.7420 | IALIIKEVKRKY | 12.95 | 0.9813 | Non-allergen | Non-toxin | |
| NP_1505570 | AIFMLVSTI | 3.269 | 0.4443 | NVSIRHLKVISL | 39.5 | 1.5742 | Non-allergen | Non-toxin |
| NVSCNYVSY | 3.345 | 1.4980 | VSYIICVKRLYN | 31.55 | 0.5581 | Non-allergen | Non-toxin | |
| NYVSYIICV | 3.867 | 0.6540 | SIRHLKVISLTY | 36.61 | 1.5129 | Non-allergen | Non-toxin | |
| AAK84969.1 | DKKGCIVEF | 2.357491 | 0.6565 | KTDLSLLKRRIQ | 28.47 | 0.9108 | Non-allergen | Non-toxin |
| DFWIKFISI | 2.84741 | 0.9557 | DLSLLKRRIQKV | 28.48 | 0.5208 | Non-allergen | Non-toxin | |
| NTDDFWIKF | 2.988478 | 0.7614 | VFIKRQDVNTVL | 45.84 | 0.5375 | Non-allergen | Non-toxin |
| Vaccine Constructs | No. of Amino Acids | Molecular Weight | Theoretical PI | Aliphatic Index | Grand Average of Hydropathicity | Instability of Index | GC Content | CAI |
|---|---|---|---|---|---|---|---|---|
| LSDV-V1 | 294 | 30.96 | 9.67 | 77.11 | −0.200 | 20.68 Stable | 51.36 | 1.0 |
| LSDV-V2 | 294 | 30.75 | 9.68 | 78.78 | −0.120 | 17.85 Stable | 49.77 | 1.0 |
| LSDV-V3 | 294 | 30.98 | 9.64 | 79.08 | −0.061 | 15.60 Stable | 50.68 | 1.0 |
| Vaccine Constructs | α-Helix | Extended Strand | β-Turns | Random Coils |
|---|---|---|---|---|
| LSDV-V1 | 39.12% | 21.43% | 8.16% | 31.29% |
| LSDV-V2 | 41.16% | 23.13% | 8.84% | 26.87% |
| LSDV-V3 | 34.69% | 27.55% | 8.16% | 29.59% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakakhel, S.; Ahmad, A.; Mahdi, W.A.; Alshehri, S.; Aiman, S.; Begum, S.; Shams, S.; Kamal, M.; Imran, M.; Shakeel, F.; et al. Annotation of Potential Vaccine Targets and Designing of mRNA-Based Multi-Epitope Vaccine against Lumpy Skin Disease Virus via Reverse Vaccinology and Agent-Based Modeling. Bioengineering 2023, 10, 430. https://doi.org/10.3390/bioengineering10040430
Kakakhel S, Ahmad A, Mahdi WA, Alshehri S, Aiman S, Begum S, Shams S, Kamal M, Imran M, Shakeel F, et al. Annotation of Potential Vaccine Targets and Designing of mRNA-Based Multi-Epitope Vaccine against Lumpy Skin Disease Virus via Reverse Vaccinology and Agent-Based Modeling. Bioengineering. 2023; 10(4):430. https://doi.org/10.3390/bioengineering10040430
Chicago/Turabian StyleKakakhel, Sehrish, Abbas Ahmad, Wael A. Mahdi, Sultan Alshehri, Sara Aiman, Sara Begum, Sulaiman Shams, Mehnaz Kamal, Mohd. Imran, Faiyaz Shakeel, and et al. 2023. "Annotation of Potential Vaccine Targets and Designing of mRNA-Based Multi-Epitope Vaccine against Lumpy Skin Disease Virus via Reverse Vaccinology and Agent-Based Modeling" Bioengineering 10, no. 4: 430. https://doi.org/10.3390/bioengineering10040430