
Anti-Inflammatory and Anti-Thrombogenic Properties of Arterial Elastic Laminae
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Molecular Structure of Elastic Laminae
2.1. Elastin Gene
2.2. Tropoelastin
2.3. Elastic Fibers and Elastic Laminae
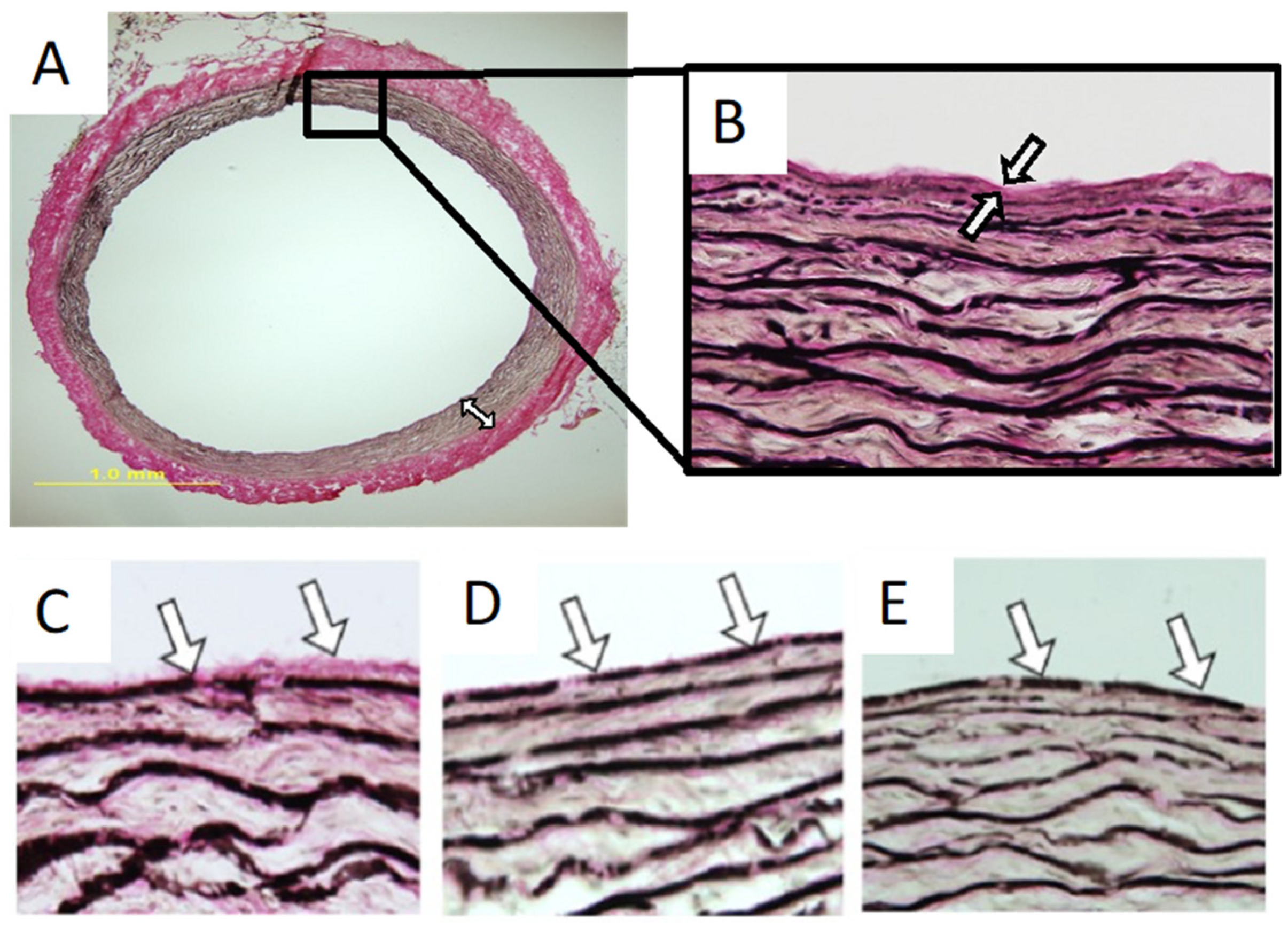
3. Mechanical Properties of Elastic Laminae
4. Fundamental Pathogenic Processes in Reconstructed Arteries
4.1. Inflammation
4.2. Thrombosis
4.3. Intimal Hyperplasia
5. Anti-Inflammatory and Anti-Thrombogenic Activities of Elastic Laminae
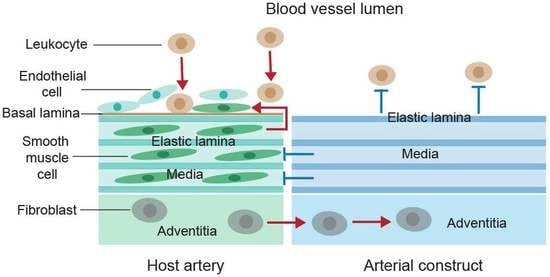
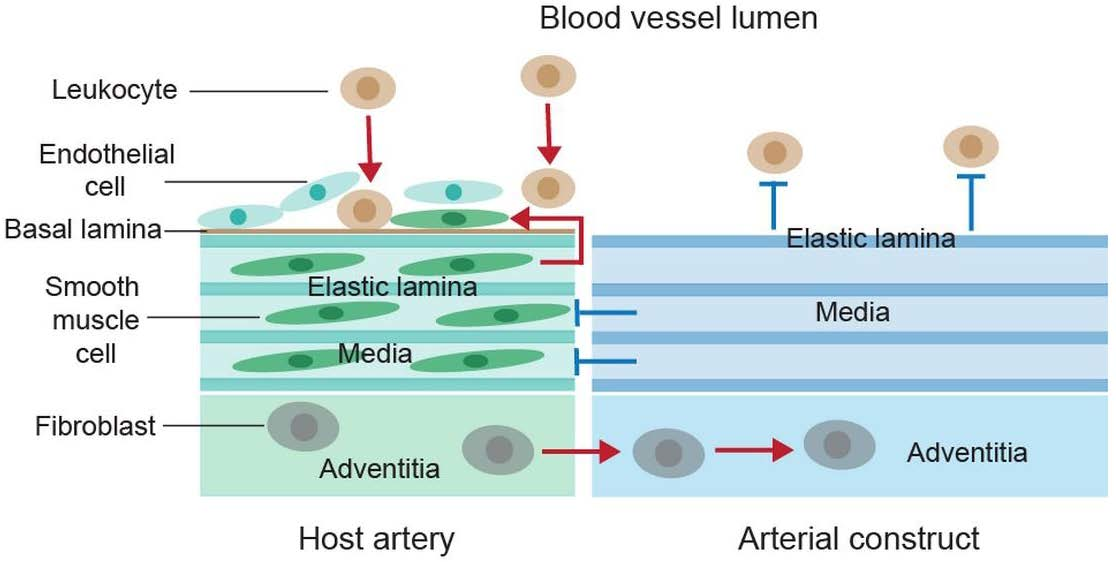
5.1. Elastic Laminae-Based Protection against Arterial Inflammation
5.2. Elastic Lamina-Mediated Prevention of Vascular Smooth Muscle Cell Proliferation and Neointima Formation
5.3. Elastin-Enhanced Actin Filament Generation in Vascular Smooth Muscle Cells
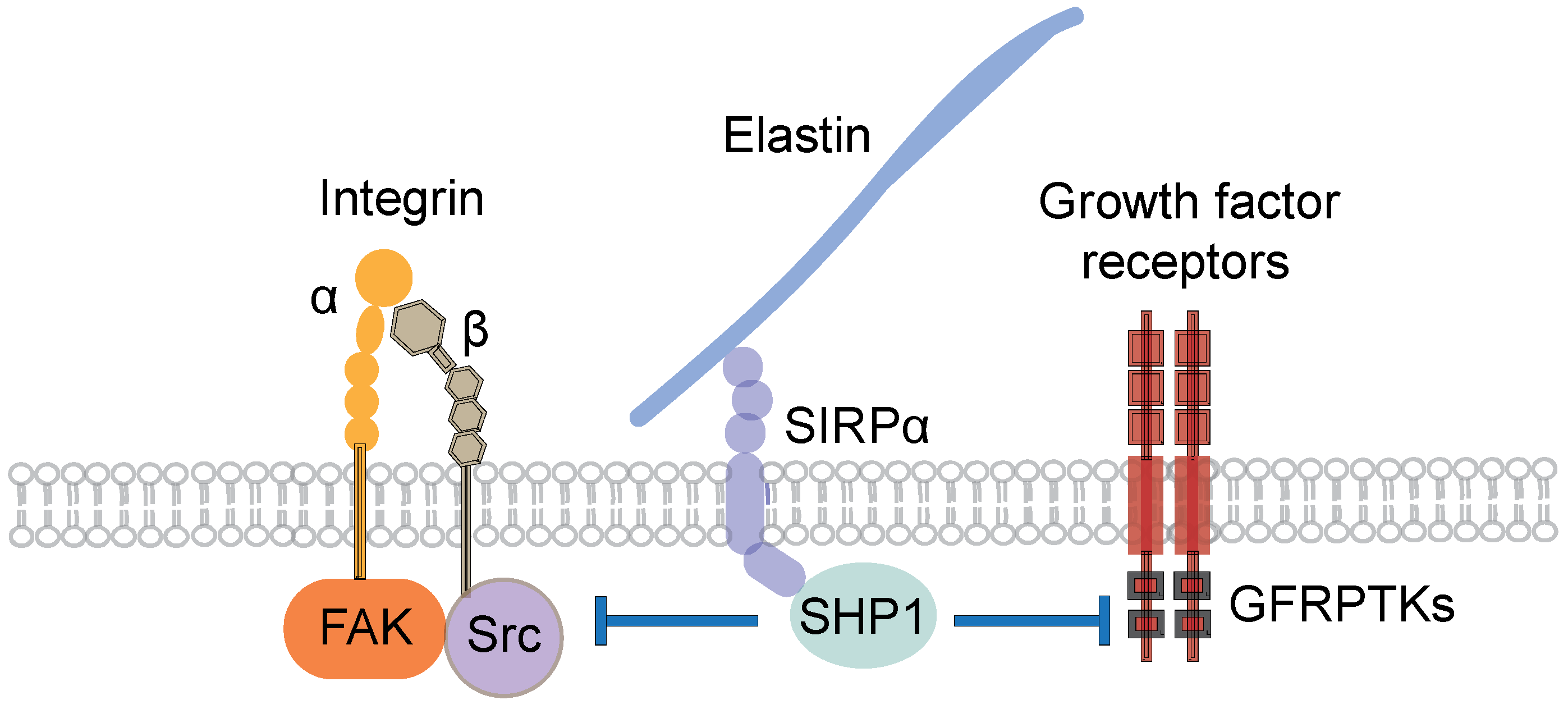
5.4. Mechanisms of the Inhibitory Action of Elastic Laminae
6. Application of Elastic Laminae and Elastin-Based Materials to Arterial Reconstruction
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, S.Q. Cardiovascular Engineering: A Protective Approach, 1st ed.; McGraw-Hill: New York, NY, USA, 2020. [Google Scholar]
- Galkina, E.; Ley, K. Immune and Inflammatory Mechanisms of Atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Robertson, A.-L.; Söderberg-Nauclér, C. Inflammation and atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 297–329. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995, 57, 791–804. [Google Scholar] [CrossRef] [PubMed]
- de Fougerolles, A.R.; Sprague, A.G.; Nickerson-Nutter, C.L.; Chi-Rosso, G.; Rennert, P.D.; Gardner, H.; Gotwals, P.J.; Lobb, R.R.; Koteliansky, V.E. Regulation of inflammation by collagen-binding integrins α1β1 and α2β1 in models of hypersensitivity and arthritis. J. Clin. Investig. 2000, 105, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldman, R.; Harvey, J.; Hogg, N. VLA-2 is the integrin used as a collagen receptor by leukocytes. Eur. J. Immunol. 1992, 22, 1109–1114. [Google Scholar] [CrossRef]
- Lahti, M.; Heino, J.; Käpylä, J. Leukocyte integrins αLβ2, αMβ2 and αXβ2 as collagen receptors—Receptor activation and recognition of GFOGER motif. Int. J. Biochem. Cell Biol. 2013, 45, 1204–1211. [Google Scholar] [CrossRef]
- Liu, S.Q.; Tieche, C.; Alkema, P.K. Neointima Formation on Elastic Lamina and Collagen Matrix Scaffolds Implanted in the Rat Aorta. Biomaterials 2004, 25, 1869–1882. [Google Scholar] [CrossRef]
- Liu, S.Q.; Alkema, P.K.; Tieche, C.; Tefft, B.J.; Liu, D.Z.; Sumpio, B.E.; Caprini, J.A.; Li, Y.C.; Paniagua, M. Negative Regulation of Monocyte Adhesion to Arterial Elastic Laminae by Signal-Regulatory Protein Alpha and SH2 Domain-Containing Protein Tyrosine Phosphatase-1. J. Biolog. Chem. 2005, 280, 39294–39301. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Q.; Tefft, B.J.; Zhang, A.; Zhang, L.-Q.; Wu, Y.H. Formation of smooth muscle α actin filaments in CD34-positive bone marrow cells in elastic lamina-dominantmatrix ofarteries. Matrix Biol. 2008, 27, 282–294. [Google Scholar] [CrossRef]
- Cocciolone, A.J.; Hawes, J.Z.; Staiculescu, M.C.; Johnson, E.O.; Murshed, M.; Wagenseil, J.E. Elastin, arterial mechanics, and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H189–H205. [Google Scholar] [CrossRef] [Green Version]
- Collins, R.T., 2nd. Cardiovascular disease in Williams syndrome. Circulation 2013, 127, 2125–2134. [Google Scholar] [PubMed] [Green Version]
- Karnik, S.K.; Brooke, B.S.; Bayes-Genis, A.; Sorensen, L.; Wythe, J.D.; Schwartz, R.S.; Keating, M.T.; Li, D.Y. A critical role for elastin signaling in vascular morphogenesis and disease. Development 2003, 130, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasio, M.L.D.; Kozel, B.A. Elastin-Driven Genetic Diseases. Matrix Biol. 2018, 71–72, 144–160. [Google Scholar] [CrossRef]
- Li, D.Y.; Brooke, B.; Davis, E.C.; Mecham, R.P.; Sorensen, L.K.; Boak, B.B.; Eichwald, E.; Keating, M.T. Elastin is an essential determinant of arterial morphogenesis. Nature 1998, 393, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Li, D.Y.; Faury, G.; Taylor, D.G.; Davis, E.C.; Boyle, W.A.; Mecham, R.P.; Stenzel, P.; Boak, B.; Keating, M.T. Novel arterial pathology in mice and humans hemizygous for elastin. J. Clin. Investig. 1998, 102, 1783–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-J.; Cocciolone, A.J.; Wagenseil, J.E. Elastin, arterial mechanics, and stenosis. Am. J. Physiol. Cell Physiol. 2022, 322, C875–C886. [Google Scholar] [CrossRef]
- Liu, S.Q. Bioregenerative Engineering: Principles and Applications; Wiley Interscience: Hoboken, NJ, USA, 2007. [Google Scholar]
- Pober, B.R.; Johnson, M.; Urban, Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J. Clin. Investig. 2008, 118, 1606–1615. [Google Scholar] [CrossRef]
- Tieche, C.; Alkema, P.K.; Liu, S.Q. Vascular elastic laminae: Anti-inflammatory properties and potential applications to arterial reconstruction. Front. Biosci. 2004, 9, 2205–2217. [Google Scholar] [CrossRef] [Green Version]
- Urban, Z.; Riazi, S.; Seidl, T.L.; Katahira, J.; Smoot, L.B.; Chitayat, D.; Boyd, C.D.; Hinek, A. Connection between elastin haploinsufficiency increased cell proliferation in patients with supravalvular aortic stenosis Williams-Beuren syndrome. Am. J. Hum. Genet. 2002, 71, 30–44. [Google Scholar] [CrossRef] [Green Version]
- Ascione, R.; Lloyd, C.T.; Underwood, M.J.; Lotto, A.A.; Pitsis, A.A.; Angelini, G.D. Inflammatory response after coronary revascularization with or without cardiopulmonary bypass. Ann. Thorac. Surg. 2000, 69, 1198–1204. [Google Scholar] [CrossRef]
- de Vries, M.R.; Quax, P.H.A. Inflammation in Vein Graft Disease. Front. Cardiovasc. Med. 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Astudillo, R.; Ivert, T.; Hjemdahl, P. Biphasic pro-thrombotic and inflammatory responses after coronary artery bypass surgery. J. Thromb. Haemost. 2003, 1, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Eaton, J.W. Inflammatory Responses to Biomaterials. Am. J. Clin. Pathol. 1995, 103, 466–471. [Google Scholar] [CrossRef]
- Ward, A.O.; Angelini, G.D.; Caputo, M.; Evans, P.C.; Johnson, J.L.; Suleiman, M.S.; Tulloh, R.M.; George, S.J.; Zakkar, M. NF-κB inhibition prevents acute shear stress-induced inflammation in the saphenous vein graft endothelium. Sci. Rep. 2020, 10, 15133. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Chen, C.; Bush, R.L.; Yao, Q.; Lumsden, A.; Hanson, S.R. Small-caliber heparin-coated ePTFE grafts reduce platelet deposition and neointimal hyperplasia in a baboon model. J. Vasc. Surg. 2004, 39, 1322–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Sales, K.M.; Hamilton, G.; Seifalian, A.M. Addressing thrombogenicity in vascular graft construction. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 82, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Tucker, E.I.; Marzec, U.M.; White, T.C.; Hurst, S.; Rugonyi, S.; McCarty, O.J.T.; Gailani, D.; Gruber, A.; Hanson, S.R. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood 2009, 113, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Cooley, B.C. Murine Model of Neointimal Formation and Stenosis in Vein Grafts. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.G.; Owens, E.L.; Mason, D.P.; Lea, H.; Tran, P.K.; Vergel, S.; Hawkins, S.A.; Hart, C.E.; Clowes, A.W. Effect of Platelet-Derived Growth Factor Receptor-α and -β Blockade on Flow-Induced Neointimal Formation in Endothelialized Baboon Vascular Grafts. Circ. Res. 2000, 86, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Q. Prevention of focal intimal hyperplasia in rat vein grafts by using a tissue engineering approach. Atherosclerosis 1998, 140, 365–377. [Google Scholar] [CrossRef]
- Liu, S.Q. Biomechanical basis of vascular tissue engineering. Crit. Rev. Biomed. Eng. 1999, 27, 75–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Moore, M.M.; Glucksberg, M.R.; Mockros, L.F.; Grotberg, J.B.; Mok, A.P. Partial prevention of monocyte and granulocyte activation in experimental vein grafts by using a biomechanical engineering approach. J. Biomech. 1999, 32, 1165–1175. [Google Scholar] [CrossRef]
- Liu, S.Q. Focal expression of angiotensin II type 1 receptor and smooth muscle cell proliferation in the neointima of experimental vein grafts: Relation to eddy blood flow. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2630–2639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.Q.; Moore, M.M.; Yap, C. Prevention of mechanical stretch-induced endothelial and smooth muscle cell injury in experimental vein grafts. J. Biomech. Eng. 2000, 122, 31–38. [Google Scholar] [CrossRef]
- Moore, M.M.; Goldman, J.; Patel, A.; Chien, S.; Liu, S.Q. Role of tensile stress and strain in the induction of cell death in experimental vein grafts. J. Biomech. 2001, 34, 289–297. [Google Scholar] [CrossRef]
- Xu, Q. Mouse Models of Arteriosclerosis: From Arterial Injuries to Vascular Grafts. Am. J. Pathol. 2004, 165, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, J.; Abrams, W.R.; Indik, Z.; Yeh, H.; Ornstein-Goldstein, N.; Bashir, M.M. Structure of the elastin gene. Ciba Found. Symp. 1995, 192, 59–74. [Google Scholar] [PubMed]
- Bashir, M.M.; Indik, Z.; Yeh, H.; Ornstein-Goldstein, N.; Rosenbloom, J.C.; Abrams, W.; Fazio, M.; Uitto, J.; Rosenbloom, J. Characterization of the complete human elastin gene. Delineation of unusual features in the 5′-flanking region. J. Biol. Chem. 1989, 264, 8887–8891. [Google Scholar] [CrossRef]
- Davis, E.C. Stability of elastin in the developing mouse aorta: A quantitative radioautographic study. Histochemistry 1993, 100, 17–26. [Google Scholar] [CrossRef]
- Lefevre, M.; Rucker, R.B. Aorta elastin turnover in normal and hypercholesterolemic Japanese quail. Biochim. Biophys. Acta 1980, 630, 519–529. [Google Scholar] [CrossRef]
- Keeley, F.W.; Hussain, R.A.; Johnson, D.J. Pattern of accumulation of elastin and the level of mRNA for elastin in aortic tissue of growing chickens. Arch. Biochem. Biophys. 1990, 282, 226–232. [Google Scholar] [CrossRef]
- Hew, Y.; Grzelczak, Z.; Lau, C.; Keeley, F.W. Identification of a large region of secondary structure in the 3’-untranslated region of chicken elastin mRNA with implications for the regulation of mRNA stability. J. Biol. Chem. 1999, 14, 14415–14421. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.J.; Robson, P.; Hew, Y.; Keeley, F.W. Decreased elastin synthesis in normal development and in long-term aortic organ and cell cultures is related to rapid and selective destabilization of mRNA for elastin. Circ. Res. 1995, 77, 1107–1113. [Google Scholar] [CrossRef]
- Indik, Z.; Yeh, H.; Ornstein-Goldstein, N.; Sheppard, P.; Anderson, N.; Rosenbloom, J.C.; Peltonen, L.; Rosenbloom, J. Alternative splicing of human elastin mRNA indicated by sequence analysis of cloned genomic complementary, DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 5680–5684. [Google Scholar] [CrossRef] [Green Version]
- Indik, Z.; Yeh, H.; Ornstein-Goldstein, N.; Kucich, U.; Abrams, W.; Rosenbloom, J.C.; Rosenbloom, J. Structure of the elastin gene and alternative splicing of elastin mRNA: Implications for human disease. Am. J. Med. Genet. 1989, 34, 81–90. [Google Scholar] [CrossRef]
- Miao, M.; Reichheld, S.E.; Muiznieks, L.D.; Sitarz, E.E.; Sharpe, S.; Keeley, F.W. Single nucleotide polymorphisms and domain/splice variants modulate assembly and elastomeric properties of human elastin. Implications for tissue specificity and durability of elastic tissue. Biopolymers 2017, 107, e23007. [Google Scholar] [CrossRef]
- Gray, W.R.; Sandberg, L.B.; Foster, J.A. Molecular model for elastin structure and function. Nature 1973, 246, 461–466. [Google Scholar] [CrossRef]
- Foster, J.A.; Bruenger, E.; Gray, W.R.; Sandberg, L.B. Isolation and amino acid sequences of tropoelastin peptides. J. Biol. Chem. 1973, 248, 2876–2879. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, L.B.; Weissman, N.; Gray, W.R. Structural features of tropoelastin related to the sites of cross-links in aortic elastin. Biochemistry 1971, 10, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.A.; Starcher, B.C.; Urry, D.W. Communication: Coacervation of tropoelastin results in fiber formation. J. Biol. Chem. 1974, 249, 997–998. [Google Scholar] [CrossRef]
- Vrhovski, B.; Weiss, A.S. Biochemistry of tropoelastin. Eur. J. Biochem. 1998, 258, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.C.; Keeley, F.W.; Weiss, A.S. Coacervation of tropoelastin. Adv. Colloid Interface Sci. 2011, 167, 94–103. [Google Scholar] [CrossRef]
- Lucero, H.A.; Kagan, H.M. Lysyl oxidase: An oxidative enzyme and effector of cell function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. [Google Scholar] [CrossRef] [PubMed]
- Baldock, C.; Oberhauser, A.F.; Ma, L.; Lammie, D.; Siegler, V.; Mithieux, S.M.; Tu, Y.; Chow, J.Y.H.; Suleman, F.; Malfois, M.; et al. Shape of tropoelastin, the highly extensible protein that controls human tissue elasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 4322–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenseil, J.E.; Mecham, R.P. New insights into elastic fiber assembly. Birth Defects Res. C Embryo Today 2007, 81, 229–240. [Google Scholar] [CrossRef]
- Ozsvar, J.; Yang, C.; Cain, S.A.; Baldock, C.; Tarakanova, A.; Weiss, A.S. Tropoelastin and Elastin Assembly. Front. Bioeng. Biotechnol. 2021, 9, 643110. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J. Clin. Investig. 1991, 87, 1828–1834. [Google Scholar] [CrossRef] [Green Version]
- Hungerford, J.E.; Owens, G.K.; Argraves, W.S.; Little, C.D. Development of the aortic vessel wall as defined by vascular smooth muscle and extracellular matrix markers. Dev. Biol. 1996, 178, 375–392. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, C.A.; Shadwick, R.E. Functional similarities in the mechanical design of the aorta in lower vertebrates and mammals. Experientia 1989, 45, 1083–1088. [Google Scholar] [CrossRef]
- Aaron, B.B.; Gosline, J.M. Elastin as a random-network elastomer: A mechanical and optical analysis of single elastin fibers. Biopolymers 1981, 20, 1247–1260. [Google Scholar] [CrossRef]
- Koenders, M.M.; Yang, L.; Wismans, R.G.; van der Werf, K.O.; Reinhardt, D.P.; Daamen, W.; Bennink, M.L.; Dijkstra, P.J.; van Kuppevelt, T.H.; Feijen, J. Microscale mechanical properties of single elastic fibers: The role of fibrillin-microfibrils. Biomaterials 2009, 30, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Lillie, M.A.; David, G.J.; Gosline, J.M. Mechanical role of elastin-associated microfibrils in pig aortic elastic tissue. Connect. Tissue Res. 1998, 37, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Sherebrin, M.H.; Song, S.H.; Roach, M.R. Mechanical anisotropy of purified elastin from the thoracic aorta of dog and sheep. Can. J. Physiol. Pharmacol. 1983, 61, 539–545. [Google Scholar] [CrossRef]
- Gautieri, A.; Vesentini, S.; Redaelli, A.; Buehler, M.J. Hierarchical structure and nanomechanics of collagen microfibrils from the atomistic scale up. Nano Lett. 2011, 11, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Heim, A.J.; Matthews, W.G.; Koob, T.J. Determination of the elastic modulus of native collagen fibrils via radial indentation. Appl. Phys. Lett. 2006, 89, 181902. [Google Scholar] [CrossRef]
- Farand, P.; Garon, A.; Plante, G.E. Structure of large arteries: Orientation of elastin in rabbit aortic internal elastic lamina and in the elastic lamellae of aortic media. Microvasc. Res. 2007, 73, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Lillie, M.A.; Gosline, J.M. Mechanical properties of elastin along the thoracic aorta in the pig. J. Biomech. 2007, 40, 2214–2221. [Google Scholar] [CrossRef]
- Yu, X.; Turcotte, R.; Seta, F.; Zhang, Y. Micromechanics of elastic lamellae: Unravelling the role of structural inhomogeneity in multi-scale arterial mechanics. J. R. Soc. Interface 2018, 15, 20180492. [Google Scholar] [CrossRef]
- Trachet, B.; Ferraro, M.; Lovric, G.; Aslanidou, L.; Logghe, G.; Segers, P.; Stergiopulos, N. Synchrotron-based visualization and segmentation of elastic lamellae in the mouse carotid artery during quasi-static pressure inflation. J. R. Soc. Interface 2019, 16, 20190179. [Google Scholar] [CrossRef] [Green Version]
- Kamenskiy, A.V.; Dzenis, Y.A.; Kazmi, S.; Pemberton, M.A.; Pipinos, I.I.; Phillips, N.Y.; Herber, K.; Woodford, T.; Bowen, R.E.; Lomneth, C.S.; et al. Biaxial mechanical properties of the human thoracic and abdominal aorta, common carotid, subclavian, renal and common iliac arteries. Biomech. Model. Mechanobiol. 2014, 13, 1341–1359. [Google Scholar] [CrossRef]
- Holzapfel, G.A.; Sommer, G.; Gasser, C.T.; Regitnig, P. Determination of layer-specific mechanical properties of human coronary arteries with nonatherosclerotic intimal thickening and related constitutive modeling. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2048–H2058. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Kassab, G.S. Microstructure-based biomechanics of coronary arteries in health and disease. J. Biomech. 2016, 49, 2548–2559. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.M.; Liao, W.S. Molecular analysis of the differential hepatic expression of rat kininogen family genes. Mol Cell Biol. 1993, 13, 6766–6777. [Google Scholar] [PubMed] [Green Version]
- Zhao, A.; Lew, J.-L.; Huang, L.; Yu, J.; Zhang, T.; Hrywna, Y.; Thompson, J.R.; de Pedro, N.; Blevins, R.A.; Peláez, F.; et al. Human Kininogen Gene Is Transactivated by the Farnesoid X Receptor. J. Biol. Chem. 2003, 278, 28765–28770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.P.; Joseph, K.; Silverberg, M. Molecular mechanisms in allergy and clinical immunology. Pathw. Bradykinin Form. Inflamm. Dis. 2002, 109, 195–209. [Google Scholar]
- Wong, M.K.S. Bradykinin. In Handbook of Hormones, Comparative Endocrinology for Basic and Clinical Research; Takei, Y., Ando, H., Tsutsui, K., Eds.; Academic Press: Tokyo, Japan, 2015. [Google Scholar]
- Borriello, F.; Iannone, R.; Marone, G. Histamine Release from Mast Cells and Basophils. Handb. Exp. Pharmacol. 2017, 241, 121–139. [Google Scholar] [PubMed]
- Moriguchi, T.; Takai, J. Histamine and histidine decarboxylase: Immunomodulatory functions and regulatory mechanisms. Genes. Cells 2020, 25, 443–449. [Google Scholar] [CrossRef]
- Commins, S.P.; Borish, L.; Steinke, J.W. Immunologic Messenger Molecules: Cytokines, Interferons, and Chemokines. J. Allergy Clin. Immunol. 2010, 125, S53–S72. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Tang, D.; Tieche, C.; Alkema, P. Pattern formation of vascular smooth muscle cells subject to non-uniform fluid shear stress: Mediation by cell density gradients. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1071–H1080. [Google Scholar]
- Davie, E.W.; Kulman, J.D. An overview of the structure and function of thrombin. Semin. Thromb. Hemost. 2006, 32 (Suppl. 1), 3–15. [Google Scholar] [CrossRef]
- Cohen, C.T.; Turner, N.A.; Moake, J.L. Human endothelial cells and fibroblasts express and produce the coagulation proteins necessary for thrombin generation. Sci. Rep. 2021, 11, 21852. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sim, M.M.S.; Wood, J.P. Recent Insights Into the Regulation of Coagulation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e119–e125. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M., Sr.; Brass, L.F.; Stalker, T.J. Regulation of platelet activation and coagulation and its role in vascular injury and arterial thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kattula, S.; Byrnes, J.R.; Wolberg, A.S. Fibrinogen and Fibrin in Hemostasis and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e13–e21. [Google Scholar] [CrossRef] [Green Version]
- Luyendyk, J.P.; Schoenecker, J.G.; Flick, M.J. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood 2019, 133, 511–520. [Google Scholar] [CrossRef]
- Groves, P.H.; Penny, W.J.; Cheadle, H.A.; Lewis, M.J. Exogenous nitric oxide inhibits in vivo platelet adhesion following balloon angioplasty Get access Arrow. Cardiovasc. Res. 1992, 26, 615–619. [Google Scholar] [CrossRef] [Green Version]
- Konishi, H.; Katoh, Y.; Takaya, N.; Kashiwakura, Y.; Itoh, S.; Ra, C.; Daida, H. Platelets Activated by Collagen Through Immunoreceptor Tyrosine-Based Activation Motif Play Pivotal Role in Initiation and Generation of Neointimal Hyperplasia after Vascular Injury. Circulation 2002, 105, 912–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.-H.; Irvine, S.; Tagalakis, A.D.; McAnulty, R.J.; McEwan, J.R.; Hart, S.L. Inhibition of neointimal hyperplasia in a rabbit vein graft model following non-viral transfection with human iNOS cDNA. Gene Ther. 2013, 20, 979–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torsney, E.; Mayr, U.; Zou, Y.; Thompson, W.D.; Hu, Y.; Xu, Q. Thrombosis and Neointima Formation in Vein Grafts Are Inhibited by Locally Applied Aspirin Through Endothelial Protection. Circ. Res. 2004, 94, 1466–1473. [Google Scholar] [CrossRef] [Green Version]
- Kohler, T.R.; Kirkman, T.R.; Kraiss, L.W.; Zierler, B.K.; Clowes, A.W. Increased blood flow inhibits neointimal hyperplasia in endothelialized vascular grafts. Circ. Res. 1991, 69, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Gayral, S.; Garnotel, R.; Castaing-Berthou, A.; Blaise, S.; Fougerat, A.; Berge, E.; Montheil, A.; Malet, N.; Wymann, M.P.; Maurice, P.; et al. Elastin-derived peptides potentiate atherosclerosis through the immune Neu1-PI3Kγ pathway. Cardiovasc. Res. 2014, 102, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Metter, E.J.; Earley, C.J.; Kemper, M.K.; Becker, L.C.; Lakatta, E.G.; Fleg, J.L. Increased Carotid Artery Intimal-Medial Thickness in Asymptomatic Older Subjects With Exercise-Induced Myocardial Ischemia. Circulation 1998, 98, 1504–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virmani, R.; Avolio, A.P.; Mergner, W.J.; Robinowitz, M.; Herderick, E.E.; Cornhill, J.F.; Guo, S.Y.; Liu, T.H.; Ou, D.Y.; O’Rourke, M. Effect of aging on aortic morphology in populations with high and low prevalence of hypertension and atherosclerosis. Comparison between occidental and Chinese communities. Am. J. Pathol. 1991, 139, 1119–1129. [Google Scholar] [PubMed]
- Karnik, S.K.; Wythe, J.D.; Sorensen, L.; Brooke, B.J.; Urness, L.D.; Li, D.Y. Elastin induces myofibrillogenesis via a specific domain, VGVAPG. Matrix Biol. 2003, 22, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Papageorgopoulou, C.P.; Nikolakopoulos, K.M.; Ntouvas, I.G.; Papadoulas, S. Chronic Limb Ischemia due to Thrombosis of an Aneurysmal Degeneration of the Autogenous Vein Graft: A Case Report. Aorta 2022, 10, 77–79. [Google Scholar] [CrossRef]
- Kulik, A. Commentary: Yin and Yang: Antiplatelet and lipid-lowering therapies to prevent vein graft thrombosis and atherosclerosis after coronary artery bypass graft surgery. J. Thorac. Cardiovasc. Surg. 2022, 163, 1042–1043. [Google Scholar] [CrossRef]
- Yeo, J.W.; Law, M.S.N.; Lim, J.C.L.; Ng, C.H.; Tan, D.J.H.; Tay, P.W.L.; Syn, N.; Tham, H.Y.; Huang, D.Q.; Siddiqui, M.S.; et al. Meta-analysis and systematic review: Prevalence, graft failure, mortality, and post-operative thrombosis in liver transplant recipients with pre-operative portal vein thrombosis. Clin. Transplant. 2022, 36, e14520. [Google Scholar] [CrossRef]
- Kato, T.; Fuke, M.; Nagai, F.; Nomi, H.; Kanzaki, Y.; Yui, H.; Maruyama, S.; Nagae, A.; Sakai, T.; Saigusa, T.; et al. Successful endovascular treatment with a stent graft for chronic deep vein thrombosis with multiple arteriovenous fistulas: A case report. J. Med. Case Rep. 2022, 16, 257. [Google Scholar] [CrossRef]
- Esenboga, K.; Baskovski, E.; Ates, B.N.; Ozyuncu, N.; Turhan, S.; Tutar, E. Thrombotic Complication of COVID-19: A Case Report of Acute Saphenous Vein Graft Thrombosis in a Newly Diagnosed Patient. Turk Kardiyol. Dern. Ars. 2022, 50, 228–230. [Google Scholar] [CrossRef]
- Tatterton, M.; Wilshaw, S.P.; Ingham, E.; Homer-Vanniasinkam, S. The use of antithrombotic therapies in reducing synthetic small-diameter vascular graft thrombosis. Vasc. Endovasc. Surg. 2012, 46, 212–222. [Google Scholar] [CrossRef]
- De Visscher, G.; Mesure, L.; Meuris, B.; Ivanova, A.; Flameng, W. Improved endothelialization and reduced thrombosis by coating a synthetic vascular graft with fibronectin and stem cell homing factor SDF-1alpha. Acta Biomater. 2012, 8, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Mallis, P.; Kostakis, A.; Stavropoulos-Giokas, C.; Michalopoulos, E. Future Perspectives in Small-Diameter Vascular Graft Engineering. Bioengineering 2020, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Radke, D.; Jia, W.; Sharma, D.; Fena, K.; Wang, G.; Goldman, J.; Zhao, F. Tissue Engineering at the Blood-Contacting Surface: A Review of Challenges and Strategies in Vascular Graft Development. Adv. Healthc. Mater. 2018, 7, e1701461. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Ellman, D.G.; Andersen, D.C. Review: Tissue Engineering of Small-Diameter Vascular Grafts and Their In Vivo Evaluation in Large Animals and Humans. Cells 2021, 10, 713. [Google Scholar] [CrossRef]
- Imashiro, C.; Shimizu, T. Fundamental Technologies and Recent Advances of Cell-Sheet-Based Tissue Engineering. Int. J. Mol. Sci. 2021, 22, 425. [Google Scholar] [CrossRef]
- McAllister, T.N.; Maruszewski, M.; Garrido, S.A.; Wystrychowski, W.; Dusserre, N.; Marini, A.; Zagalski, K.; Fiorillo, A.; Avila, H.; Manglano, X.; et al. Effectiveness of haemo-dialysis access with an autologous tissue-engineered vascular graft: A multicentre co-hort study. Lancet 2009, 373, 1440–1446. [Google Scholar] [CrossRef]
- Wystrychowski, W.; Garrido, S.A.; Marini, A.; Dusserre, N.; Radochonski, S.; Zagalski, K.; Antonelli, J.; Canalis, M.; Sammartino, A.; Darocha, Z.; et al. Long-term results of autologous scaffold-free tissue-engineered vascular graft for hemodialysis access. J. Vasc. Access. 2022. [Google Scholar] [CrossRef]
- Kristofik, N.J.; Qin, L.; Calabro, N.E.; Dimitrievska, S.; Li, G.; Tellides, G.; Niklason, L.E.; Kyriakides, T.R. Improving in vivo outcomes of decellularized vascular grafts via incorporation of a novel extracellular matrix. Biomaterials 2017, 141, 63–73. [Google Scholar] [CrossRef]
- Gui, L.; Muto, A.; Chan, S.A.; Breuer, C.K.; Niklason, L.E. Development of Decellularized Human Umbilical Arteries as Small-Diameter Vascular Grafts. Tissue Eng. Part A 2009, 15, 9. [Google Scholar] [CrossRef]
- Wolfe, J.T.; Shradhanjali, A.; Tefft, B.J. Strategies for Improving Endothelial Cell Adhesion to Blood-Contacting Medical Devices. Tissue Eng. Part. B Rev. 2022, 28, 1067–1092. [Google Scholar] [CrossRef]
- Cooley, B.C. Collagen-induced thrombosis in murine arteries and veins. Thromb. Res. 2013, 131, 49–54. [Google Scholar] [CrossRef]
- Sa, Q.; Hoover-Plow, J.L. EMILIN2 (Elastin microfibril interface located protein), potential modifier of thrombosis. Thromb. J. 2011, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawecki, C.; Hezard, N.; Bocquet, O.; Poitevin, G.; Rabenoelina, F.; Kauskot, A.; Duca, L.; Blaise, S.; Romier, B.; Martiny, L.; et al. Elastin-derived peptides are new regulators of thrombosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2570–2578. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Liu, L.; Mithieux, S.M.; Weiss, A.S. Fabricating Organized Elastin in Vascular Grafts. Trends Biotechnol 2021, 39, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Agarwal, R.; Tara, S.; Yi, T.; Lee, Y.U.; Breuer, C.K.; Weiss, A.S.; Shinoka, T. Tropoelastin inhibits intimal hyperplasia of mouse bioresorbable arterial vascular grafts. Acta Biomater. 2017, 52, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Ryan, A.J.; O’Brien, F.J. Insoluble elastin reduces collagen scaffold stiffness, improves viscoelastic properties, and induces a contractile phenotype in smooth muscle cells. Biomaterials 2015, 73, 296–307. [Google Scholar] [CrossRef]
- Jordan, S.W.; Haller, C.A.; Sallach, R.E.; Apkarian, R.P.; Hanson, S.R.; Chaikof, E.L. The effect of a recombinant elastin-mimetic coating of an ePTFE prosthesis on acute thrombogenicity in a baboon arteriovenous shunt. Biomaterials 2007, 28, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Koens, M.J.; Krasznai, A.G.; Hanssen, A.E.; Hendriks, T.; Praster, R.; Daamen, W.F.; van der Vliet, J.A.; van Kuppevelt, T.H. Vascular replacement using a layered elastin-collagen vascular graft in a porcine model: One week patency versus one month occlusion. Organogenesis 2015, 11, 105–121. [Google Scholar] [CrossRef] [Green Version]
- McKenna, K.A.; Hinds, M.T.; Sarao, R.C.; Wu, P.C.; Maslen, C.L.; Glanville, R.W.; Babcock, D.; Gregory, K.W. Mechanical property characterization of electrospun recombinant human tropoelastin for vascular graft biomaterials. Acta Biomater. 2012, 8, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Ibanez-Fonseca, A.; Flora, T.; Acosta, S.; Rodriguez-Cabello, J.C. Trends in the design and use of elastin-like recombinamers as biomaterials. Matrix Biol. 2019, 84, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Vassalli, M.; Sbrana, F.; Laurita, A.; Papi, M.; Bloise, N.; Visai, L.; Bochicchio, B. Biological and structural characterization of a naturally inspired material engineered from elastin as a candidate for tissue engineering applications. Langmuir 2013, 29, 15898–15906. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mithieux, S.M.; Vindin, H.; Wang, Y.; Zhang, M.; Liu, L.; Zbinden, J.; Blum, K.M.; Yi, T.; Matsuzaki, Y.; et al. Rapid Regeneration of a Neoartery with Elastic Lamellae. Adv. Mater. 2022, 34, e2205614. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.W.; Ahrens, D.C.; Joda, D.; Curtis, T.E.; Bowen, P.K.; Guillory, R.J., 2nd; Liu, S.Q.; Zhao, F.; Frost, M.C.; Goldman, J. Fabrication and Short-Term in Vivo Performance of a Natural Elastic Lamina-Polymeric Hybrid Vascular Graft. ACS Appl. Mater. Interfaces 2015, 7, 16202–16212. [Google Scholar] [CrossRef] [PubMed]
- Babapulle, M.N.; Eisenberg, M.J. Coated Stents for the Prevention of Restenosis: Part, I. Circulation 2002, 106, 2734–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nusca, A.; Viscusi, M.M.; Piccirillo, F.; De Filippis, A.; Nenna, A.; Spadaccio, C.; Nappi, F.; Chello, C.; Mangiacapra, F.; Grigioni, F.; et al. In Stent Neo-Atherosclerosis: Pathophysiology, Clinical Implications, Prevention, and Therapeutic Approaches. Life 2022, 12, 393. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldman, J.; Liu, S.Q.; Tefft, B.J. Anti-Inflammatory and Anti-Thrombogenic Properties of Arterial Elastic Laminae. Bioengineering 2023, 10, 424. https://doi.org/10.3390/bioengineering10040424
Goldman J, Liu SQ, Tefft BJ. Anti-Inflammatory and Anti-Thrombogenic Properties of Arterial Elastic Laminae. Bioengineering. 2023; 10(4):424. https://doi.org/10.3390/bioengineering10040424
Chicago/Turabian StyleGoldman, Jeremy, Shu Q. Liu, and Brandon J. Tefft. 2023. "Anti-Inflammatory and Anti-Thrombogenic Properties of Arterial Elastic Laminae" Bioengineering 10, no. 4: 424. https://doi.org/10.3390/bioengineering10040424