
The Mechanism Underlying the Amylose-Zein Complexation Process and the Stability of the Molecular Conformation of Amylose-Zein Complexes in Water Based on Molecular Dynamics Simulation
Abstract
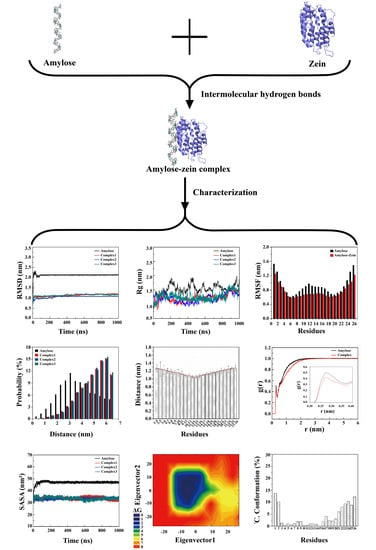
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Force Field
2.3. Molecular Dynamics Simulation Parameters
2.4. Statistical Analysis
3. Results and Discussion
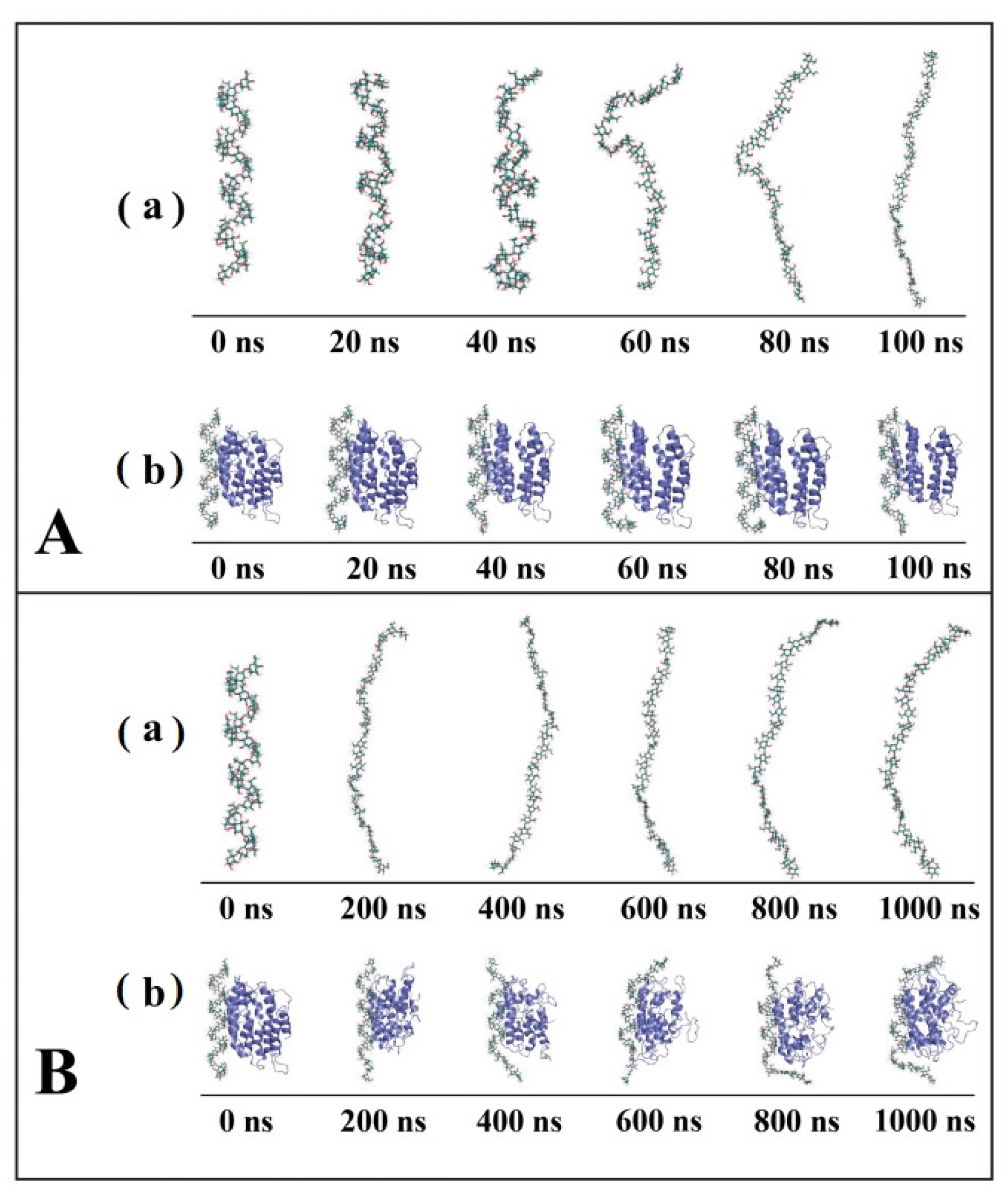
3.1. Conformational Transitions of the Amylose Molecule and Amylose–Zein Complexes in Water
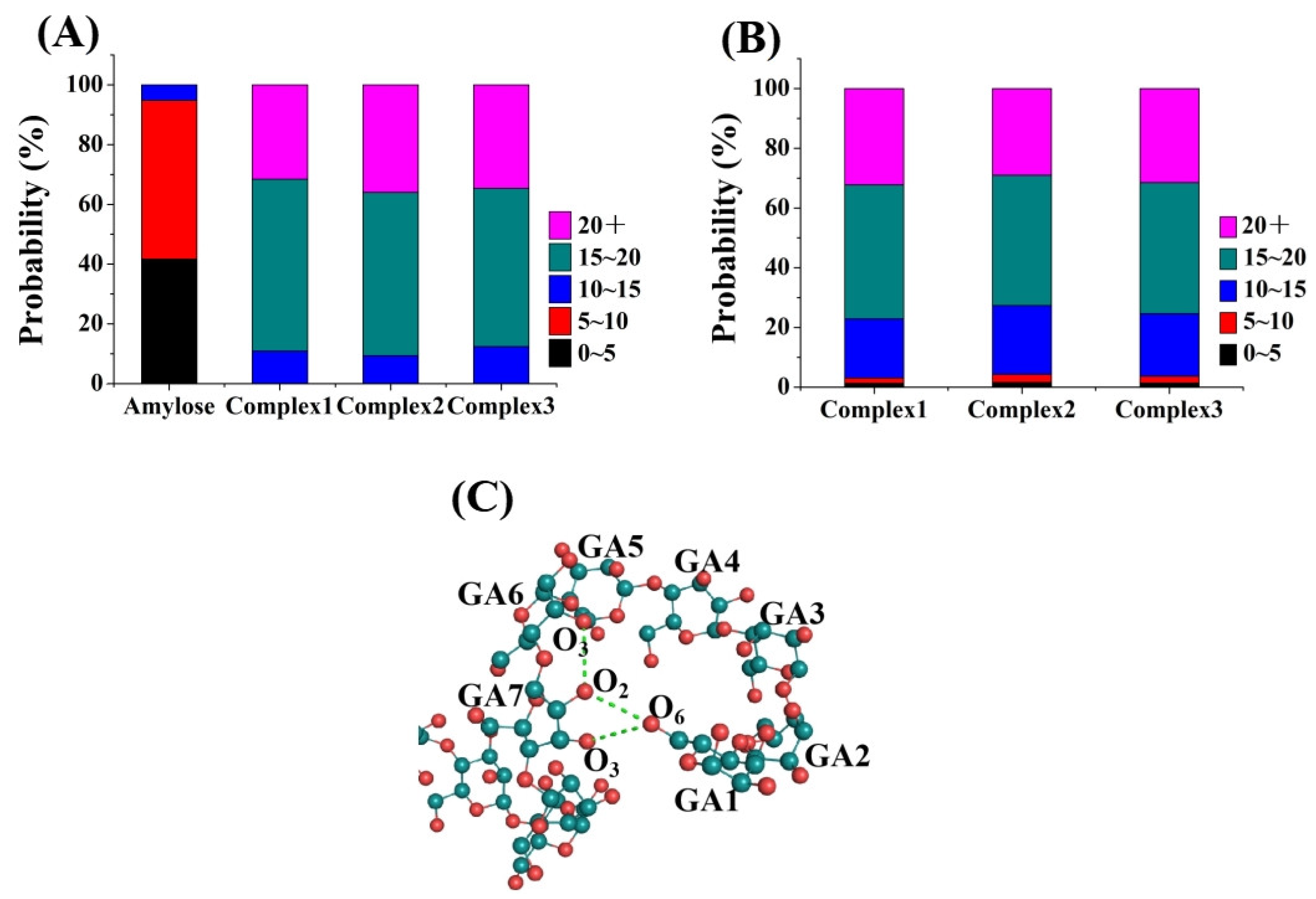
3.2. Intramolecular and Intermolecular Hydrogen Bonds
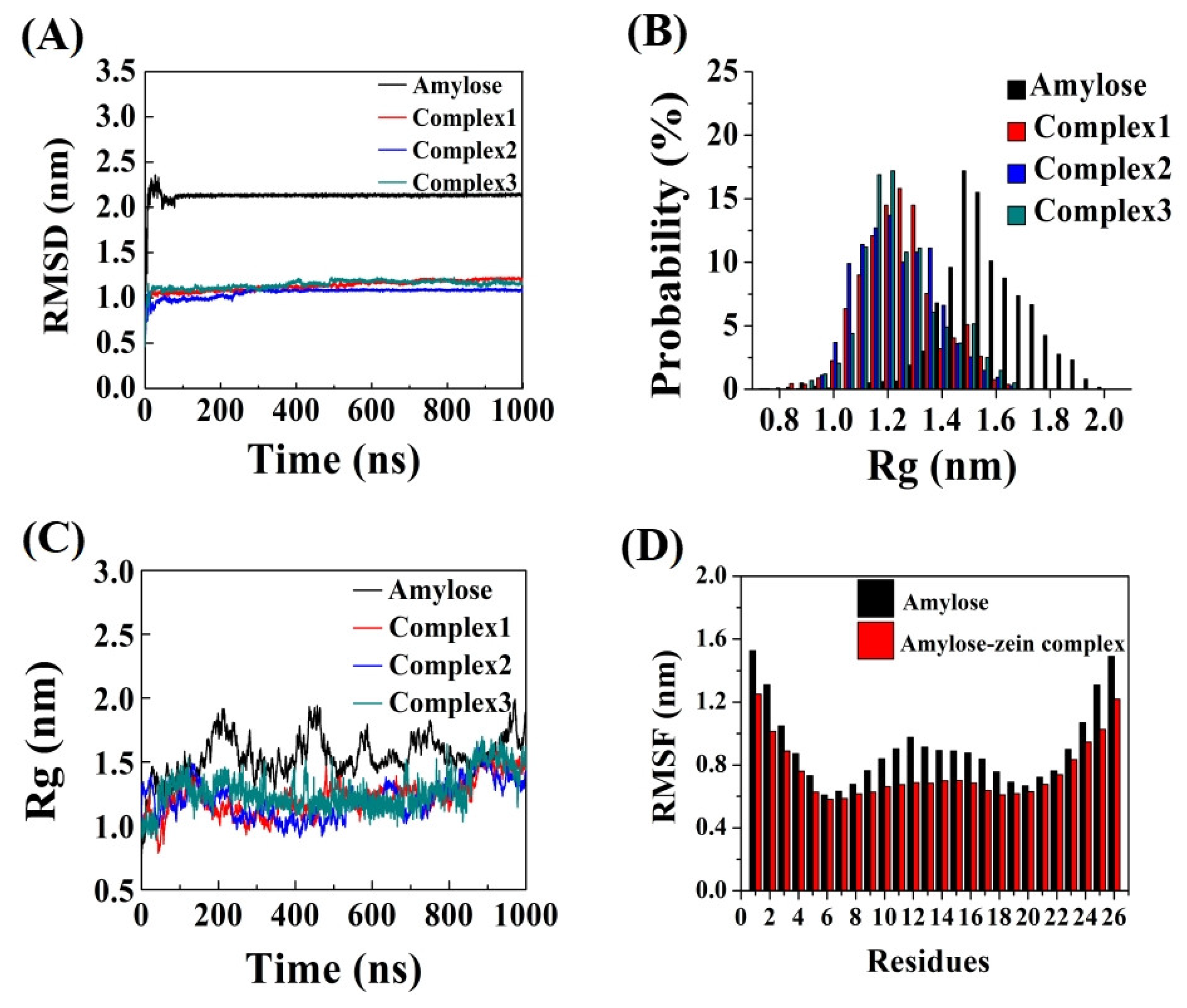
3.3. Root Mean Square Deviation (RMSD)
3.4. Radius of Gyration (Rg)
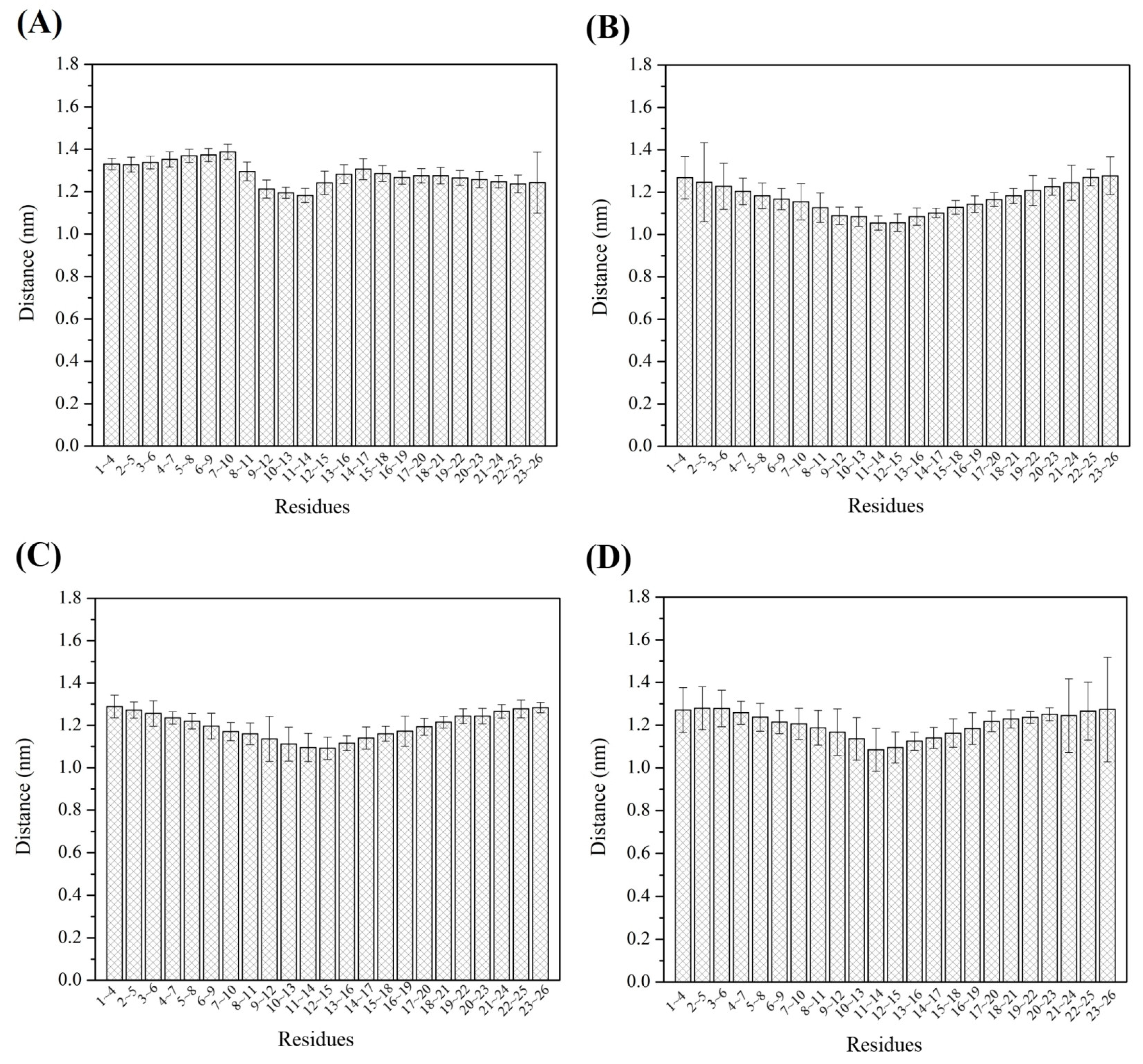
3.5. Root Mean Square Fluctuation (RMSF)
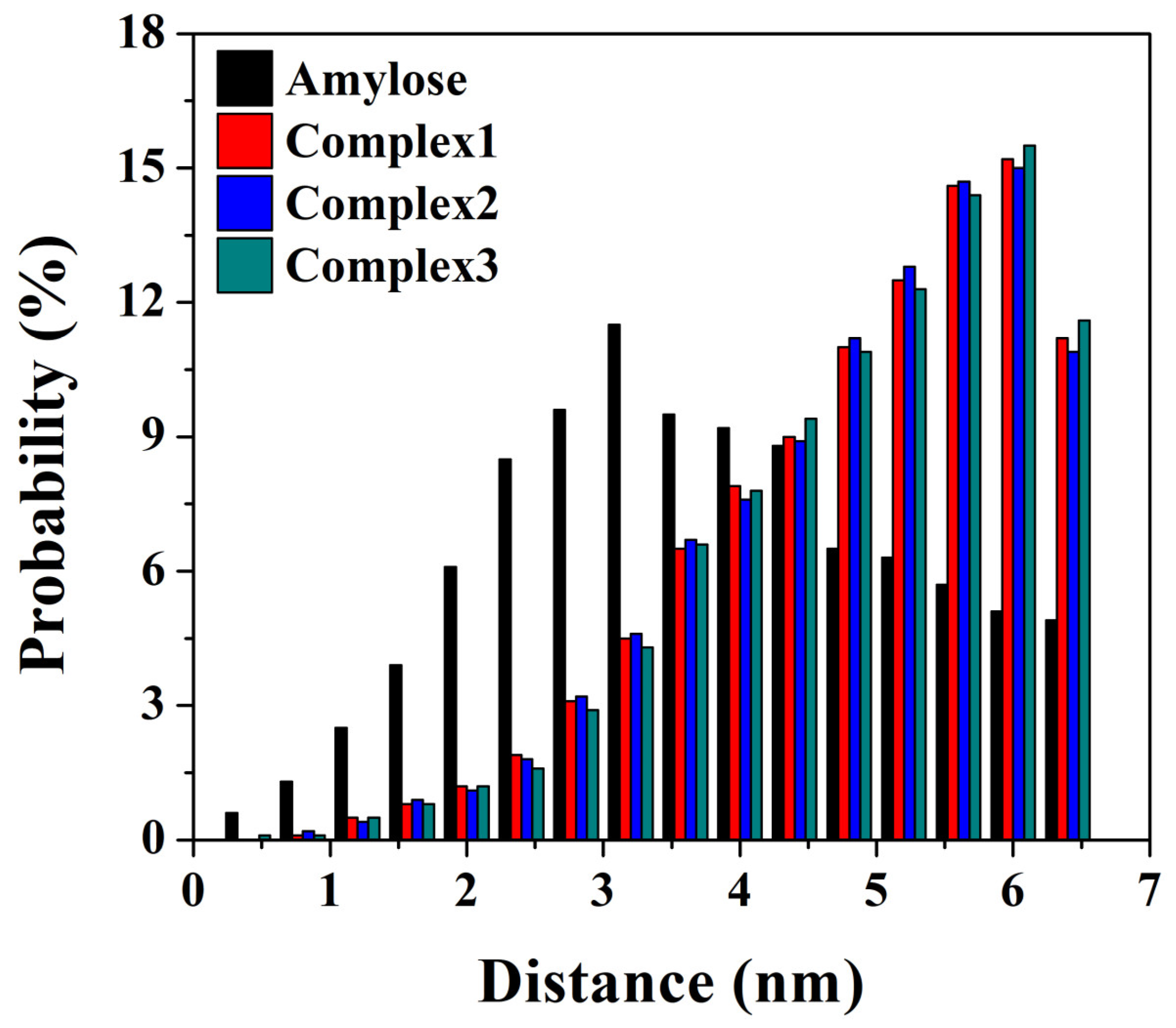
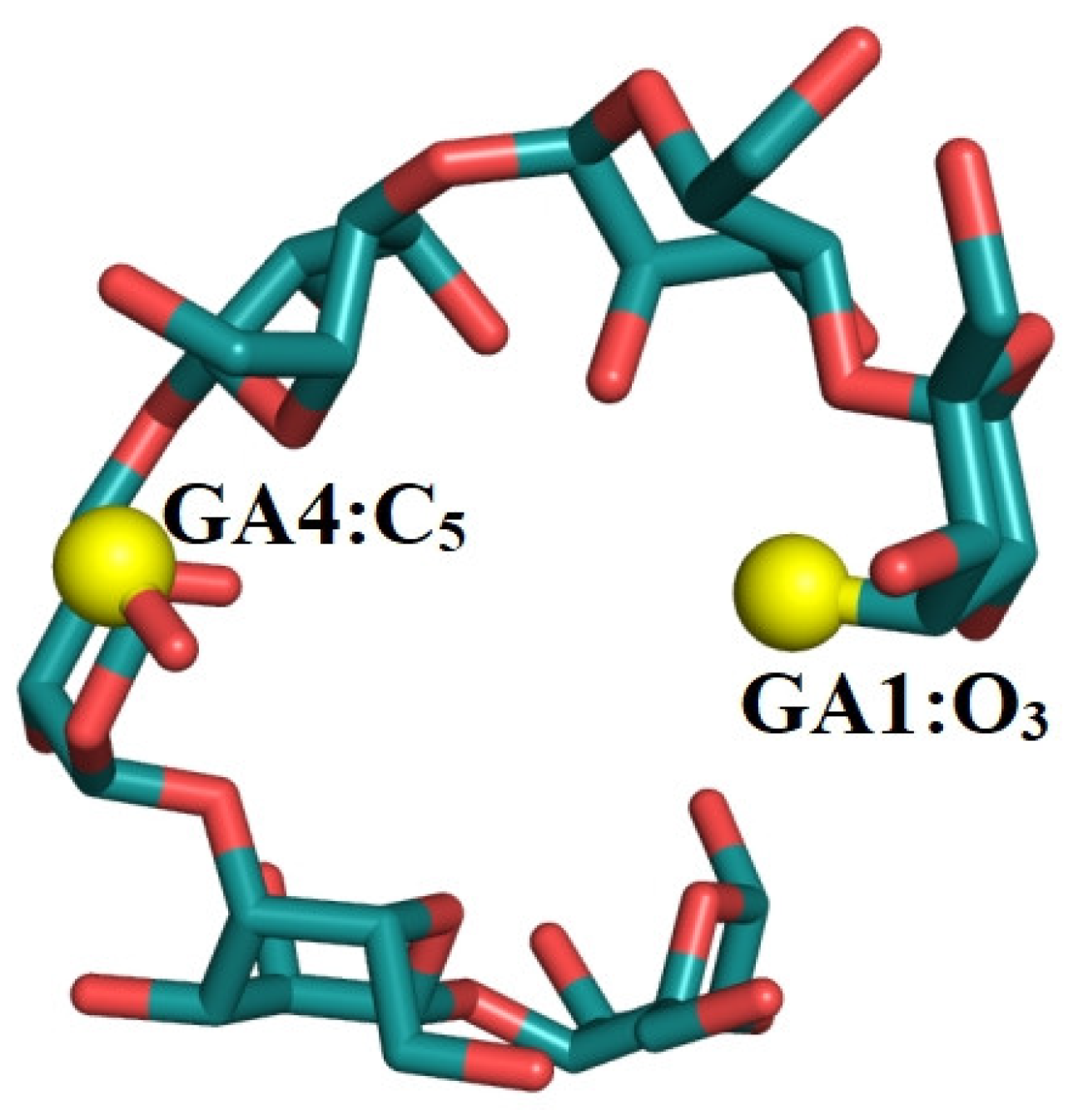
3.6. End-to-End Distance and Atom Distance
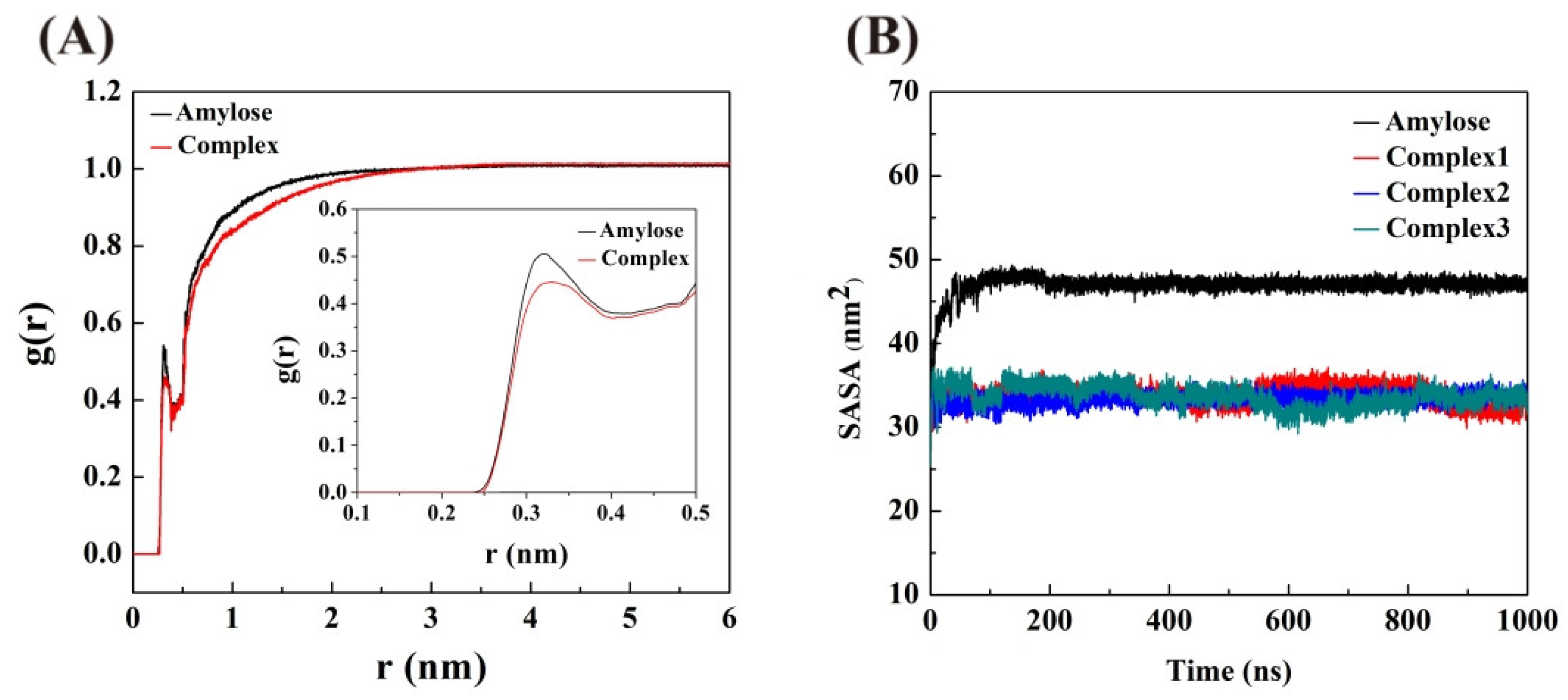
3.7. Radial Distribution Functions (RDF)
3.8. Solvent Accessible Surface Area (SASA)
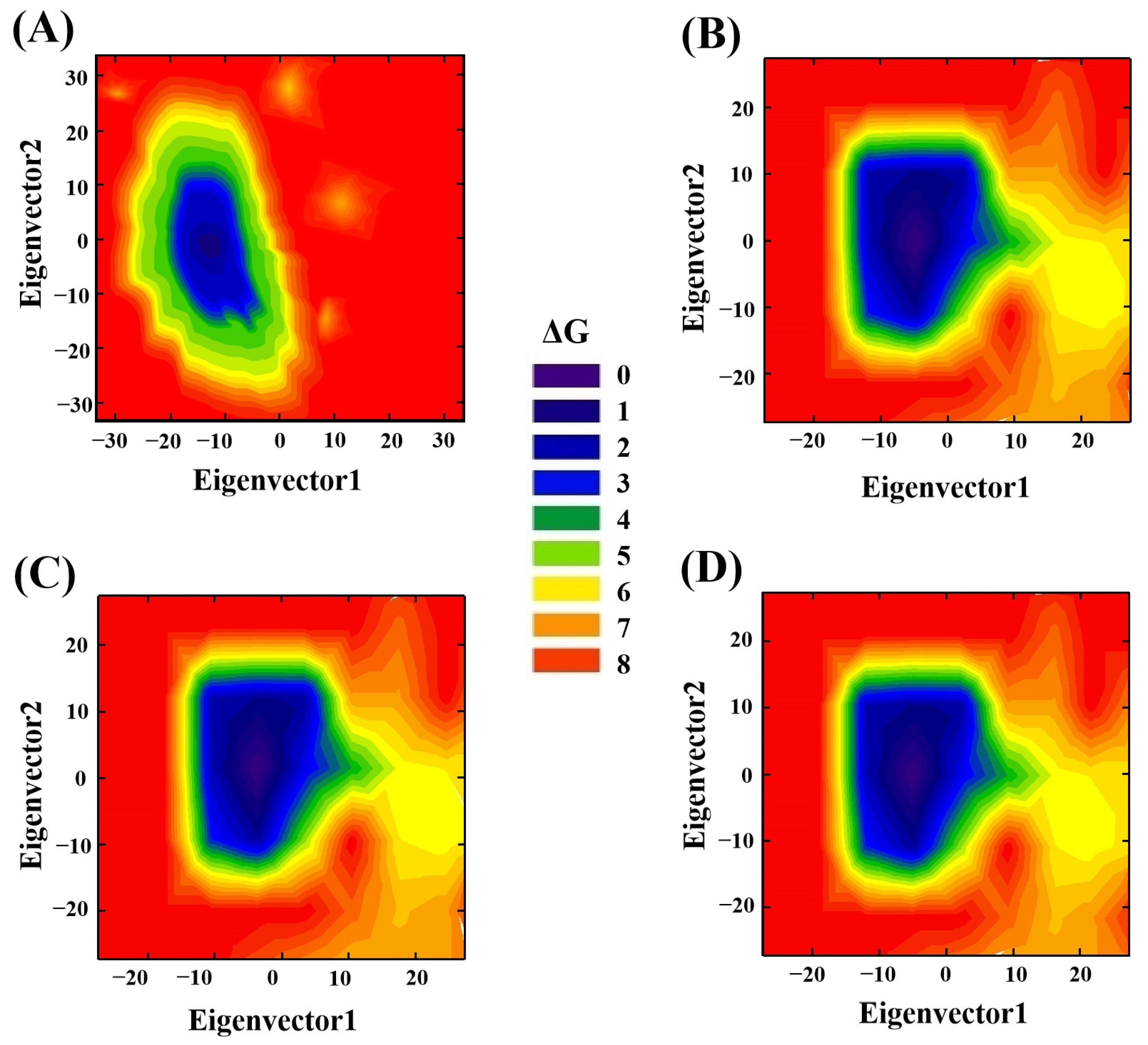
3.9. Principal Component Analysis (PCA)
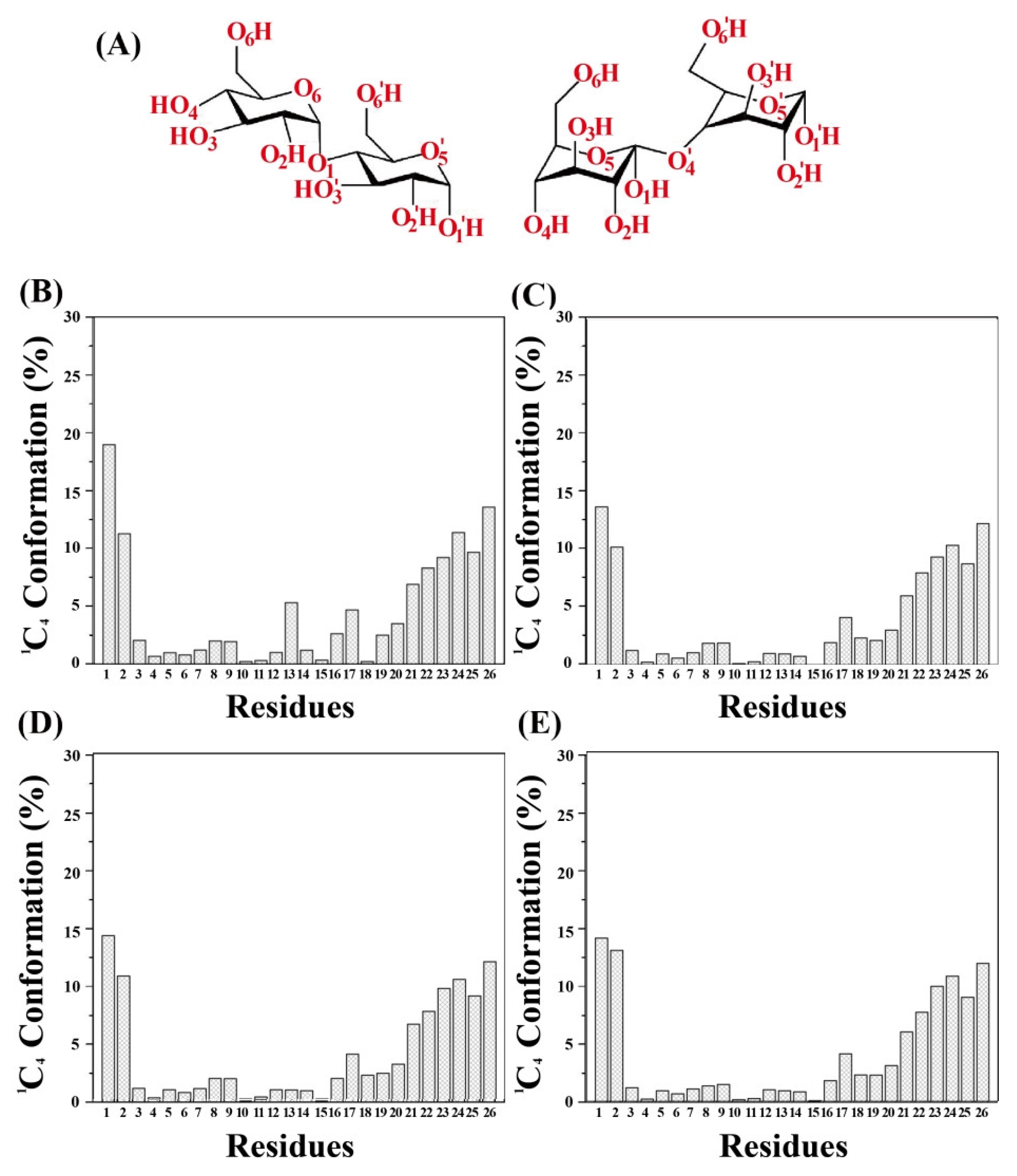
3.10. The Distribution of the Proportion of the 1C4 Chair Conformation
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, Q. Understanding Starches and Their Role in Foods. In Food Carbohydrates: Chemistry, Physical Properties and Applications; Taylor & Francis: Abingdon, UK, 2005; Volume 340. [Google Scholar]
- Tamura, M.; Okazaki, Y.; Kumagai, C.; Ogawa, Y. The importance of an oral digestion step in evaluating simulated in vitro digestibility of starch from cooked rice grain. Food Res. Int. 2017, 94, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.H.; Donner, E.; Yada, R.Y.; Liu, Q. Physicochemical properties and in vitro starch digestibility of potato starch/protein blends. Carbohydr. Polym. 2016, 154, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Yu, X.; Gao, Y.; Mei, L.; Zhu, Z.; Du, X. Physicochemical properties and in vitro starch digestibility of wheat starch/rice protein hydrolysate complexes. Food Hydrocoll. 2022, 125, 107348. [Google Scholar] [CrossRef]
- López-Barón, N.; Gu, Y.; Vasanthan, T.; Hoover, R. Plant proteins mitigate in vitro wheat starch digestibility. Food Hydrocoll. 2017, 69, 19–27. [Google Scholar] [CrossRef]
- Zhang, B.; Qiao, D.; Zhao, S.; Lin, Q.; Wang, J.; Xie, F. Starch-based food matrices containing protein: Recent understanding of morphology, structure, and properties. Trends Food Sci. Technol. 2021, 114, 212–231. [Google Scholar] [CrossRef]
- Kumari, I.; Sandhu, P.; Ahmed, M.; Akhter, Y. Molecular dynamics simulations, challenges and opportunities: A Biologist’s prospective. Curr. Protein Pept. Sci. 2017, 18, 1163–1179. [Google Scholar] [CrossRef]
- Han, J.A.; Lim, S.T. Structural changes of corn starches by heating and stirring in DMSO measured by SEC-MALLS-RI system. Carbohydr. Polym. 2004, 55, 265–272. [Google Scholar] [CrossRef]
- Zhong, F.; Yokoyama, W.; Wang, Q.; Shoemaker, C.F. Rice starch, amylopectin, and amylose: Molecular weight and solubility in dimethyl sulfoxide-based solvents. J. Agric. Food Chem. 2006, 54, 2320–2326. [Google Scholar] [CrossRef]
- Tusch, M.; Krüger, J.; Fels, G. Structural stability of V-amylose helices in water-DMSO mixtures analyzed by molecular dynamics. J. Chem. Theory Comput. 2011, 7, 2919–2928. [Google Scholar] [CrossRef]
- Śledź, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef]
- Feng, T.; Zhu, X.; Campanella, O. Molecular modeling tools to characterize the structure and complexation behavior of carbohydrates. Curr. Opin. Food Sci. 2016, 9, 62–69. [Google Scholar] [CrossRef]
- Chen, X.; Cui, F.; Zi, H.; Zhou, Y.; Liu, H.; Xiao, J. Development and characterization of a hydroxypropyl starch/zein bilayer edible film. Int. J. Biol. Macromol. 2019, 141, 1175–1182. [Google Scholar] [CrossRef]
- Cheng, L.; Feng, T.; Zhang, B.; Zhu, X.; Hamaker, B.; Zhang, H.; Campanella, O. A molecular dynamics simulation study on the conformational stability of amylose-linoleic acid complex in water. Carbohydr. Polym. 2018, 196, 56–65. [Google Scholar] [CrossRef]
- Kawai, K.; Takato, S.; Sasaki, T.; Kajiwara, K. Complex formation, thermal properties, and in-vitro digestibility of gelatinized potato starch–fatty acid mixtures. Food Hydrocoll. 2012, 27, 228–234. [Google Scholar] [CrossRef]
- Wang, C.; Qin, K.; Sun, Q.; Qiao, X. Preparation of natural food-grade core-shell starch/zein microparticles by antisolvent exchange and transglutaminase crosslinking for reduced digestion of starch. Front. Nutr. 2022, 9, 879757. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, S.; Yu, W. Revealing the mechanism beneath the effects of starch-amino acids interactions on starch physicochemical properties by molecular dynamic simulations. Food Hydrocoll. 2022, 124, 107359. [Google Scholar] [CrossRef]
- Tamura, M.; Singh, J.; Kaur, L.; Ogawa, Y. Impact of the degree of cooking on starch digestibility of rice—An in vitro study. Food Chem. 2016, 191, 98–104. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Q.; Shi, K.; Huang, Q. Scaling behaviors of α-zein in acetic acid solutions. J. Phys. Chem. B 2011, 115, 9695–9702. [Google Scholar] [CrossRef]
- Kony, D.; Damm, W.; Stoll, S.; Van Gunsteren, W.F. An improved OPLS—AA force field for carbohydrates. J. Comput. Chem. 2002, 23, 1416–1429. [Google Scholar] [CrossRef]
- Ong, E.E.; Liow, J.L. The temperature-dependent structure, hydrogen bonding and other related dynamic properties of the standard TIP3P and CHARMM-modified TIP3P water models. Fluid Phase Equilib. 2019, 481, 55–65. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Diem, M.; Oostenbrink, C. The Effect of Using a Twin-Range Cutoff Scheme for Nonbonded Interactions: Implications for Force-Field Parametrization? J. Chem. Theory Comput. 2020, 16, 5985–5990. [Google Scholar] [CrossRef]
- Pitera, J.W.; van Gunsteren, W.F. The importance of solute−solvent van der Waals interactions with interior atoms of biopolymers. J. Am. Chem. Soc. 2001, 123, 3163–3164. [Google Scholar] [CrossRef]
- Cheng, L.; Zhu, X.; Hamaker, B.R.; Zhang, H.; Campanella, O.H. Complexation process of amylose under different concentrations of linoleic acid using molecular dynamics simulation. Carbohydr. Polym. 2019, 216, 157–166. [Google Scholar] [CrossRef]
- Khatami, M.H.; Barber, W.; de Haan, H.W. Using geometric criteria to study helix-like structures produced in molecular dynamics simulations of single amylose chains in water. RSC Adv. 2021, 11, 11992–12002. [Google Scholar] [CrossRef]
- Yang, C.; Zhong, F.; Goff, H.D.; Li, Y. Study on starch-protein interactions and their effects on physicochemical and digestible properties of the blends. Food Chem. 2019, 280, 51–58. [Google Scholar] [CrossRef]
- Cazals, F.; Tetley, R. Characterizing molecular flexibility by combining least root mean square deviation measures. Proteins 2019, 87, 380–389. [Google Scholar] [CrossRef]
- Ma, R.; Huang, D.; Zhang, T.; Luo, T. Determining influential descriptors for polymer chain conformation based on empirical force-fields and molecular dynamics simulations. Chem. Phys. Lett. 2018, 704, 49–54. [Google Scholar] [CrossRef]
- Mitternacht, S. FreeSASA: An open source C library for solvent accessible surface area calculations. F1000Research 2016, 5, 189. [Google Scholar] [CrossRef]
- Marinopoulou, A.; Papastergiadis, E.; Raphaelides, S.N.; Kontominas, M.G. Structural characterization and thermal properties of amylose-fatty acid complexes prepared at different temperatures. Food Hydrocoll. 2016, 58, 224–234. [Google Scholar] [CrossRef]
- Xu, X.; Liu, W.; Zhong, J.; Luo, L.; Liu, C.; Luo, S.; Chen, L. Binding interaction between rice glutelin and amylose: Hydrophobic interaction and conformational changes. Int. J. Biol. Macromol. 2015, 81, 942–950. [Google Scholar] [CrossRef]
- Lelong, G.; Saboungi, M.L.; Brady, J.W. Free energy landscapes of the α-d-and β-d-glucopyranose conformations in both vacuum and aqueous solution. Mol. Simul. 2012, 38, 1186–1197. [Google Scholar] [CrossRef]
- Barnett, C.B.; Naidoo, K.J. Free Energies from Adaptive Reaction Coordinate Forces (FEARCF): An application to ring puckering. Mol. Phys. 2009, 107, 1243–1250. [Google Scholar] [CrossRef]
- Casset, F.; Imberty, A.; Haser, R.; Payan, F.; Perez, S. Molecular modelling of the interaction between the catalytic site of pig pancreatic α-amylase and amylose fragments. Eur. J. Biochem. 1995, 232, 284–293. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time (ns) | The Number of O2-O3 Intramolecular Hydrogen Bonds | |||
|---|---|---|---|---|
| Amylose | Complex1 | Complex2 | Complex3 | |
| 0 | 23 | 23 | 23 | 23 |
| 0–200 | 5.42 ± 0.51 | 16.03 ± 1.22 | 15.72 ± 0.93 | 15.54 ± 0.82 |
| 200–400 | 5.58 ± 0.62 | 15.34 ± 1.03 | 14.84 ± 1.12 | 14.73 ± 1.01 |
| 400–600 | 5.31 ± 0.42 | 13.52 ± 0.81 | 12.93 ± 0.82 | 13.74 ± 0.93 |
| 600–800 | 5.17 ± 0.31 | 12.44 ± 0.92 | 11.89 ± 0.91 | 12.13 ± 0.82 |
| 800–1000 | 4.73 ± 0.55 | 11.53 ± 1.12 | 11.14 ± 0.92 | 10.81 ± 1.22 |
| Time (ns) | The Number of O6-O2/O3 Intramolecular Hydrogen Bonds | |||
|---|---|---|---|---|
| Amylose | Complex1 | Complex2 | Complex3 | |
| 0 | 15 | 15 | 15 | 15 |
| 0–0.25 | 6.12 ± 1.43 | 11.63 ± 1.21 | 11.93 ± 1.31 | 11.24 ± 0.91 |
| 0.25–0.5 | 6.82 ± 1.51 | 10.14 ± 1.01 | 10.92 ± 0.92 | 10.43 ± 1.12 |
| 0.5–0.75 | 6.64 ± 1.81 | 9.24 ± 0.91 | 9.74 ± 1.11 | 9.54 ± 0.82 |
| 0.75–1.0 | 4.23 ± 1.62 | 7.83 ± 1.11 | 8.03 ± 1.21 | 8.23 ± 0.91 |
| 1.0–1.5 | 3.83 ± 1.11 | 6.44 ± 0.72 | 7.62 ± 0.83 | 7.14 ± 0.92 |
| 1.5–2.0 | 2.14 ± 0.61 | 6.53 ± 0.91 | 6.92 ± 1.12 | 6.23 ± 1.21 |
| 2.0–3.0 | 0.83 ± 0.31 | 6.13 ± 1.22 | 5.94 ± 1.25 | 6.84 ± 1.33 |
| 3.0–4.0 | 0 | 6.82 ± 1.13 | 6.53 ± 1.12 | 6.44 ± 1.22 |
| 4.0–100 | 0 | 5.54 ± 0.93 | 6.24 ± 1.21 | 5.83 ± 0.91 |
| 100–500 | 0 | 4.93 ± 0.81 | 5.53 ± 1.31 | 5.33 ± 1.12 |
| 500–1000 | 0 | 3.92 ± 0.93 | 4.72 ± 1.14 | 4.84 ± 0.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Ji, N.; Dai, L.; Qin, Y.; Shi, R.; Xiong, L.; Sun, Q. The Mechanism Underlying the Amylose-Zein Complexation Process and the Stability of the Molecular Conformation of Amylose-Zein Complexes in Water Based on Molecular Dynamics Simulation. Foods 2023, 12, 1418. https://doi.org/10.3390/foods12071418
Wang C, Ji N, Dai L, Qin Y, Shi R, Xiong L, Sun Q. The Mechanism Underlying the Amylose-Zein Complexation Process and the Stability of the Molecular Conformation of Amylose-Zein Complexes in Water Based on Molecular Dynamics Simulation. Foods. 2023; 12(7):1418. https://doi.org/10.3390/foods12071418
Chicago/Turabian StyleWang, Chaofan, Na Ji, Lei Dai, Yang Qin, Rui Shi, Liu Xiong, and Qingjie Sun. 2023. "The Mechanism Underlying the Amylose-Zein Complexation Process and the Stability of the Molecular Conformation of Amylose-Zein Complexes in Water Based on Molecular Dynamics Simulation" Foods 12, no. 7: 1418. https://doi.org/10.3390/foods12071418