
Identification of Small-Molecule Bioactive Constituents from the Leaves of Vaccinium bracteatum Confirms It as a Potential Functional Food with Health Benefits

Abstract
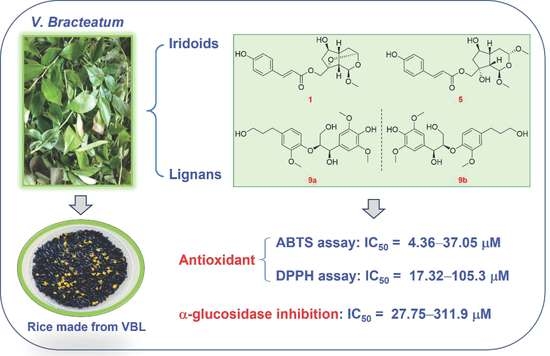
:
1. Introduction
2. Materials and Methods
2.1. Instrumentation and Reagents
2.2. Plant Materials
2.3. Extraction and Isolation
2.3.1. Compound 1
2.3.2. Compound 2
2.3.3. Compound 3
2.3.4. Compound 4
2.3.5. Compound 5
2.3.6. Compound 6
2.3.7. Compound 7
2.3.8. Compound 8
2.3.9. Compound 9
2.3.10. Compound 21
2.4. Alkaline Hydrolysis of Compounds 5 and 8
2.5. Preparation of (S) and (R)-MTPA Esters of 5 and 8
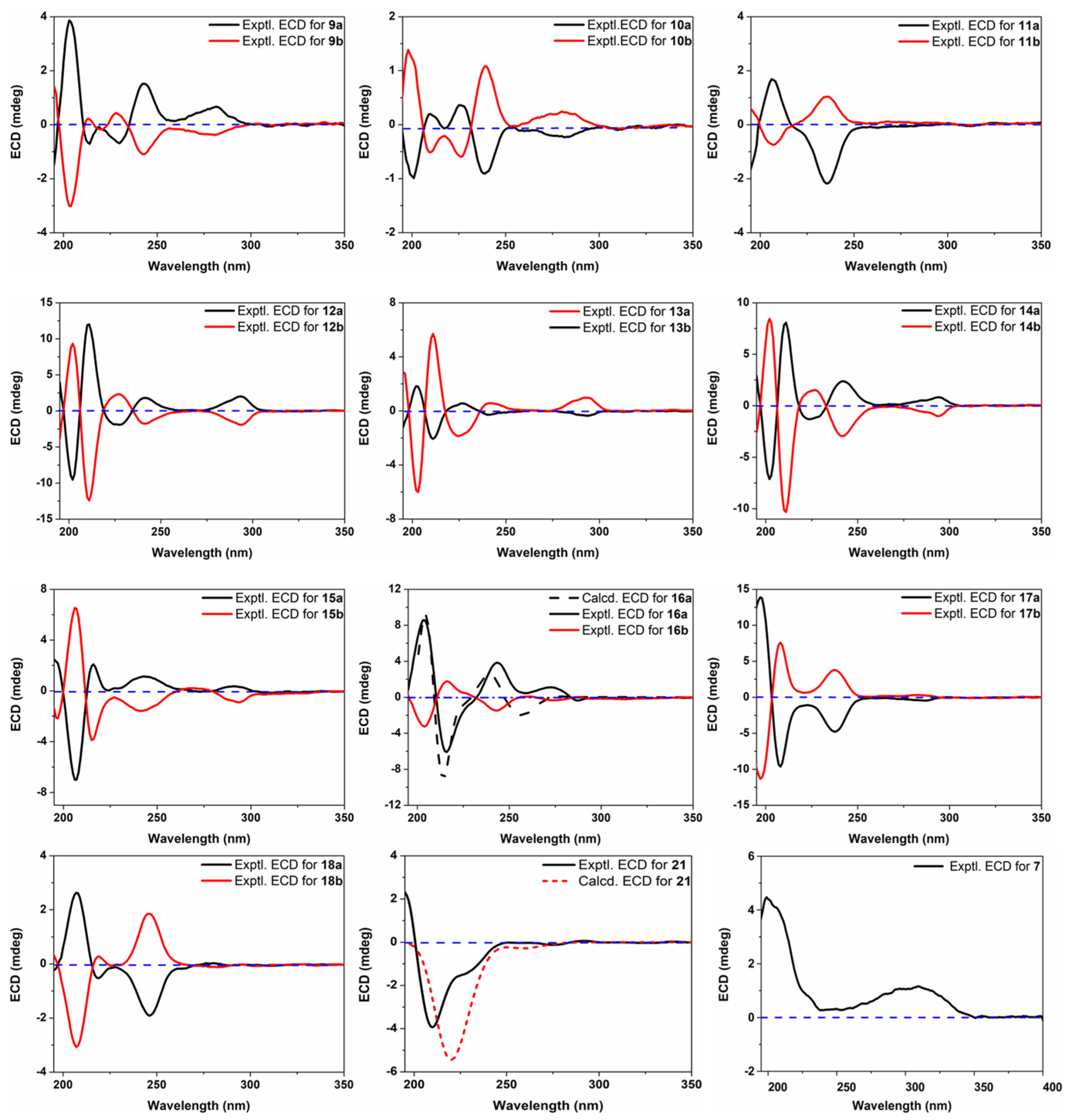
2.6. ECD Calculation
2.7. Bioassays
2.7.1. Antioxidant Assay (ABTS and DPPH Radical Scavenging Assays)
2.7.2. NO Production Inhibitory Assay
2.7.3. Acetylcholinesterase Inhibitory Assay
2.7.4. α-Glucosidase Inhibitory Assay
2.7.5. Antibacterial Assay
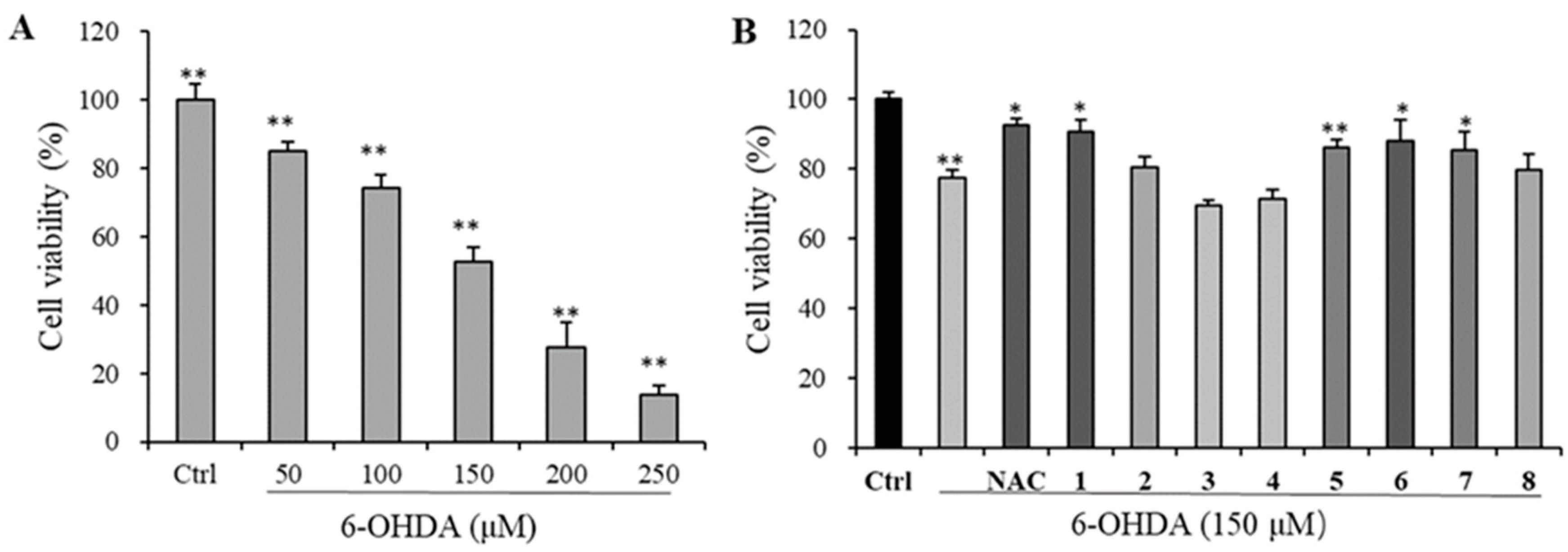
2.7.6. Neuroprotective Assay
3. Results
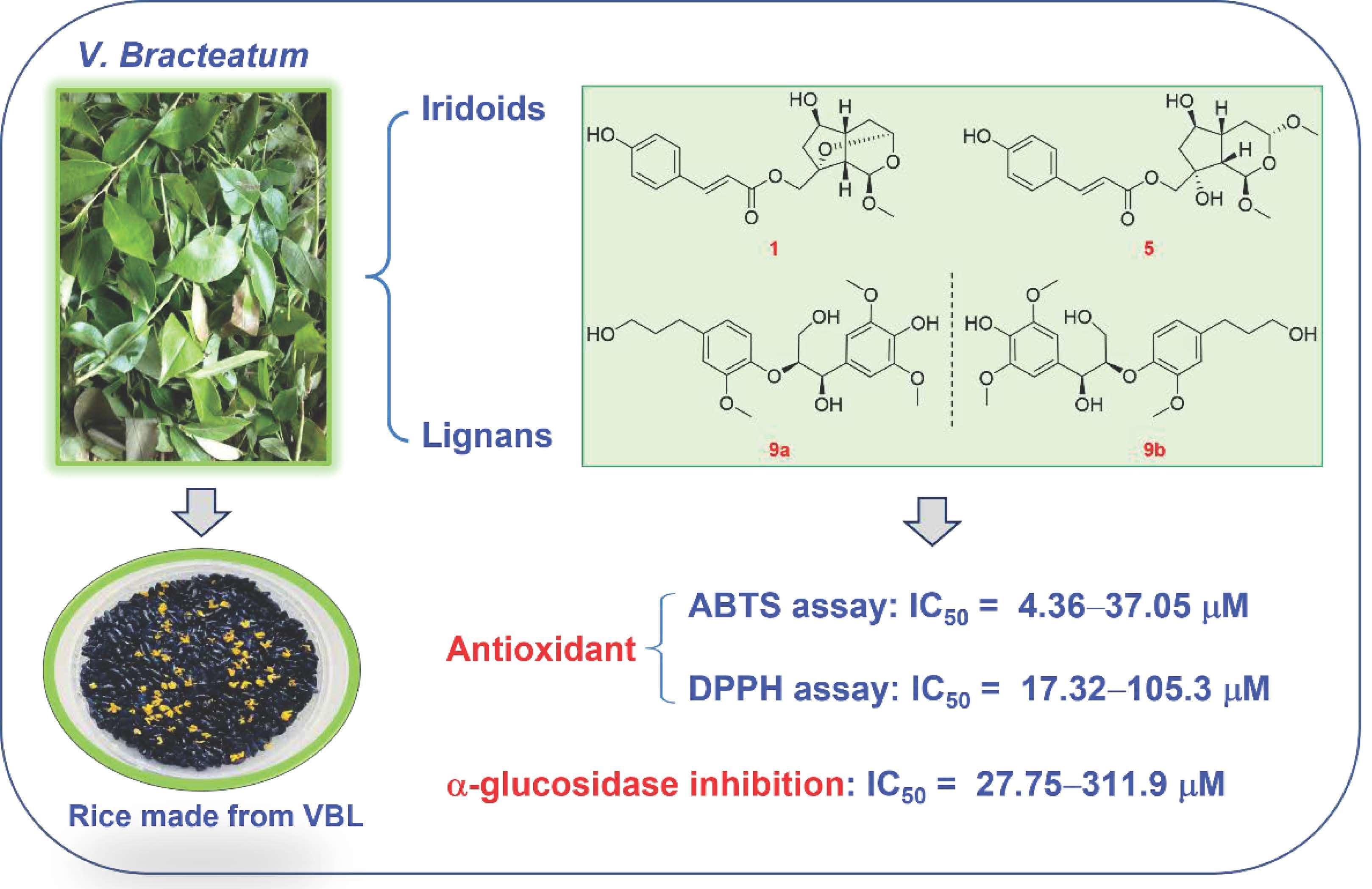
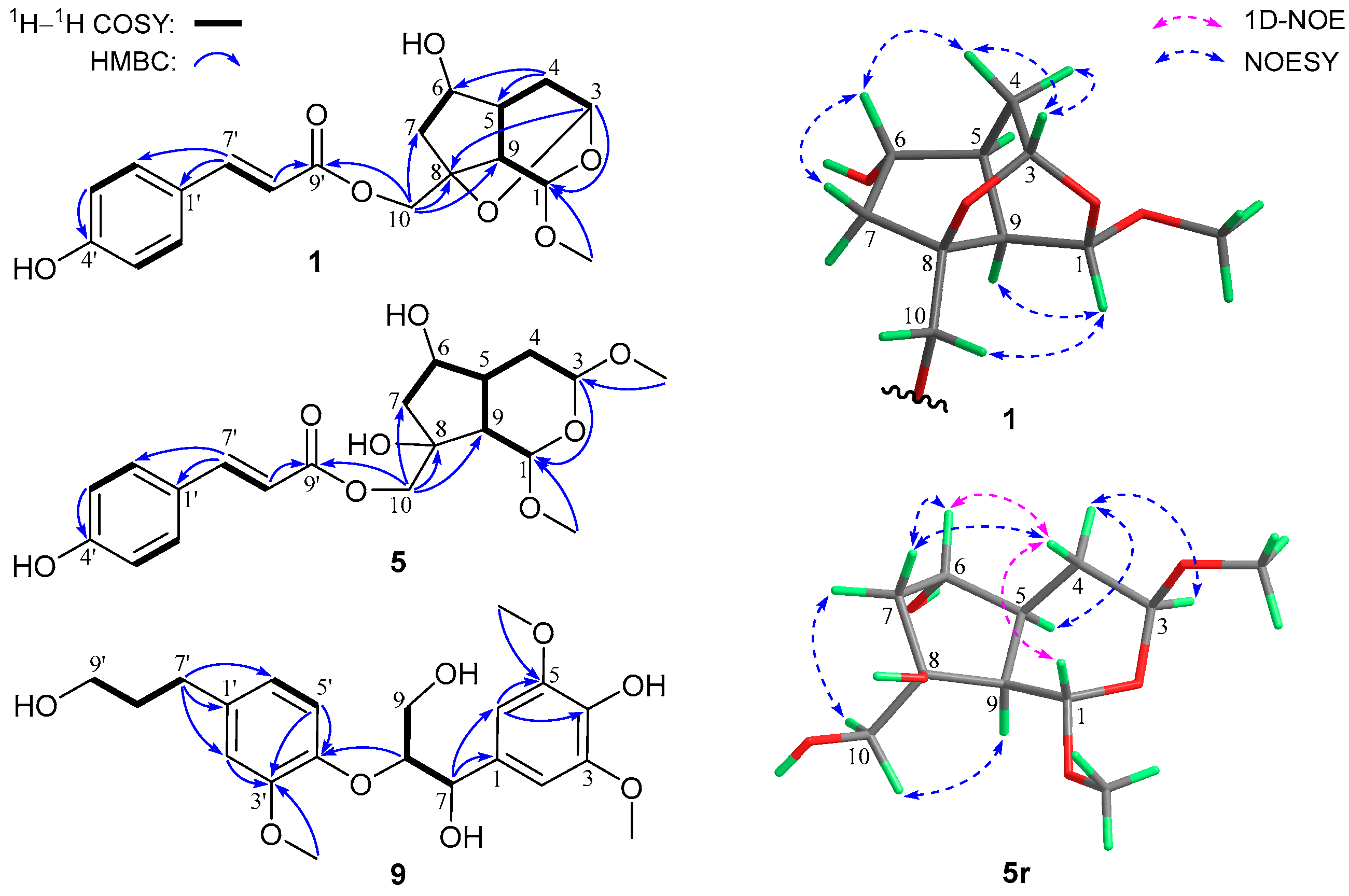
3.1. Structure Characterization of New Compounds
3.2. Structure Characterization of Known Compounds
3.3. Biological Evaluations
3.3.1. Antioxidant Evaluation
3.3.2. Neuroprotective Evaluation
3.3.3. α-Glucosidase Inhibitory Evaluation
3.3.4. Anti-Inflammatory Evaluation
3.3.5. Antimicrobial and AChE Inhibitory Evaluations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flora of China Editorial Committee of Chinese Academy of Sciences. Flora of China; Science Press: Beijing, China, 1991; Volume 57, p. 75. [Google Scholar]
- Fan, M.C.; Li, T.T.; Li, Y.; Qian, H.F.; Zhang, H.; Rao, Z.M.; Wang, L. Vaccinium bracteatum Thunb. as a promising resource of bioactive compounds with health benefits: An updated review. Food Chem. 2021, 356, 129738. [Google Scholar] [CrossRef] [PubMed]
- Flora of China Editorial Committee of Chinese Academy of Sciences. Flora of China; Science Press: Beijing, China, 1999; Volume 57, p. 107. [Google Scholar]
- Editorial Committee of the Administration Bure of Traditional Chinese Medicine. Chinese Materia Medica (Zhonghua Bencao); Shanghai Science & Technology Press: Shanghai, China, 1998; Volume 6, pp. 45–47. [Google Scholar]
- Fan, M.C.; Lian, W.J.; Li, T.T.; Rao, Z.M.; Li, Y.; Qian, H.F.; Zhang, H.; Qi, X.G.; Wang, L. Characterization of promising natural blue pigment from Vaccinium bracteatum thunb. leaves: Insights of the stability and the inhibition of α-amylase. Food Chem. 2020, 326, 126962. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.C.; Lian, W.J.; Li, Y.; Qian, H.F.; Zhang, H.; Rao, Z.M.; Wang, L. Evaluation of the physicochemical properties and in vitro digestibility of the complex formed between rice starch and a novel pigment from Vaccinium bracteatum Thunb. leaf. Food Chem. 2022, 374, 131627. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jung, E.S.; Do, S.G.; Jung, G.Y.; Song, G.; Song, J.M.; Lee, C.H. Correlation between Species-Specific Metabolite Profiles and Bioactivities of Blueberries (Vaccinium spp.). J. Agric. Food Chem. 2014, 62, 2126–2133. [Google Scholar] [CrossRef]
- Cao, X.X.; Sun, J.Y.; Liu, C.; Zhang, J.S.; Zhang, H. Antiradical Aromatic Constituents from Pleurotus eryngii. Rec. Nat. Prod. 2021, 15, 169–174. [Google Scholar] [CrossRef]
- Jungfer, E.; Zimmermann, B.F.; Ruttkat, A.; Galensa, R. Comparing Procyanidins in Selected Vaccinium Species by UHPLC-MS2 with Regard to Authenticity and Health Effects. J. Agric. Food Chem. 2012, 60, 9688–9696. [Google Scholar] [CrossRef]
- Minh, T.N.; Van, T.M.; Khanh, T.D.; Xuan, T.D. Isolation and Identification of Constituents Exhibiting Antioxidant, Antibacterial, and Antihyperuricemia Activities in Piper methysticum Root. Foods 2022, 11, 3889. [Google Scholar] [CrossRef]
- Zhang, J.S.; Cao, X.X.; Yu, J.H.; Yu, Z.P.; Zhang, H. Diarylheptanoids with NO Production Inhibitory Activity from Amomum kravanh. Bioorg. Med. Chem. Lett. 2020, 30, 127026. [Google Scholar] [CrossRef]
- Liu, C.; Tian, J.L.; An, T.; Lyu, F.N.; Jia, P.F.; Zhou, M.J.; Liu, Z.X.; Feng, Y.L. Secondary Metabolites from Solanum rostratum and Their Antifeedant Defense Mechanisms against Helicoverpa armigera. J. Agric. Food Chem. 2020, 68, 88–96. [Google Scholar] [CrossRef]
- Hou, Z.W.; Chen, C.H.; Ke, J.P.; Zhang, Y.Y.; Qi, Y.; Liu, S.Y.; Yang, Z.; Ning, J.M.; Bao, G.H. α—Glucosidase Inhibitory Activities and the Interaction Mechanism of Novel Spiro-Flavoalkaloids from YingDe Green Tea. J. Agric. Food Chem. 2022, 70, 136–148. [Google Scholar] [CrossRef]
- Bao, J.; Zhai, H.J.; Zhu, K.K.; Yu, J.H.; Zhang, Y.Y.; Wang, Y.Y.; Zhang, H. Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium sp. UJNMF0008. Mar. Drugs 2018, 16, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergi, D.; Gélinas, A.; Beaulieu, J.; Renaud, J.; Tardif-Pellerin, E.; Guillard, J.; Martinoli, M.-G. Anti-Apoptotic and Anti-Inflammatory Role of Trans ε-Viniferin in a Neuron– Glia Co-Culture Cellular Model of Parkinson’s Disease. Foods 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Mandi, A.; Kurtan, T. Applications of OR/ECD/VCD to the structure elucidation of natural products. Nat. Prod. Rep. 2019, 36, 889–918. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear Magnetic Resonance Enantiomer Regents. Configurational Correlations via Nuclear Magnetic Resonance Chemical Shifts of Diastereomeric Mandelate, O-Methylmandelate, and α-Methoxy-α-trifluoromethylphenylacetate (MTPA) Esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Bari, L.D.; Pratelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of Absolute Configuration of Acyclic 1,2-Diols with Mo2(OAc)4.1. Snatzke’s Method Revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.B.; Li, Y.; Ma, S.G.; Li, L.; Qu, J.; Zhang, D.; Jiang, D.J.; Yu, S.S. Phenolic Constituents from the roots of Alangium chinense. Chin. Chem. Lett. 2017, 28, 32–36. [Google Scholar] [CrossRef]
- Yang, Y.N.; Han, B.; Yang, P.F.; Feng, Z.M.; Jiang, J.S.; Zhang, P.C. A concise approach for determining the relative configuration of H-7 and H-8 in 8,4′-oxyneolignans by 1H NMR spectroscopy. Org. Chem. Front. 2019, 6, 886–891. [Google Scholar] [CrossRef]
- Gan, M.L.; Zhang, Y.L.; Lin, S.; Liu, M.T.; Song, W.X.; Zi, J.C.; Yang, Y.C.; Fan, X.N.; Shi, J.G.; Hu, J.F.; et al. Glycosides from the root of Iodescirrhosa. J. Nat. Prod. 2008, 71, 647–654. [Google Scholar] [CrossRef]
- Yu, J.H.; Yu, Z.P.; Capon, R.J.; Zhang, H. Natural Enantiomers: Occurrence, Biogenesis and Biological Properties. Molecules 2022, 27, 1279. [Google Scholar] [CrossRef]
- Arnoldi, A.; Merlini, L. Asymmetric Synthesis of 3-Methyl-2-phenyl-1,4-benzodioxanes. Absolute Configuration of the Neolignans Eusiderin and Eusiderin C and D. J. Chem. Soc. Perkin Trans. I 1985, 12, 2555–2557. [Google Scholar] [CrossRef]
- Wang, J.P.; Guan, Z.Y.; Dong, C.F.; Gao, L.; Luo, S.D.; Wang, Y.F. Chemical constituents of Illicium burmanicum. Zhongguo Zhongyao Zazhi 2014, 39, 2526–2530. [Google Scholar]
- Agrawal, P.K.; Agarwal, S.K.; Rastogi, R.P. A New Neolignan and Other Phenolic Constituents from Cedrus Deodara. Phytochemistry 1980, 19, 1260–1261. [Google Scholar] [CrossRef]
- Kim, T.H.; Ito, H.; Hayashi, K.; Hasegawa, T.; Machiguchi, T.; Yoshida, T. Aromatic Constituents from the Heartwood of Santalum album L. Chem. Pharm. Bull. 2005, 53, 641–644. [Google Scholar] [CrossRef] [Green Version]
- Van Dyck, S.M.O.; Lemiere, G.L.F.; Jonckers, T.H.M.; Dommisse, R. Synthesis of 4-O-Methylcedrusin. Selective Protection of Catechols with Diphenyl Carbonate. Molecules 2000, 5, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Seidel, V.; Bailleul, F.; Waterman, P.G. Novel Oligorhamnosides from the Stem Bark of Cleistopholis glauca. J. Nat. Prod. 2000, 63, 6–11. [Google Scholar] [CrossRef]
- Chin, Y.W.; Chai, H.B.; Keller, W.J.; Kinghorn, A.D. Lignans and Other Constituents of the Fruits of Euterpe oleracea (Acai) with Antioxidant and Cytoprotective Activities. J. Agric. Food Chem. 2008, 56, 7759–7764. [Google Scholar] [CrossRef]
- Sadhu, S.K.; Phattanawasin, P.; Choudhuri, M.S.K.; Ohtsuki, T.; Ishibashi, M. A new lignan from Aphanamixis polystachya. J. Nat. Med. 2006, 60, 258–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöttner, M.; Reiner, J.; Tayman, F.S. (+)-neo-olivil from roots of Urtica dioica. Phytochemisty 1997, 46, 1107–1109. [Google Scholar] [CrossRef]
- Matsushita, H.; Miyase, T.; Ueno, A. Lignan and terpene glycosides from Epimedium Sagittatum. Phytochemisty 1991, 30, 2025–2027. [Google Scholar] [CrossRef]
- Liu, L.; Zou, M.; Yin, Q.; Zhang, Z.; Zhang, X. Phenylpropanoids from Liparis nervosa and their in vitro antioxidant and α-glucosidase inhibitory activities. Med. Chem. Res. 2021, 30, 1005–1010. [Google Scholar] [CrossRef]
- Takeda, Y.; Okada, Y.; Masuda, T.; Hirata, E.; Shinzato, T.; Takushi, A.; Yu, Q.; Otsuka, H. New Megastigmane and Tetraketide from the Leaves of Euscaphis japonica. Chem. Pharm. Bull. 2000, 48, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Jutiviboonsuk, A.; Zhang, H.J.; Tan, G.T.; Ma, C.; Hung, N.V.; Cuong, N.M.; Bunyapraphatsara, N.V.; Soejarto, D.D.; Fong, H.H.S. Bioactive constituents from roots of Bursera tonkinensis. Phytochemistry 2005, 66, 2745–2751. [Google Scholar] [CrossRef] [PubMed]
- Gang, D.R.; Kasahara, H.; Xia, Z.Q.; Vander Mijnsbrugge, K.; Bauw, G.; Boerjan, W.; Montagu, M.V.; Davin, L.B.; Lewis, N.G. Evolution of plant defense mechanisms: Relationships of phenylcoumaran benzylic ether reductases to pinoresinol-lariciresinol and isoflavone reductases. J. Biol. Chem. 1999, 274, 7516–7527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.H.; Lv, J.; Mok, D.; Yao, X.S.; Wong, M.S.; Cooper, R. NMR Applications for Botanical Mixtures: The Use of HSQC Data to Determine Lignan Content in Sambucus williamsii. J. Nat. Prod. 2019, 82, 1733–1740. [Google Scholar] [CrossRef]
- Gao, G.C.; Qi, S.H.; Zhang, S.; Yin, H.; Xiao, Z.H.; Li, N.Y.; Li, Q.G. Minor compounds from the stem bark of Chinese mangrove associate Catunaregam spinosa. Pharmazie 2008, 63, 542–544. [Google Scholar] [CrossRef]
- Pan, J.Y.; Chen, S.L.; Yang, M.H.; Wu, J.; Sinkkonen, J.; Zou, K. An update on lignans: Natural products and synthesis. Nat. Prod. Rep. 2009, 26, 1251–1292. [Google Scholar] [CrossRef]
- Pieters, L.; De Bruyne, T.; Claeys, M.; Vlietinck, A.; Calomme, M.; Vanden Berghe, D. Isolation of a dihydrobenzofuran lignan from South American dragon’s blood (Croton spp.) as an inhibitor of cell proliferation. J. Nat. Prod. 1993, 56, 899–906. [Google Scholar] [CrossRef]
- Pieters, L.; De Bruyne, T.; De Groot, A.; Mei, G.; Dommisse, R.; Lemière, G.; Vlietinck, A. NMR Study of Some Dihydrobenzofuran Lignans. Magn. Reson. Chem. 1993, 31, 692–693. [Google Scholar] [CrossRef]
- Zhu, J.X.; Ren, J.; Qin, J.J.; Cheng, X.R.; Zeng, Q.; Zhang, F.; Yan, S.K.; Jin, H.Z.; Zhang, W.D. Phenylpropanoids and lignanoids from Euonymus acanthocarpus. Arch. Pharm. Res. 2012, 35, 1739–1747. [Google Scholar] [CrossRef]
- Antus, S.; Kurtán, T.; Juhász, L.; Kiss, L.; Hollósi, M.; Májer, Z.S. Chiroptical Properties of 2,3-Dihydrobenzo [b] furan and Chromane Chromophores in Naturally Occurring O-Heterocycles. Chirality 2001, 13, 493–506. [Google Scholar] [CrossRef]
- Yamauchi, H.; Kakuda, R.; Yaoita, Y.; Machida, K.; Kikuchi, M. Two New Glycosides from the Whole Plants of Glechoma hederacea L. Chem. Pharm. Bull. 2007, 55, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.D.; Qin, G.W.; Xu, R.S.; Tian, Z.Y.; Lu, Y.; Zheng, Q.T. Studies on chemical constituents of Ilex centrochinensis. II. Structures of huazhongilexol and huazhongilexin. Huaxue Xuebao 1995, 53, 98–101. [Google Scholar]
- Zhang, J.; Chu, C.J.; Li, X.L.; Yao, S.; Yan, B.; Ren, H.L.; Xu, N.Y.; Liang, Z.T.; Zhao, Z.Z. Isolation and identification of antioxidant compounds in Vaccinium bracteatum Thunb. by UHPLC-Q-TOF LC/MS and their kidney damage protection. J. Funct. Foods 2014, 11, 62–70. [Google Scholar] [CrossRef]
- Su, K.D.; Yao, S.; Li, H.R.; Jian, Z. Study on chemical constituents and antioxidant activities of the extractions of Vaccinium bracteatum Thunb. leaves. China Food Addit. (Zhongguo Shipin Tianjiaji) 2017, 7, 87–95. [Google Scholar]
- Zheng, Y.; Chen, L.; Liu, Y.; Shi, L.; Wan, S.; Wang, L. Evaluation of antimicrobial activity of water-soluble flavonoids extract from Vaccinium bracteatum Thunb. leaves. Food Sci. Biotechnol. 2019, 28, 1853–1859. [Google Scholar] [CrossRef]
- Wang, C.C.; Gong, X.; Bo, A.; Zhang, L.; Zhang, M.X.; Zang, E.; Zhang, C.H.; Li, M.H. Iridoids: Research Advances in Their Phytochemistry, Biological Activities, and Pharmacokinetics. Molecules 2020, 25, 287. [Google Scholar] [CrossRef] [Green Version]
- Kandola, K.; Bowman, A.; Birch-Machin, M.A. Oxidative stress–a key emerging impact factor in health, ageing, lifestyle and aesthetics. Int. J. Cosmet. Sci. 2015, 37 (Suppl. S2), 1–8. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.P.; Rahman, H.S. Antioxidant and Oxidative Stress: A Mutual Interplay in Age—Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [Green Version]
- Mehdi, A.; Lamiae, B.; Samira, B.; Ramchoun, M.; Abdelouahed, K.; Tamas, F.; Hicham, B. Pomegranate (Punica granatum L.) Attenuates Neuroinflammation Involved in Neurodegenerative Diseases. Foods 2022, 11, 2570. [Google Scholar] [CrossRef]
- Peñalver, P.; Zodio, S.; Lucas, L.; De-paz, M.V.; Morales, C. Neuroprotective and Anti-inflammatory Effects of Pterostilbene Metabolites in Human Neuroblastoma SH-SY5Y and RAW 264.7 Macrophage Cells. J. Agric. Food Chem. 2020, 68, 1609–1620. [Google Scholar] [CrossRef]
- Zhou, L.; Guo-Dong Yao, G.D.; Song, X.Y.; Wang, J.; Lin, B.; Wang, X.B.; Huang, X.X.; Song, S.J. Neuroprotective Effects of 1,2-Diarylpropane Type Phenylpropanoid Enantiomers from Red Raspberry against H2O2-Induced Oxidative Stress in Human Neuroblastoma SH-SY5Y Cells. J. Agric. Food Chem. 2018, 66, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Akbari, B.; Baghaei-Yazdi, N.; Bahmaie, M.; Abhari, F.M. The role of plant-derived natural antioxidants in reduction of oxidative stress. Biofactors 2022, 48, 611–633. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
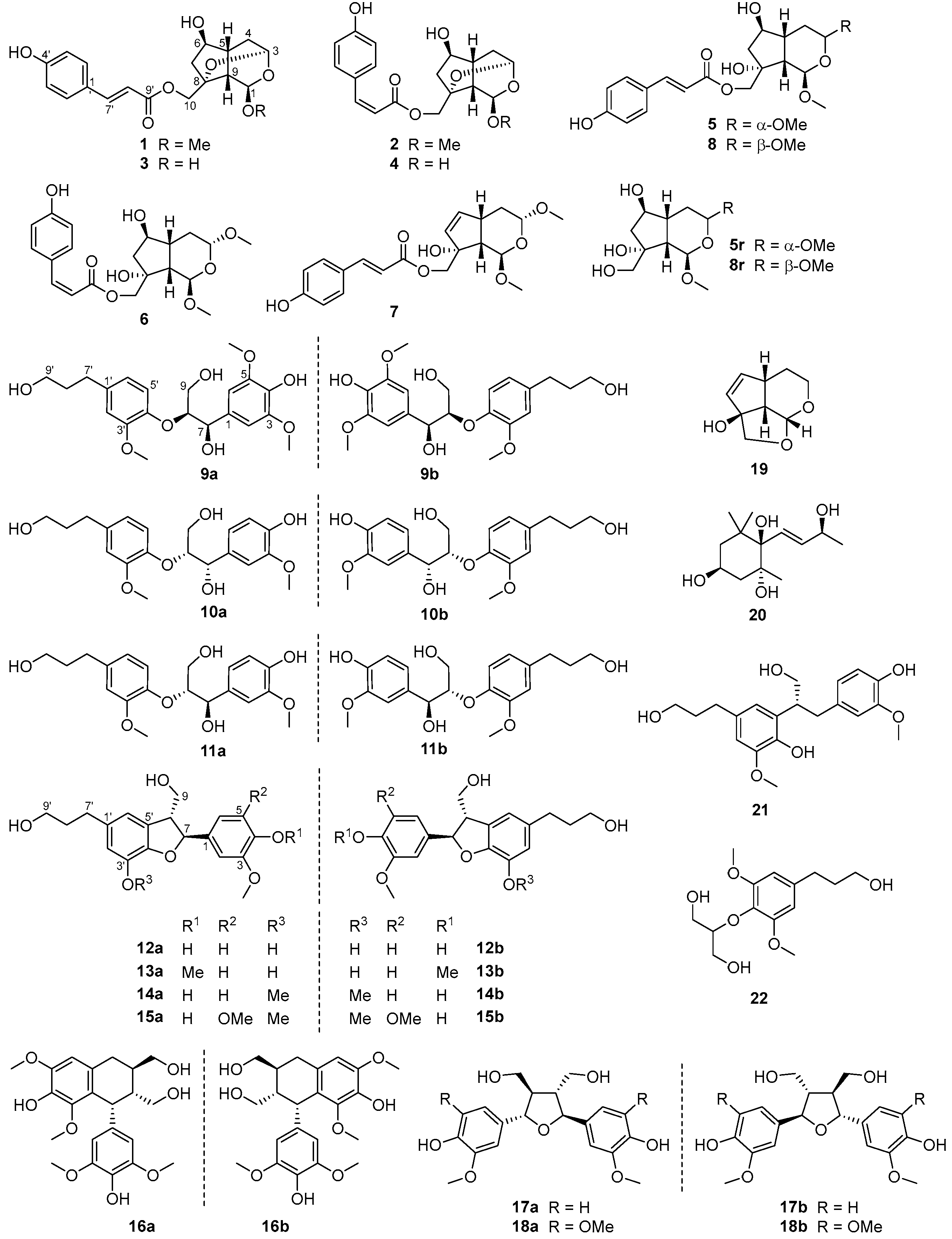{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ABTS | DPPH | Compounds | ABTS | DPPH | Compounds | ABTS | DPPH |
|---|---|---|---|---|---|---|---|---|
| 1 | 15.28 ± 1.37 | NA | 9a | 9.39 ± 0.97 | 47.49 ± 7.24 | 14a | 12.32 ± 1.82 | NA |
| 2 | 34.17 ± 3.03 | NA | 9b | 10.08 ± 0.67 | 52.62 ±6.89 | 14b | 14.07 ± 1.30 | NA |
| 3 | 35.58 ± 1.04 | NA | 10a | 19.69 ± 0.61 | NA | 15a | 17.21 ± 0.89 | 40.43 ± 6.09 |
| 4 | 19.40 ± 0.24 | NA | 10b | 16.23 ± 2.65 | NA | 15b | 24.25 ±3.92 | 55.10 ± 2.43 |
| 5 | 32.01 ± 1.76 | NA | 11a | 12.13 ± 1.42 | 98.65 ± 1.78 | 16a | 11.09 ± 3.31 | 47.92 ± 6.60 |
| 6 | 28.25 ± 0.63 | NA | 11b | 14.79 ± 1.25 | 105.3 ± 4.11 | 16b | 26.20 ± 2.24 | 96.16 ± 8.34 |
| 7 | 50.65 ± 2.97 | NA | 12a | 9.54 ± 0.64 | 31.15 ± 1.90 | 17a | 5.96 ± 0.30 | 40.79 ± 5.98 |
| 8 | 39.14 ± 2.30 | NA | 12b | 12.30 ± 0.24 | 44.28 ± 2.05 | 17b | 7.75 ± 0.71 | 62.85 ± 5.65 |
| 19 | NA | NA | 13a | 21.66 ± 3.50 | NA | 18a | 6.87 ± 0.54 | 17.32 ± 7.51 |
| 20 | NA | NA | 13b | 37.05 ± 8.17 | NA | 18b | 4.36 ± 0.30 | 17.92 ± 4.58 |
| 21 | 9.17 ± 0.24 | NA | 22 | 168.0 ± 4.34 | 20.21 ± 3.53 | Ascorbic acid | 24.29 ± 0.41 | 13.30 ± 1.86 |
| Compounds | IC50 (μM) | Compounds. | IC50 (μM) | Compounds | IC50 (μM) |
|---|---|---|---|---|---|
| 1 | NA | 10b | 39.96 ± 9.71 | 16a | 121.1 ± 3.18 |
| 2 | NA | 11a | 27.75 ± 2.82 | 16b | 243.4 ± 4.24 |
| 3 | NA | 11b | 61.80 ± 3.58 | 17a | 284.6 ± 4.53 |
| 4 | NA | 12a | 69.37 ± 3.32 | 17b | 106.31 ± 12.86 |
| 5 | NA | 12b | NA | 18a | 46.36 ± 7.92 |
| 6 | NA | 13a | 293.9 ± 26.87 | 18b | 212.9 ± 15.98 |
| 7 | NA | 13b | NA | 19 | NA |
| 8 | NA | 14a | NA | 20 | NA |
| 9a | 311.9 ± 10.18 | 14b | NA | 21 | NA |
| 9b | 304.6 ± 6.64 | 15a | NA | 22 | NA |
| 10a | 214.4 ± 16.33 | 15b | NA | Acarbose | 493.5 ± 8.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-Y.; Zhang, J.-S.; Wang, X.-X.; Tian, L.-L.; Li, Y.-P.; Wang, C.; Ma, R.-F.; Yin, Y.-K.; Bao, J.; Zhang, H. Identification of Small-Molecule Bioactive Constituents from the Leaves of Vaccinium bracteatum Confirms It as a Potential Functional Food with Health Benefits. Foods 2023, 12, 177. https://doi.org/10.3390/foods12010177
Wang Y-Y, Zhang J-S, Wang X-X, Tian L-L, Li Y-P, Wang C, Ma R-F, Yin Y-K, Bao J, Zhang H. Identification of Small-Molecule Bioactive Constituents from the Leaves of Vaccinium bracteatum Confirms It as a Potential Functional Food with Health Benefits. Foods. 2023; 12(1):177. https://doi.org/10.3390/foods12010177
Chicago/Turabian StyleWang, Yin-Yin, Jun-Sheng Zhang, Xin-Xin Wang, Lin-Lin Tian, Yu-Peng Li, Chao Wang, Ren-Fen Ma, Yi-Ke Yin, Jie Bao, and Hua Zhang. 2023. "Identification of Small-Molecule Bioactive Constituents from the Leaves of Vaccinium bracteatum Confirms It as a Potential Functional Food with Health Benefits" Foods 12, no. 1: 177. https://doi.org/10.3390/foods12010177