
IUP-BERT: Identification of Umami Peptides Based on BERT Features
 , and
, and
Abstract
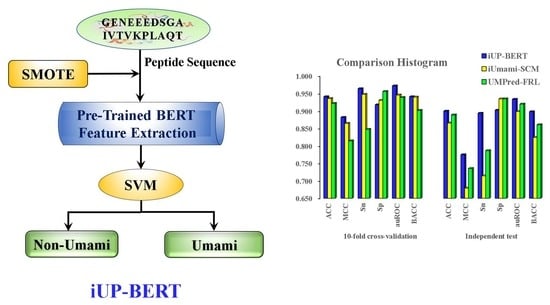
:
1. Introduction
2. Materials and Methods
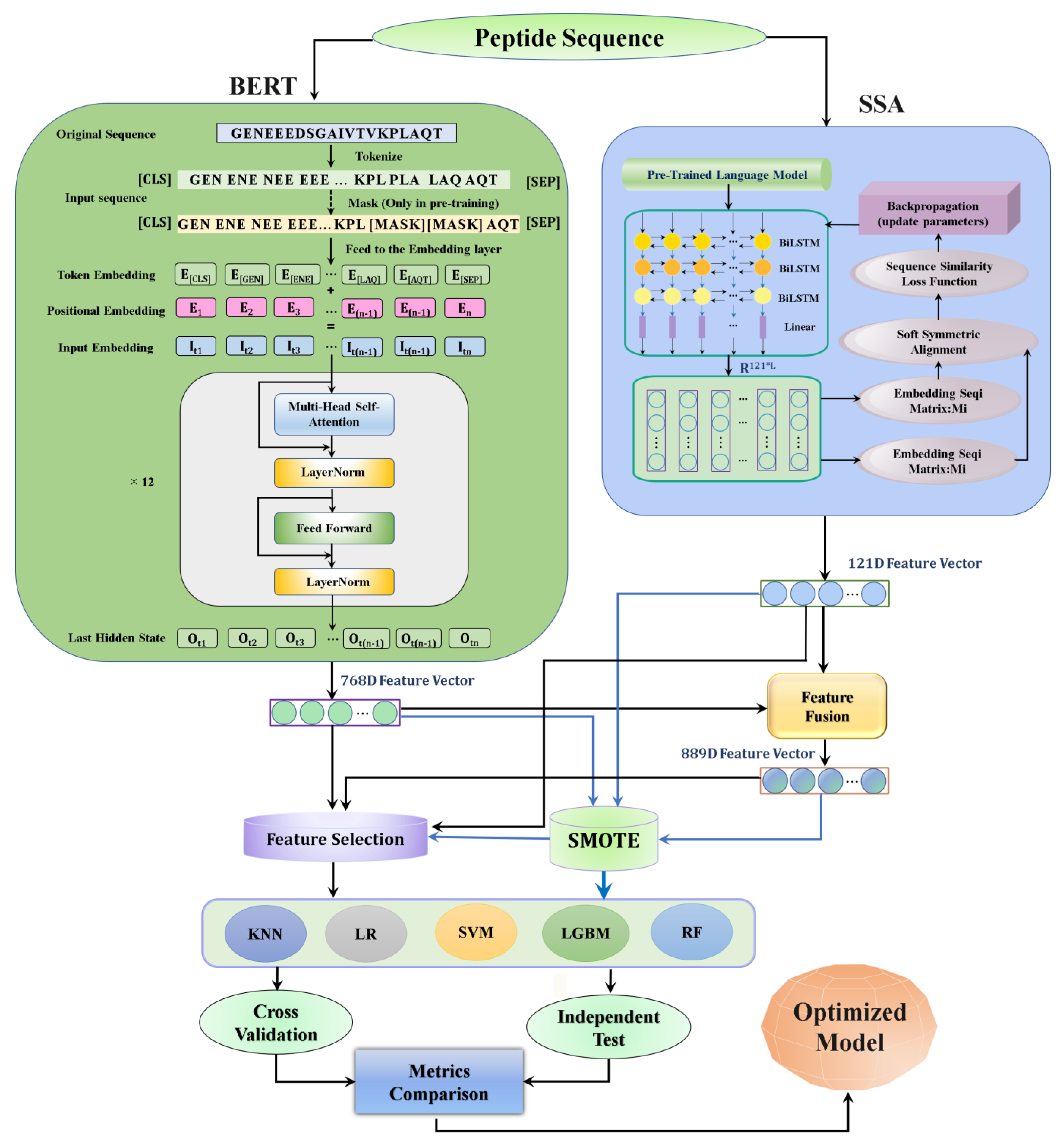
2.1. Overall Framework
- Upon the introduction of the peptide sequence, the pretrained sequence embedding technique, BERT, was used for feature extraction. For comparison, the SSA sequence embedding technique was included.
- After the feature extraction, BERT was fused with SSA to make an 889D fusion feature vector.
- The SMOTE was used to overcome the data imbalance.
- For feature space optimization, the LGBM feature technique method was used.
- Five different ML algorithms (KNN, LR, SVM, RF, and LGBM) were combined with the above techniques to build several models. The features of the BERT-SMOTE-SVM model were selected and applied to raise the prediction accuracy after optimization.
- The optimized feature representations were combined to establish the final iUP-BERT predictor.
2.2. Datasets
2.3. Feature Extraction
2.3.1. Pretrained SSA Embedding Model
2.3.2. Pretrained BERT Embedding Model
2.3.3. Feature Fusion
2.3.4. Synthetic Minority Oversampling Technique (SMOTE)
2.4. Machine Learning Methods
2.5. Performance Evaluation
3. Results and Discussion
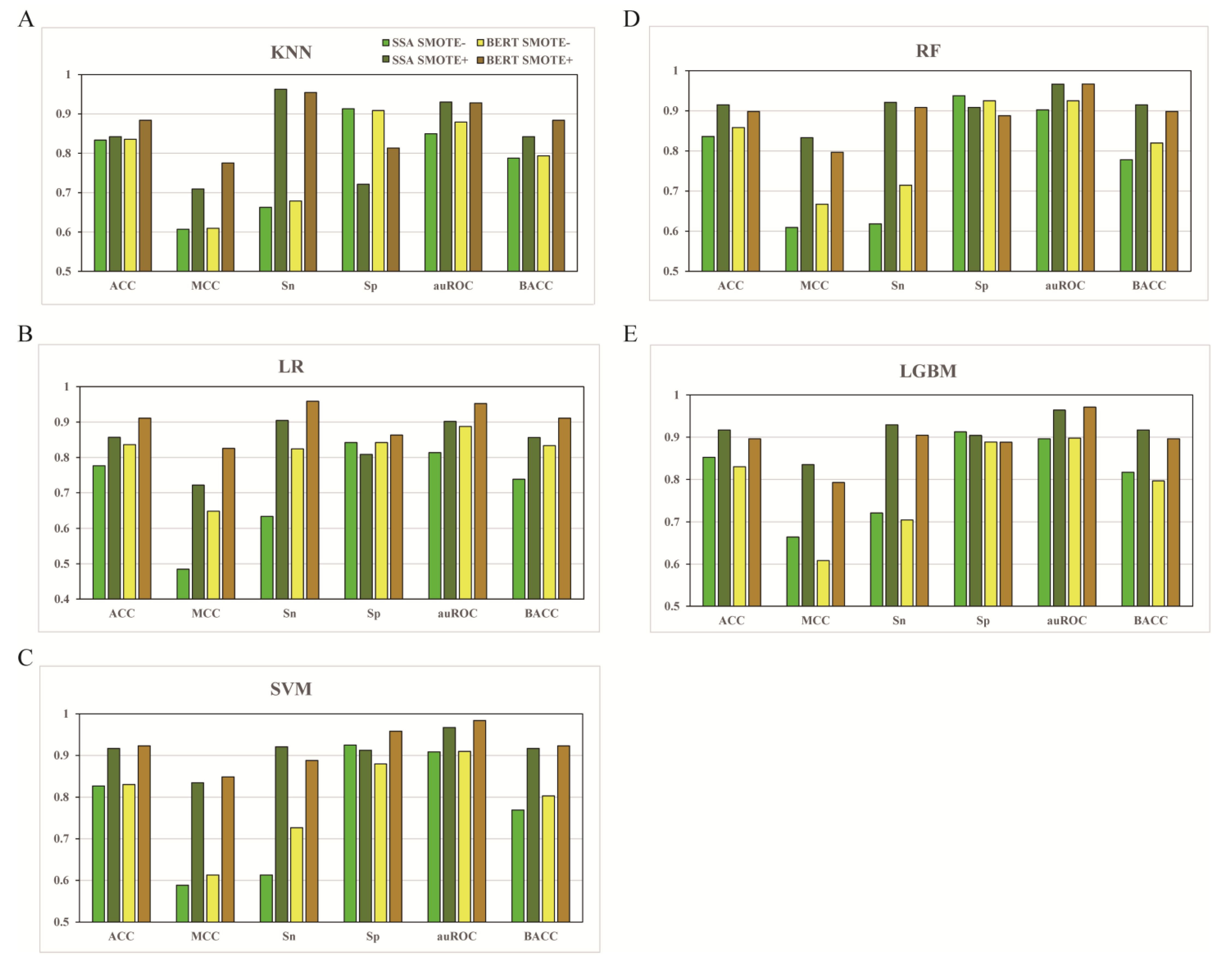
3.1. Preliminary Performance of Models Trained with or without SMOTE
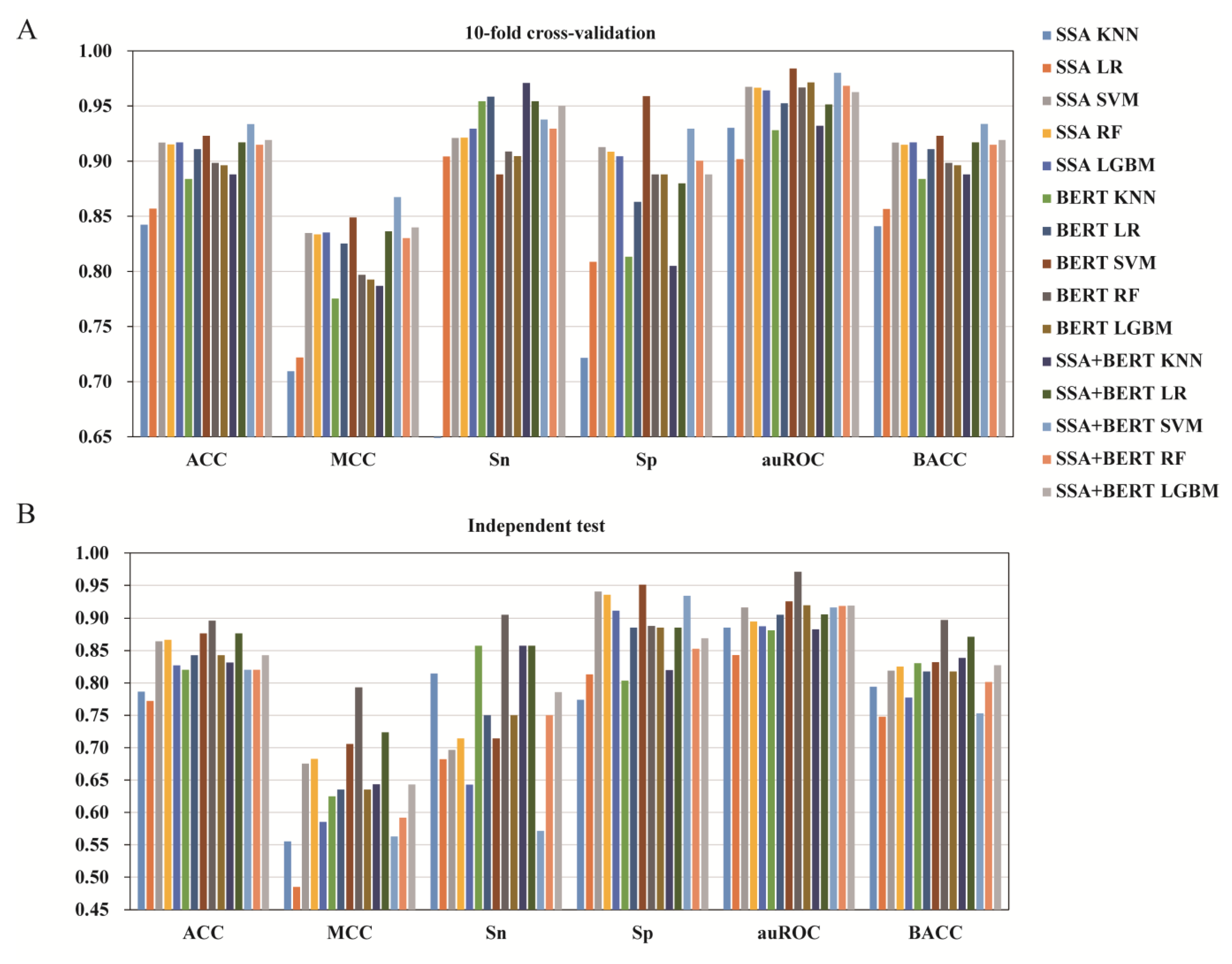
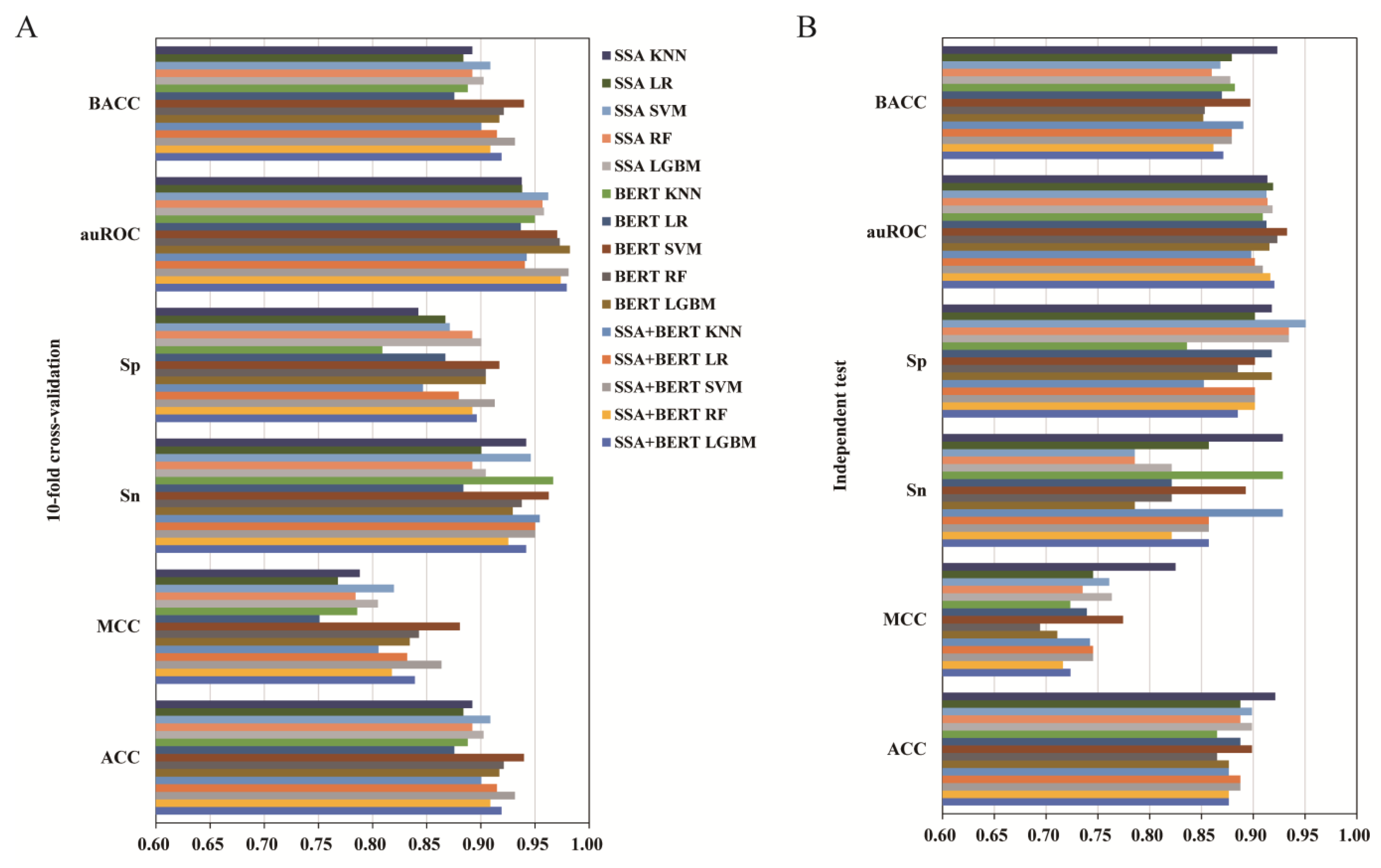
3.2. The Effect of Different Feature Types
3.3. The Effect of Feature Fusion
3.4. The Effect of Feature Selection
3.5. Comparison of iUP-BERT with Existing Models
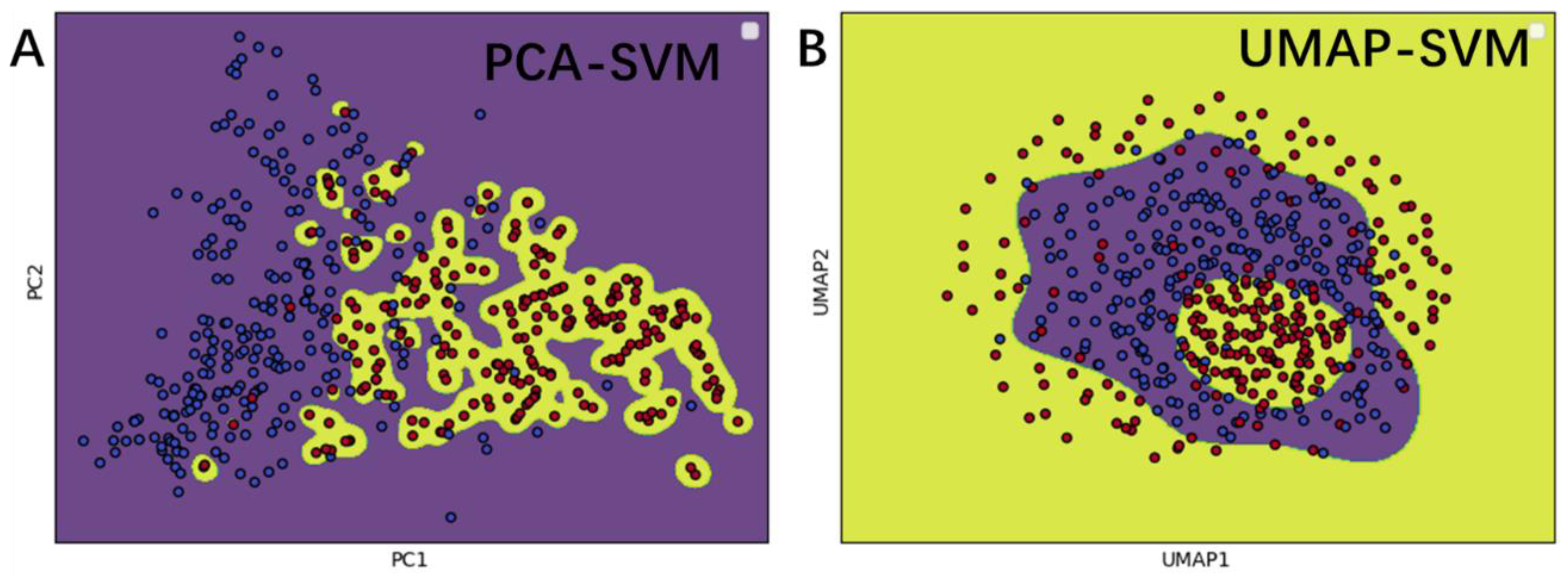
3.6. Feature Analysis Using Feature Projection and Decision Function
3.7. Construction of the Web Server of iUP-BERT
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviation
| ML | machine learning |
| BERT | bidirectional encoder representations from transformer |
| SSA | soft symmetric alignment |
| SMOTE | synthetic minority over-sampling technique |
| KNN | k-nearest neighbor |
| RF | random forest |
| SVM | support vector machine |
| LGBM | light gradient boosting machine |
| LR | logistic regression |
| SCM | scoring card method |
| ACC | accuracy |
| BACC | deduced balanced accuracy |
| Sn | sensitivity |
| Sp | specificity |
| MCC | Matthew’s coefficient correlation |
| ROC | receiver operating characteristic curve |
| auROC | area under the receiver operating characteristic curve |
| PCA | principal components analysis |
| UMAP | uniform manifold approximation and projection |
References
- Liang, L.; Zhou, C.; Zhang, J.; Huang, Y.; Zhao, J.; Sun, B.; Zhang, Y. Characteristics of umami peptides identified from porcine bone soup and molecular docking to the taste receptor T1R1/T1R3. Food Chem. 2022, 387, 132870. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Duan, W.; Zhang, J.; Huang, Y.; Zhang, Y.; Sun, B. Characterization and molecular docking study of taste peptides from chicken soup by sensory analysis combined with nano-LC-Q-TOF-MS/MS. Food Chem. 2022, 383, 132455. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhang, L.L.; Zhao, J.; Zhang, Y.Y.; Sun, B.G.; Chen, H.T. Isolation and identification of the umami peptides from shiitake mushroom by consecutive chromatography and LC-Q-TOF-MS. Food Res. Int. 2019, 121, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lioe, H.N.; Selamat, J.; Yasuda, M. Soy sauce and its umami taste: A link from the past to current situation. J. Food Sci. 2010, 75, R71–R76. [Google Scholar] [CrossRef] [PubMed]
- Shiyan, R.; Liping, S.; Xiaodong, S.; Jinlun, H.; Yongliang, Z. Novel umami peptides from tilapia lower jaw and molecular docking to the taste receptor T1R1/T1R3. Food Chem. 2021, 362, 130249. [Google Scholar] [CrossRef]
- Zhang, Y.; Venkitasamy, C.; Pan, Z.; Liu, W.; Zhao, L. Novel Umami Ingredients: Umami Peptides and Their Taste. J. Food Sci. 2017, 82, 16–23. [Google Scholar] [CrossRef]
- Temussi, P.A. The good taste of peptides. J. Pept. Sci. 2012, 18, 73–82. [Google Scholar] [CrossRef]
- Kondoh, T.; Torii, K. Brain activation by umami substances via gustatory and visceral signaling pathways, and physiological significance. Biol. Pharm. Bull. 2008, 31, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Spaggiari, G.; Di Pizio, A.; Cozzini, P. Sweet, umami and bitter taste receptors: State of the art of in silico molecular modeling approaches. Trends Food Sci. Technol. 2020, 96, 21–29. [Google Scholar] [CrossRef]
- Zhang, J.A.; Sun-Waterhouse, D.; Su, G.W.; Zhao, M.M. New insight into umami receptor, umami/umami-enhancing peptides and their derivatives: A review. Trends Food Sci. Technol. 2019, 88, 429–438. [Google Scholar] [CrossRef]
- Dang, Y.L.; Gao, X.C.; Xie, A.Y.; Wu, X.Q.; Ma, F.M. Interaction Between Umami Peptide and Taste Receptor T1R1/T1R3. Cell Biochem. Biophys. 2014, 70, 1841–1848. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Venkitasamy, C.; Pan, Z.; Ke, H.; Guo, S.; Wu, D.; Wu, W.; Zhao, L. Potential effects of umami ingredients on human health: Pros and cons. Crit. Rev. Food Sci. 2020, 60, 2294–2302. [Google Scholar] [CrossRef]
- Su, G.; Cui, C.; Zheng, L.; Yang, B.; Ren, J.; Zhao, M. Isolation and identification of two novel umami and umami-enhancing peptides from peanut hydrolysate by consecutive chromatography and MALDI-TOF/TOF MS. Food Chem. 2012, 135, 479–485. [Google Scholar] [CrossRef]
- Liu, H.; Da, L.T.; Liu, Y. Understanding the molecular mechanism of umami recognition by T1R1-T1R3 using molecular dynamics simulations. Biochem. Biophys. Res. Commun. 2019, 514, 967–973. [Google Scholar] [CrossRef]
- Dang, Y.L.; Hao, L.; Zhou, T.Y.; Cao, J.X.; Sun, Y.Y.; Pan, D.D. Establishment of new assessment method for the synergistic effect between umami peptides and monosodium glutamate using electronic tongue. Food Res. Int. 2019, 121, 20–27. [Google Scholar] [CrossRef]
- Yu, Z.; Jiang, H.; Guo, R.; Yang, B.; You, G.; Zhao, M.; Liu, X. Taste, umami-enhance effect and amino acid sequence of peptides separated from silkworm pupa hydrolysate. Food Res. Int. 2018, 108, 144–150. [Google Scholar] [CrossRef]
- Hao, L.; Gao, X.; Zhou, T.; Cao, J.; Sun, Y.; Dang, Y.; Pan, D. Angiotensin I-Converting Enzyme (ACE) Inhibitory and Antioxidant Activity of Umami Peptides after In Vitro Gastrointestinal Digestion. J. Agric. Food Chem. 2020, 68, 8232–8241. [Google Scholar] [CrossRef]
- Zhang, Y.; Pan, D.D.; Yang, Z.C.; Gao, X.C.; Dang, Y.L. Angiotensin I-Converting enzyme (ACE) inhibitory and dipeptidyl Peptidase-4 (DPP-IV) inhibitory activity of umami peptides from Ruditapes philippinarum. LWT-Food Sci. Technol. 2021, 144, 111265. [Google Scholar] [CrossRef]
- Dang, Y.L.; Gao, X.C.; Ma, F.M.; Wu, X.Q. Comparison of umami taste peptides in water-soluble extractions of Jinhua and Parma hams. LWT-Food Sci. Technol. 2015, 60, 1179–1186. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, M.; Su, G.; Lin, L. Identification and taste characteristics of novel umami and umami-enhancing peptides separated from peanut protein isolate hydrolysate by consecutive chromatography and UPLC-ESI-QTOF-MS/MS. Food Chem. 2019, 278, 674–682. [Google Scholar] [CrossRef]
- Qi, L.; Gao, X.; Pan, D.; Sun, Y.; Cai, Z.; Xiong, Y.; Dang, Y. Research progress in the screening and evaluation of umami peptides. Compr. Rev. Food Sci. Food Saf. 2022, 21, 1462–1490. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Kang, L.; Zhao, W.; Wu, S.; Ding, L.; Zheng, F.; Liu, J.; Li, J. Identification of novel umami peptides from myosin via homology modeling and molecular docking. Food Chem. 2021, 344, 128728. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Yana, J.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iUmami-SCM: A Novel Sequence-Based Predictor for Prediction and Analysis of Umami Peptides Using a Scoring Card Method with Propensity Scores of Dipeptides. J. Chem. Inf. Model. 2020, 60, 6666–6678. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Moni, M.A.; Manavalan, B.; Shoombuatong, W. UMPred-FRL: A New Approach for Accurate Prediction of Umami Peptides Using Feature Representation Learning. Int. J. Mol. Sci. 2021, 22, 13124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, X.; MacLeod, J.; Liu, J. Discerning novel splice junctions derived from RNA-seq alignment: A deep learning approach. BMC Genom. 2018, 19, 971. [Google Scholar] [CrossRef] [Green Version]
- Malebary, S.; Rahman, S.; Barukab, O.; Ash'ari, R.; Khan, S.A. iAcety-SmRF: Identification of Acetylation Protein by Using Statistical Moments and Random Forest. Membranes 2022, 12, 265. [Google Scholar] [CrossRef]
- Dai, R.; Zhang, W.; Tang, W.; Wynendaele, E.; Zhu, Q.; Bin, Y.; De Spiegeleer, B.; Xia, J. BBPpred: Sequence-Based Prediction of Blood-Brain Barrier Peptides with Feature Representation Learning and Logistic Regression. J. Chem. Inf. Model. 2021, 61, 525–534. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, Z.; Lee, T.Y. Incorporating support vector machine with sequential minimal optimization to identify anticancer peptides. BMC Bioinform. 2021, 22, 286. [Google Scholar] [CrossRef]
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.M.; Li, C.; Bernards, M.T.; Shi, Y.; Shao, Q.; He, Y. Sequence-based peptide identification, generation, and property prediction with deep learning: A review. Mol. Syst. Des. Eng. 2021, 6, 406–428. [Google Scholar] [CrossRef]
- Devlin, J.; Chang, M.-W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805L. [Google Scholar]
- Ji, Y.R.; Zhou, Z.H.; Liu, H.; Davuluri, R.V. DNABERT: Pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics 2021, 37, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Manavalan, B.; Shoombuatong, W. BERT4Bitter: A bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics 2021, 31, 2556–2562. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, J.; Zhao, L.; Zeng, X.; Liu, X. A novel antibacterial peptide recognition algorithm based on BERT. Brief. Bioinform. 2021, 22, bbab200. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, G.; Li, K.; Li, F.; Huang, L.; Duan, M.; Zhou, F. HLAB: Learning the BiLSTM features from the ProtBert-encoded proteins for the class I HLA-peptide binding prediction. Brief. Bioinform. 2022, 23, bbac173. [Google Scholar] [CrossRef]
- Bepler, T.; Berger, B. Learning protein sequence embeddings using information from structure. arXiv 2019, arXiv:1902.086613. [Google Scholar]
- Lv, Z.; Cui, F.; Zou, Q.; Zhang, L.; Xu, L. Anticancer peptides prediction with deep representation learning features. Brief. Bioinform. 2021, 22, bbab008. [Google Scholar] [CrossRef]
- Jiang, J.C.; Lin, X.X.; Jiang, Y.Q.; Jiang, L.Z.; Lv, Z.B. Identify Bitter Peptides by Using Deep Representation Learning Features. Int. J. Mol. Sci. 2022, 23, 7877. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 106. [Google Scholar] [CrossRef] [Green Version]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Database of Bioactive Peptides: Current Opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [Green Version]
- Charoenkwan, P.; Yana, J.; Schaduangrat, N.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iBitter-SCM: Identification and characterization of bitter peptides using a scoring card method with propensity scores of dipeptides. Genomics 2020, 112, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Moni, M.A.; Lio, P.; Shoombuatong, W. iBitter-Fuse: A Novel Sequence-Based Bitter Peptide Predictor by Fusing Multi-View Features. Int. J. Mol. Sci. 2021, 22, 8958. [Google Scholar] [CrossRef]
- Wang, S.; Dai, Y.; Shen, J.; Xuan, J. Research on expansion and classification of imbalanced data based on SMOTE algorithm. Sci. Rep. 2021, 11, 24039. [Google Scholar] [CrossRef]
- Akbar, S.; Ahmad, A.; Hayat, M.; Rehman, A.U.; Khan, S.; Ali, F. iAtbP-Hyb-EnC: Prediction of antitubercular peptides via heterogeneous feature representation and genetic algorithm based ensemble learning model. Comput. Biol. Med. 2021, 137, 104778. [Google Scholar] [CrossRef]
- Lin, D.; Yu, J.; Zhang, J.; He, H.; Guo, X.; Shi, S. PREDAIP: Computational Prediction and Analysis for Anti-inflammatory Peptide via a Hybrid Feature Selection Technique. Curr. Bioinform. 2021, 16, 1048–1059. [Google Scholar] [CrossRef]
- Mulpuru, V.; Semwal, R.; Varadwaj, P.K.; Mishra, N. HAMP: A Knowledgebase of Antimicrobial Peptides from Human Microbiome. Curr. Bioinform. 2021, 16, 534–540. [Google Scholar] [CrossRef]
- Sakib, M.M.H.; Nishat, A.A.; Islam, M.T.; Uddin, M.A.R.; Iqbal, M.S.; Bin Hossen, F.F.; Ahmed, M.I.; Bashir, M.S.; Hossain, T.; Tohura, U.S.; et al. Computational screening of 645 antiviral peptides against the receptor-binding domain of the spike protein in SARS-CoV-2. Comput. Biol. Med. 2021, 136, 104759. [Google Scholar] [CrossRef]
- Zhao, W.; Xu, G.; Yu, Z.; Li, J.; Liu, J. Identification of nut protein-derived peptides against SARS-CoV-2 spike protein and main protease. Comput. Biol. Med. 2021, 138, 104937. [Google Scholar] [CrossRef]
- Cao, C.; Wang, J.; Kwok, D.; Cui, F.; Zhang, Z.; Zhao, D.; Li, M.J.; Zou, Q. webTWAS: A resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res. 2022, 50, D1123–D1130. [Google Scholar] [CrossRef]
- Chen, Z.; Jiao, S.; Zhao, D.; Hesham, A.E.; Zou, Q.; Xu, L.; Sun, M.; Zhang, L. Sequence-Based Prediction with Feature Representation Learning and Biological Function Analysis of Channel Proteins. Front. Biosci. Landmark 2022, 27, 177. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Model | SMOTE | Dim | 10-Fold Cross-Validation | Independent Test | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | auROC | BACC | ACC | MCC | Sn | Sp | auROC | BACC | ||||
| SSA b | KNN c | − | 121 | 0.833 | 0.607 | 0.663 | 0.913 | 0.849 | 0.788 | 0.825 | 0.575 | 0.596 | 0.930 | 0.876 | 0.763 |
| LR c | − | 121 | 0.776 | 0.485 | 0.634 | 0.842 | 0.814 | 0.738 | 0.780 | 0.498 | 0.679 | 0.826 | 0.839 | 0.752 | |
| SVM c | − | 121 | 0.827 | 0.588 | 0.613 | 0.925 | 0.909 | 0.769 | 0.857 | 0.658 | 0.668 | 0.944 | 0.907 | 0.806 | |
| RF c | − | 121 | 0.836 | 0.609 | 0.618 | 0.938 | 0.902 | 0.778 | 0.826 | 0.578 | 0.557 | 0.949 | 0.879 | 0.753 | |
| LGBM c | − | 121 | 0.852 | 0.664 | 0.721 | 0.913 | 0.896 | 0.817 | 0.827 | 0.583 | 0.621 | 0.921 | 0.880 | 0.771 | |
| KNN c | + | 121 | 0.842 | 0.709 | 0.962a | 0.721 | 0.930 | 0.841 | 0.787 | 0.555 | 0.814 | 0.774 | 0.885 | 0.794 | |
| LR c | + | 121 | 0.857 | 0.722 | 0.904 | 0.809 | 0.902 | 0.856 | 0.772 | 0.485 | 0.682 | 0.813 | 0.843 | 0.748 | |
| SVM c | + | 121 | 0.917 | 0.835 | 0.921 | 0.913 | 0.967 | 0.917 | 0.864 | 0.675 | 0.696 | 0.941 | 0.916 | 0.819 | |
| RF c | + | 121 | 0.915 | 0.833 | 0.921 | 0.908 | 0.967 | 0.915 | 0.866 | 0.683 | 0.714 | 0.936 | 0.895 | 0.825 | |
| LGBM c | + | 121 | 0.917 | 0.835 | 0.929 | 0.904 | 0.964 | 0.917 | 0.827 | 0.585 | 0.643 | 0.911 | 0.887 | 0.777 | |
| BERT b | KNN c | − | 768 | 0.836 | 0.610 | 0.679 | 0.908 | 0.879 | 0.794 | 0.807 | 0.537 | 0.618 | 0.893 | 0.872 | 0.756 |
| LR c | − | 768 | 0.836 | 0.649 | 0.824 | 0.842 | 0.888 | 0.833 | 0.855 | 0.660 | 0.743 | 0.907 | 0.912 | 0.825 | |
| SVM c | − | 768 | 0.830 | 0.613 | 0.727 | 0.880 | 0.910 | 0.803 | 0.820 | 0.599 | 0.775 | 0.841 | 0.875 | 0.808 | |
| RF c | − | 768 | 0.859 | 0.667 | 0.714 | 0.925 | 0.925 | 0.820 | 0.819 | 0.567 | 0.643 | 0.900 | 0.900 | 0.771 | |
| LGBM c | − | 768 | 0.830 | 0.609 | 0.705 | 0.889 | 0.898 | 0.797 | 0.830 | 0.596 | 0.668 | 0.905 | 0.915 | 0.786 | |
| KNN c | + | 768 | 0.884 | 0.775 | 0.954 | 0.813 | 0.928 | 0.884 | 0.820 | 0.625 | 0.857 | 0.803 | 0.881 | 0.830 | |
| LR c | + | 768 | 0.911 | 0.825 | 0.959 | 0.863 | 0.952 | 0.911 | 0.843 | 0.635 | 0.750 | 0.885 | 0.905 | 0.818 | |
| SVM c | + | 768 | 0.923 | 0.849 | 0.888 | 0.959 | 0.984 | 0.923 | 0.876 | 0.706 | 0.714 | 0.951 | 0.926 | 0.832 | |
| RF c | + | 768 | 0.898 | 0.797 | 0.909 | 0.888 | 0.967 | 0.898 | 0.896 | 0.793 | 0.905 | 0.888 | 0.971 | 0.897 | |
| LGBM c | + | 768 | 0.896 | 0.793 | 0.905 | 0.888 | 0.971 | 0.896 | 0.843 | 0.635 | 0.750 | 0.885 | 0.920 | 0.818 | |
| Feature | Model | SMOTE | Dim | 10-Fold Cross-Validation | Independent Test | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | auROC | BACC | ACC | MCC | Sn | Sp | auROC | BACC | ||||
| SSA b + BERT b | KNN c | − | 889 | 0.836 | 0.610 | 0.679 | 0.909 | 0.908 | 0.794 | 0.820 | 0.576 | 0.679 | 0.885 | 0.900 | 0.782 |
| LR c | − | 889 | 0.844 | 0.640 | 0.750 | 0.888 | 0.900 | 0.819 | 0.876a | 0.716 | 0.821 | 0.902 | 0.910 | 0.862 | |
| SVM c | − | 889 | 0.858 | 0.667 | 0.732 | 0.917 | 0.921 | 0.825 | 0.854 | 0.658 | 0.750 | 0.902 | 0.906 | 0.826 | |
| RF c | − | 889 | 0.841 | 0.620 | 0.643 | 0.934 | 0.906 | 0.788 | 0.831 | 0.599 | 0.679 | 0.902 | 0.906 | 0.790 | |
| LGBM c | − | 889 | 0.813 | 0.553 | 0.625 | 0.900 | 0.892 | 0.763 | 0.831 | 0.606 | 0.714 | 0.885 | 0.921 | 0.800 | |
| KNN c | + | 889 | 0.888 | 0.787 | 0.971 | 0.805 | 0.932 | 0.888 | 0.831 | 0.643 | 0.857 | 0.820 | 0.883 | 0.838 | |
| LR c | + | 889 | 0.917 | 0.836 | 0.954 | 0.880 | 0.951 | 0.917 | 0.876 | 0.724 | 0.857 | 0.885 | 0.906 | 0.871 | |
| SVM c | + | 889 | 0.934 | 0.867 | 0.938 | 0.929 | 0.980 | 0.934 | 0.820 | 0.563 | 0.571 | 0.934 | 0.916 | 0.753 | |
| RF c | + | 889 | 0.915 | 0.830 | 0.929 | 0.900 | 0.968 | 0.915 | 0.820 | 0.592 | 0.750 | 0.852 | 0.919 | 0.801 | |
| LGBM c | + | 889 | 0.919 | 0.840 | 0.950 | 0.888 | 0.963 | 0.919 | 0.843 | 0.643 | 0.786 | 0.869 | 0.919 | 0.827 | |
| Feature | Model | SMOTE | Dim | 10-Fold Cross-Validation | Independent Test | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | auROC | BACC | ACC | MCC | Sn | Sp | auROC | BACC | ||||
| SSA b | KNN c | + | 43 | 0.892 | 0.788 | 0.942 | 0.842 | 0.938 | 0.892 | 0.921a | 0.825 | 0.929 | 0.918 | 0.914 | 0.923 |
| LR c | + | 41 | 0.884 | 0.768 | 0.900 | 0.867 | 0.938 | 0.884 | 0.888 | 0.745 | 0.857 | 0.902 | 0.919 | 0.879 | |
| SVM c | + | 29 | 0.909 | 0.820 | 0.946 | 0.871 | 0.962 | 0.909 | 0.899 | 0.761 | 0.786 | 0.951 | 0.913 | 0.868 | |
| RF c | + | 30 | 0.892 | 0.784 | 0.892 | 0.892 | 0.957 | 0.892 | 0.888 | 0.735 | 0.786 | 0.934 | 0.914 | 0.860 | |
| LGBM c | + | 39 | 0.902 | 0.805 | 0.905 | 0.900 | 0.958 | 0.902 | 0.899 | 0.763 | 0.821 | 0.934 | 0.919 | 0.878 | |
| BERT b | KNN c | + | 163 | 0.888 | 0.786 | 0.967 | 0.809 | 0.950 | 0.888 | 0.865 | 0.723 | 0.929 | 0.836 | 0.909 | 0.882 |
| LR c | + | 29 | 0.876 | 0.751 | 0.884 | 0.867 | 0.937 | 0.876 | 0.888 | 0.739 | 0.821 | 0.918 | 0.913 | 0.870 | |
| SVM c | + | 139 | 0.940 | 0.881 | 0.963 | 0.917 | 0.971 | 0.940 | 0.899 | 0.774 | 0.893 | 0.902 | 0.933 | 0.897 | |
| RF c | + | 79 | 0.921 | 0.843 | 0.938 | 0.905 | 0.973 | 0.921 | 0.865 | 0.694 | 0.821 | 0.885 | 0.923 | 0.853 | |
| LGBM c | + | 174 | 0.917 | 0.834 | 0.929 | 0.905 | 0.982 | 0.917 | 0.876 | 0.711 | 0.786 | 0.918 | 0.916 | 0.852 | |
| SSA b + BERT b | KNN c | + | 65 | 0.900 | 0.806 | 0.954 | 0.846 | 0.942 | 0.900 | 0.876 | 0.742 | 0.929 | 0.852 | 0.898 | 0.891 |
| LR c | + | 74 | 0.915 | 0.832 | 0.950 | 0.880 | 0.941 | 0.915 | 0.888 | 0.745 | 0.857 | 0.902 | 0.902 | 0.879 | |
| SVM c | + | 39 | 0.932 | 0.864 | 0.950 | 0.913 | 0.981 | 0.932 | 0.888 | 0.745 | 0.857 | 0.902 | 0.909 | 0.879 | |
| RF c | + | 168 | 0.909 | 0.818 | 0.925 | 0.892 | 0.974 | 0.909 | 0.876 | 0.716 | 0.821 | 0.902 | 0.917 | 0.862 | |
| LGBM c | + | 114 | 0.919 | 0.839 | 0.942 | 0.896 | 0.979 | 0.919 | 0.876 | 0.724 | 0.857 | 0.885 | 0.920 | 0.871 | |
| Classifier | 10-Fold Cross-Validation | Independent Test | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | auROC | BACC | ACC | MCC | Sn | Sp | auROC | BACC | |
| iUP-BERT | 0.940a | 0.881 | 0.963 | 0.917 | 0.971 | 0.940 | 0.899 | 0.774 | 0.893 | 0.902 | 0.933 | 0.897 |
| iUmami-SCM | 0.935 | 0.864 | 0.947 | 0.930 | 0.945 | 0.939 | 0.865 | 0.679 | 0.714 | 0.934 | 0.898 | 0.824 |
| UMPred-FRL | 0.921 | 0.814 | 0.847 | 0.955 | 0.938 | 0.901 | 0.888 | 0.735 | 0.786 | 0.934 | 0.919 | 0.860 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/)
Share and Cite
Jiang, L.; Jiang, J.; Wang, X.; Zhang, Y.; Zheng, B.; Liu, S.; Zhang, Y.; Liu, C.; Wan, Y.; Xiang, D.; et al. IUP-BERT: Identification of Umami Peptides Based on BERT Features. Foods 2022, 11, 3742. https://doi.org/10.3390/foods11223742
Jiang L, Jiang J, Wang X, Zhang Y, Zheng B, Liu S, Zhang Y, Liu C, Wan Y, Xiang D, et al. IUP-BERT: Identification of Umami Peptides Based on BERT Features. Foods. 2022; 11(22):3742. https://doi.org/10.3390/foods11223742
Chicago/Turabian StyleJiang, Liangzhen, Jici Jiang, Xiao Wang, Yin Zhang, Bowen Zheng, Shuqi Liu, Yiting Zhang, Changying Liu, Yan Wan, Dabing Xiang, and et al. 2022. "IUP-BERT: Identification of Umami Peptides Based on BERT Features" Foods 11, no. 22: 3742. https://doi.org/10.3390/foods11223742