
Adsorption of Metal Atoms on SiC Monolayer
Abstract
:1. Introduction
2. Computational Details
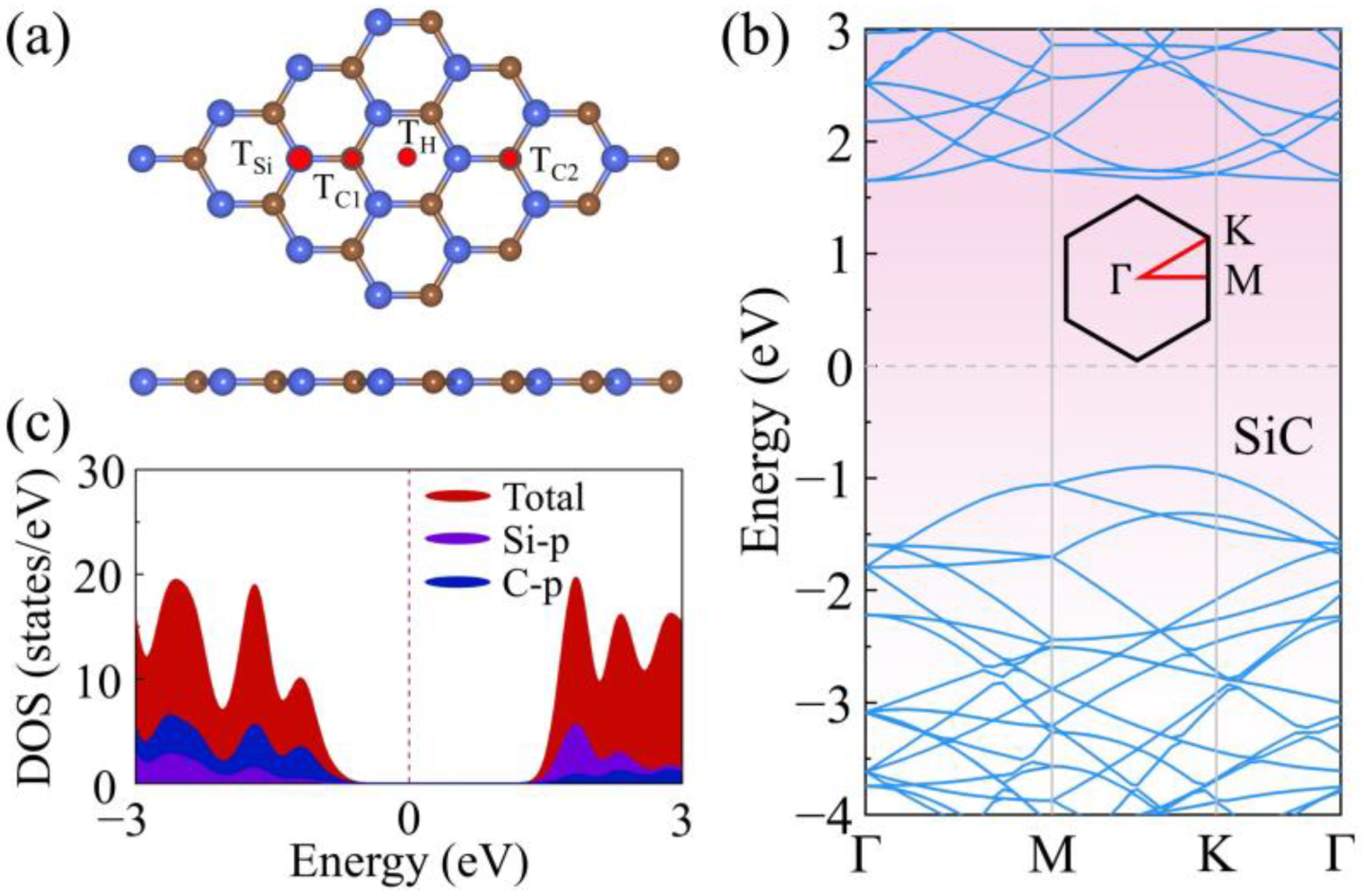
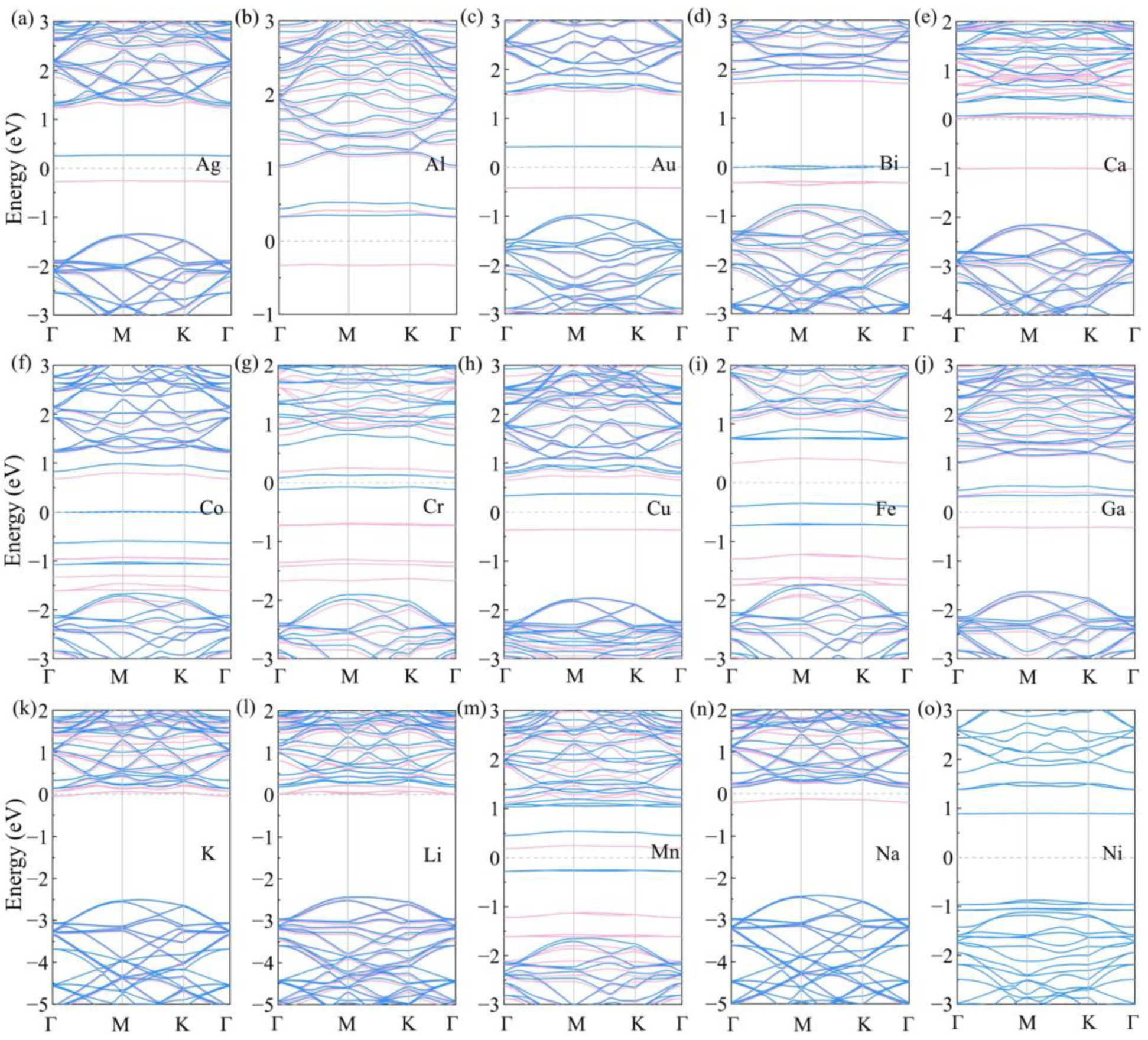
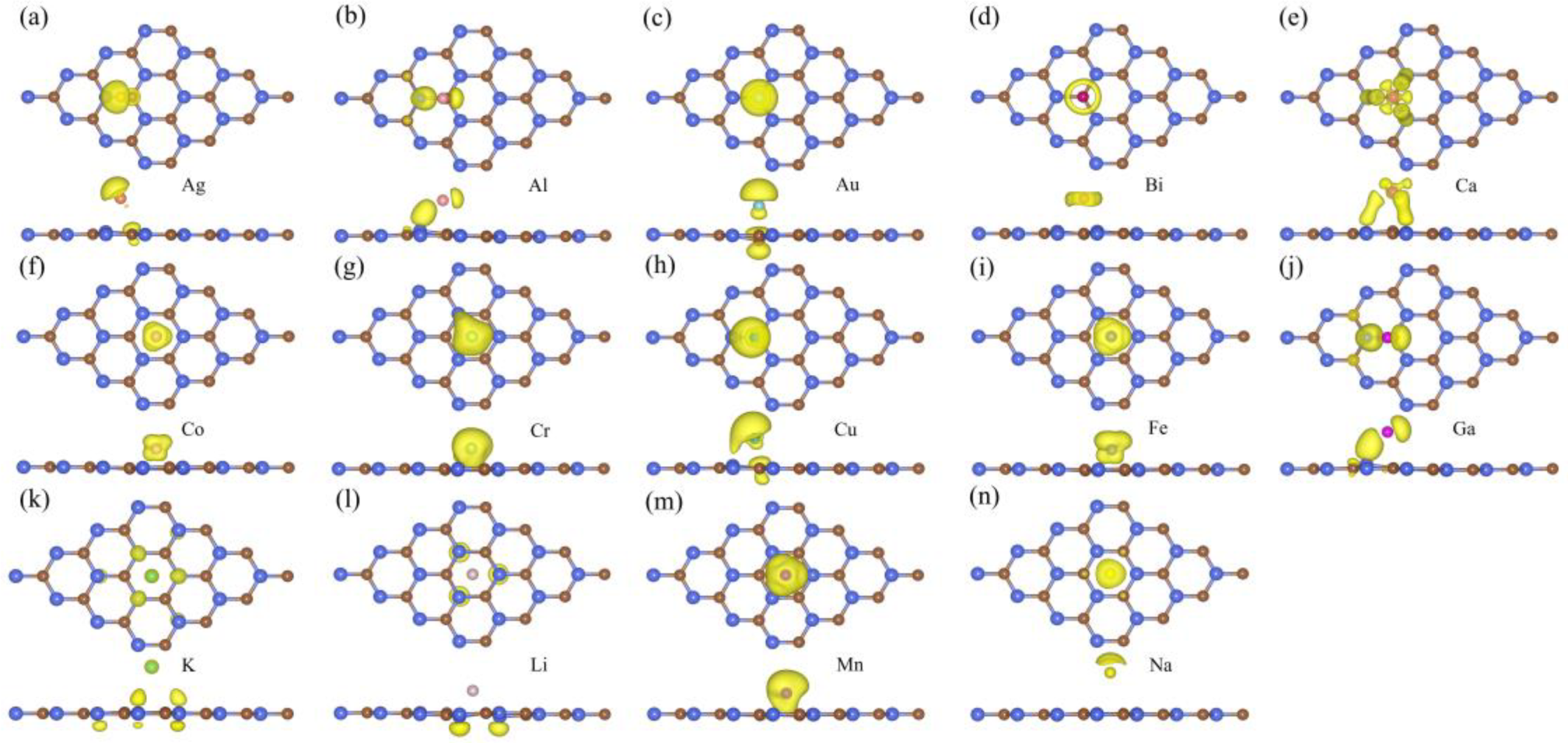
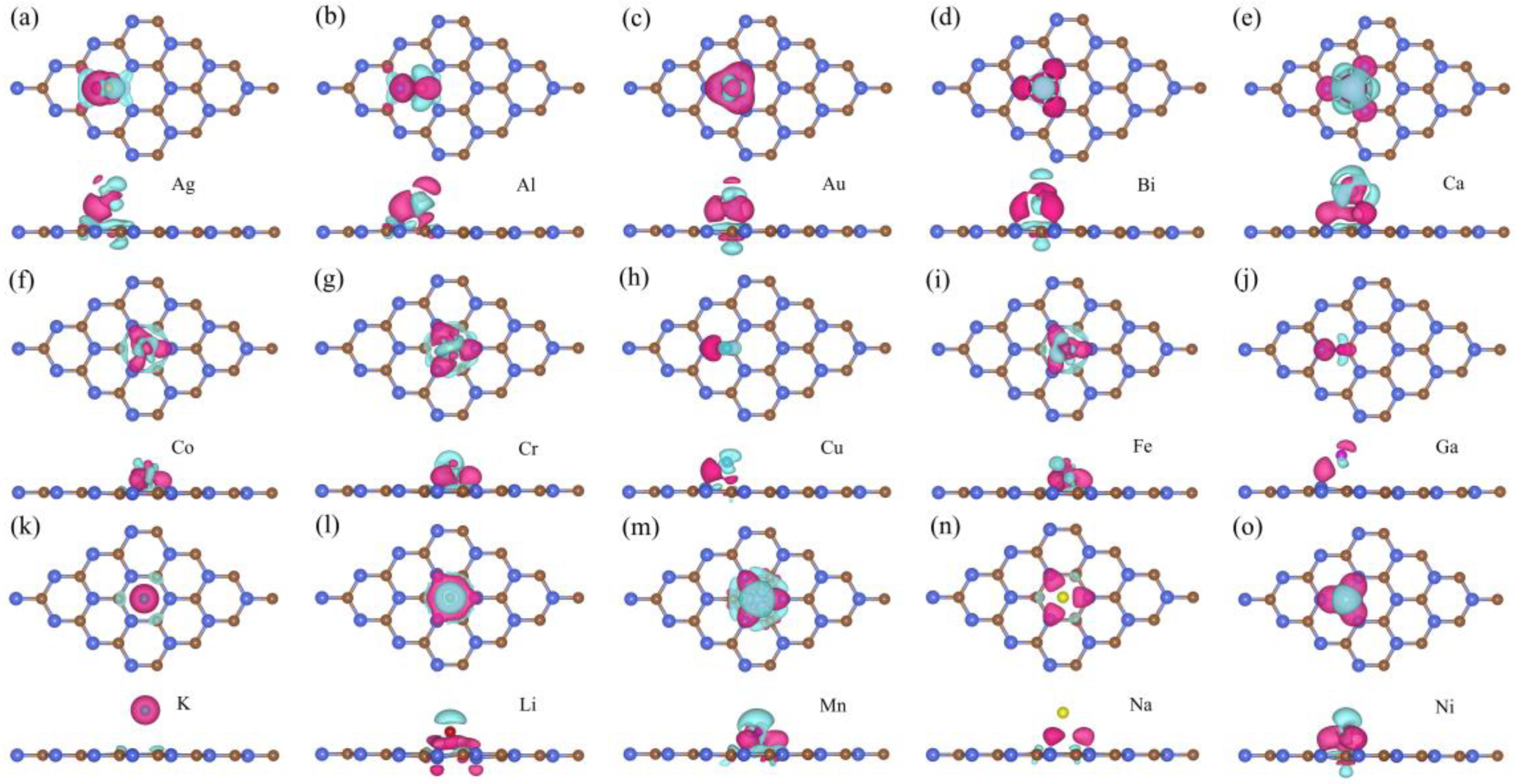
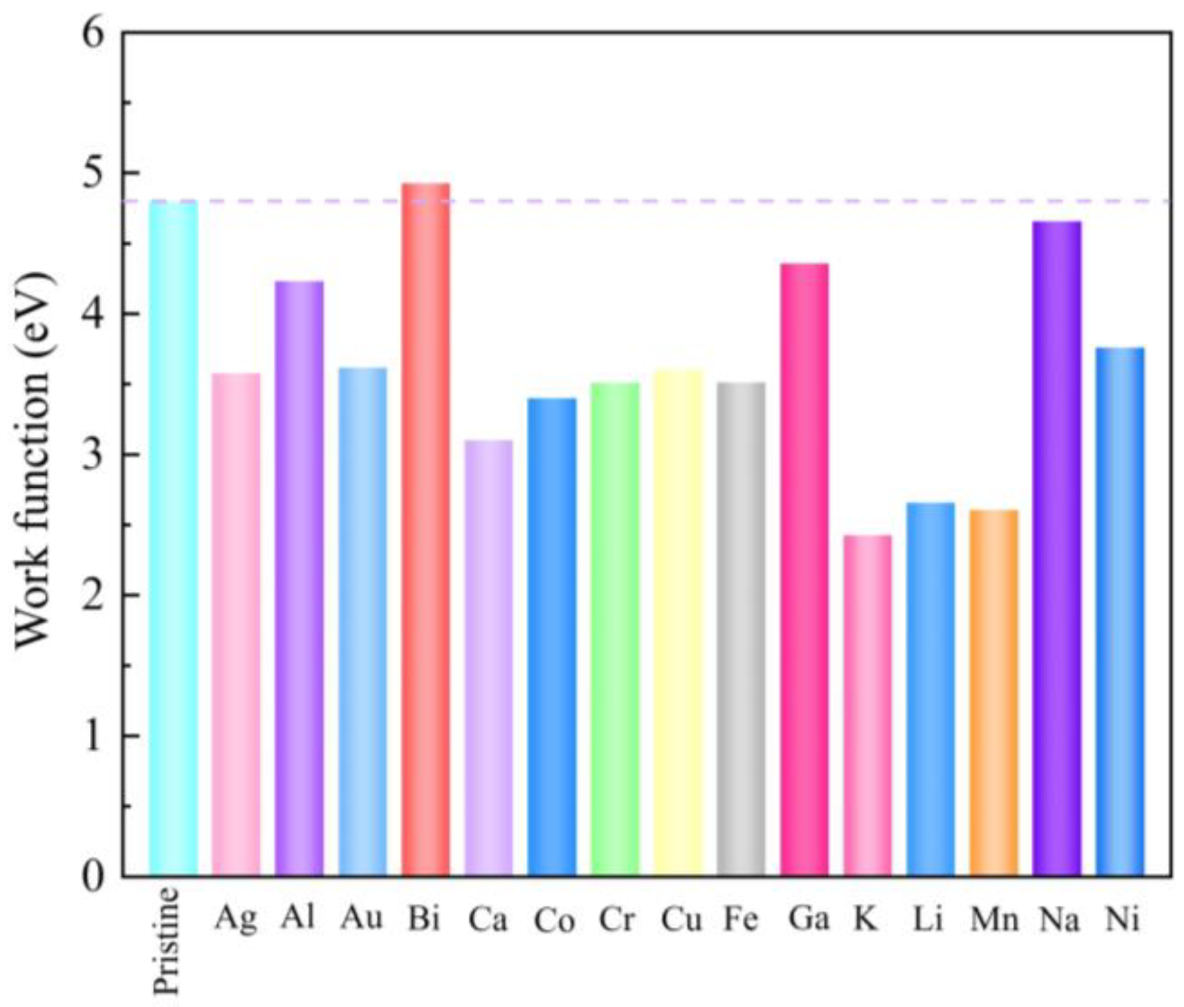
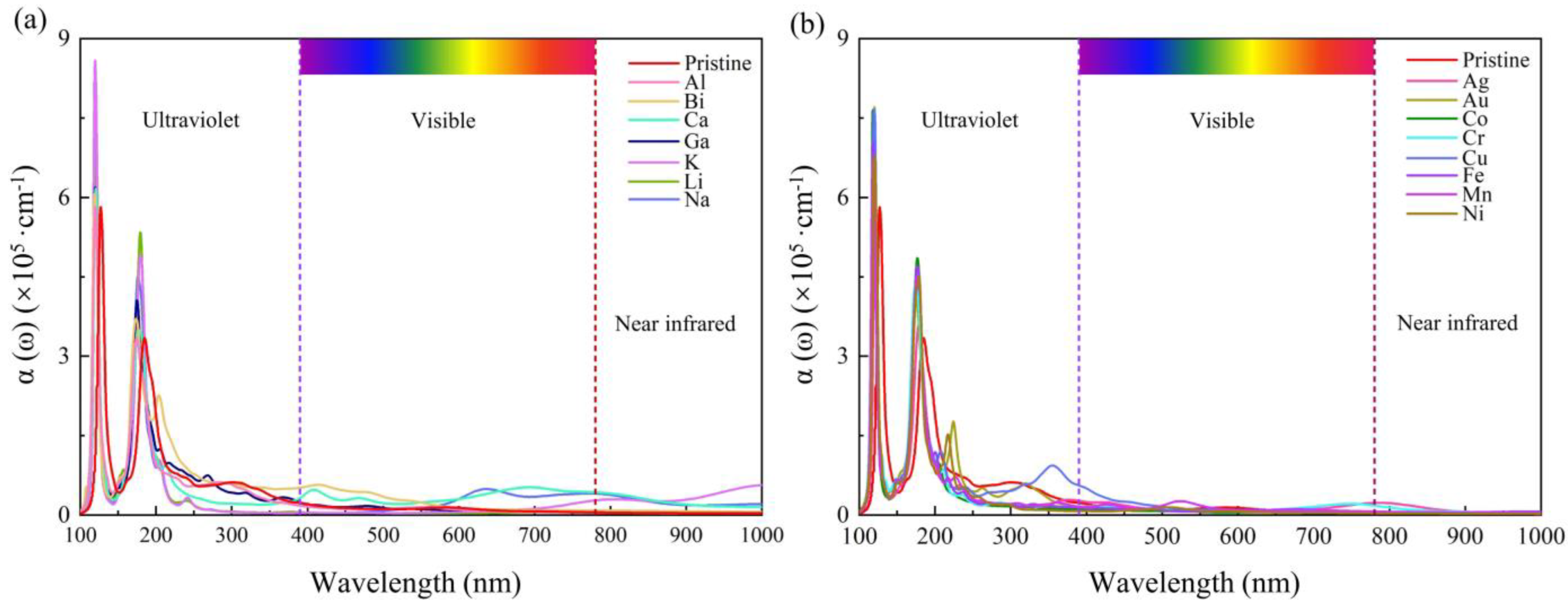
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef]
- Cui, Z.; Yang, K.; Shen, Y.; Yuan, Z.; Dong, Y.; Yuan, P.; Li, E. Toxic gas molecules adsorbed on intrinsic and defective WS2: Gas sensing and detection. Appl. Surf. Sci. 2023, 613, 155978. [Google Scholar] [CrossRef]
- Xia, S.; Wang, Y.; Shi, H.; Diao, Y.; Kan, C. Structural and electronic properties of nanoclusters (Xn, X = Au, Ag, Al; n = 1–4) adsorption on GaN/WS2 van der Waals heterojunction: A first principle study. Appl. Surf. Sci. 2022, 605, 154716. [Google Scholar] [CrossRef]
- Sanders, N.; Bayerl, D.; Shi, G.; Mengle, K.A.; Kioupakis, E. Electronic and optical properties of two-dimensional GaN from first-principles. Nano Lett. 2017, 17, 7345–7349. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dai, J.; Li, J.; Zhang, S.; Zhou, J.; Zhang, L.; Chu, W.; Chen, D.; Zhao, H.; Yang, J. Electronic structures and magnetic properties of GaN sheets and nanoribbons. J. Phys. Chem. C 2010, 114, 11390–11394. [Google Scholar] [CrossRef]
- Dong, Y.; Li, E.; Cui, Z.; Shen, Y.; Ma, D.; Wang, F.; Yuan, Z.; Yang, K. Electronic properties and photon scattering of buckled and planar few-layer 2D GaN. Vacuum 2023, 210, 111861. [Google Scholar] [CrossRef]
- Tang, Q.; Bao, J.; Li, Y.; Zhou, Z.; Chen, Z. Tuning band gaps of BN nanosheets and nanoribbons via interfacial dihalogen bonding and external electric field. Nanoscale 2014, 6, 8624–8634. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Li, S.; Zhao, P.; Xing, S.; Zhuo, R.; Liang, J. Effects of strain and Al doping on monolayer h-BN: First-principles calculations. Phys. E 2023, 146, 115546. [Google Scholar] [CrossRef]
- Ling, X.; Wang, H.; Huang, S.; Xia, F.; Dresselhaus, M.S. The renaissance of black phosphorus. Proc. Nat. Acad. Sci. USA 2015, 112, 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Han, R.; Feng, S.; Sun, D.-M.; Cheng, H.-M. Properties and photodetector applications of two-dimensional black arsenic phosphorus and black phosphorus. Sci. China Inform. Sci. 2021, 64, 140402. [Google Scholar] [CrossRef]
- Wang, S.; Ren, C.; Tian, H.; Yu, J.; Sun, M. MoS2/ZnO van der Waals heterostructure as a high-efficiency water splitting photocatalyst: A first-principles study. Phys. Chem. Chem. Phys. 2018, 20, 13394–13399. [Google Scholar] [CrossRef]
- Xia, S.; Diao, Y.; Kan, C. Electronic and optical properties of two-dimensional GaN/ZnO heterojunction tuned by different stacking configurations. J. Colloid Interface Sci. 2022, 607, 913–921. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, Z. Electronic, Magnetic, and Optical Performances of Non-Metals Doped Silicon Carbide. Front. Chem. 2022, 10, 898174. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, M.; Chou, J.-P.; Wei, J.; Xing, H.; Hu, A. First-principles calculations of the electronic properties of SiC-based bilayer and trilayer heterostructures. Phys. Chem. Chem. Phys. 2018, 20, 24726–24734. [Google Scholar] [CrossRef]
- Zhan, J.; Yao, X.; Li, W.; Zhang, X. Tensile mechanical properties study of SiC/graphene composites based on molecular dynamics. Comp. Mater. Sci. 2017, 131, 266–274. [Google Scholar] [CrossRef]
- Yan, W.-J.; Xie, Q.; Qin, X.-M.; Zhang, C.-H.; Zhang, Z.-Z.; Zhou, S.-Y. First-principle analysis of photoelectric properties of silicon-carbon materials with graphene-like honeycomb structure. Comp. Mater. Sci. 2017, 126, 336–343. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, Z.; Kutana, A.; Yakobson, B.I. Predicting two-dimensional silicon carbide monolayers. ACS Nano 2015, 9, 9802–9809. [Google Scholar] [CrossRef] [PubMed]
- Chabi, S.; Chang, H.; Xia, Y.; Zhu, Y. From graphene to silicon carbide: Ultrathin silicon carbide flakes. Nanotechnology 2016, 27, 075602. [Google Scholar] [CrossRef] [PubMed]
- Lin, S. Light-emitting two-dimensional ultrathin silicon carbide. J. Phys. Chem. C 2012, 116, 3951–3955. [Google Scholar] [CrossRef]
- Zhao, Q.; Xiong, Z.; Qin, Z.; Chen, L.; Wu, N.; Li, X. Tuning magnetism of monolayer GaN by vacancy and nonmagnetic chemical doping. J. Phys. Chem. Solids 2016, 91, 1–6. [Google Scholar] [CrossRef]
- Zhao, Q.; Xiong, Z.; Luo, L.; Sun, Z.; Qin, Z.; Chen, L.; Wu, N. Design of a new two-dimensional diluted magnetic semiconductor: Mn-doped GaN monolayer. Appl. Surf. Sci. 2017, 396, 480–483. [Google Scholar] [CrossRef]
- Chernozatonskii, L.A.; Katin, K.P.; Kochaev, A.I.; Maslov, M.M. Moiré and non-twisted sp3-hybridized structures based on hexagonal boron nitride bilayers: Ab initio insight into infrared and Raman spectra, bands structures and mechanical properties. Appl. Surf. Sci. 2022, 606, 154909. [Google Scholar] [CrossRef]
- Cai, X.; Deng, S.; Li, L.; Hao, L. A first-principles theoretical study of the electronic and optical properties of twisted bilayer GaN structures. J. Comput. Electron. 2020, 19, 910–916. [Google Scholar] [CrossRef]
- Mu, Y. Chemical functionalization of GaN monolayer by adatom adsorption. J. Phys. Chem. C 2015, 119, 20911–20916. [Google Scholar] [CrossRef]
- Tang, W.; Sun, M.; Yu, J.; Chou, J.-P. Magnetism in non-metal atoms adsorbed graphene-like gallium nitride monolayers. Appl. Surf. Sci. 2018, 427, 609–612. [Google Scholar] [CrossRef]
- Islam, S.; Lee, K.; Verma, J.; Protasenko, V.; Rouvimov, S.; Bharadwaj, S.; Xing, H.; Jena, D. MBE-grown 232–270 nm deep-UV LEDs using monolayer thin binary GaN/AlN quantum heterostructures. Appl. Phys. Lett. 2017, 110, 041108. [Google Scholar] [CrossRef]
- Cui, Z.; Ren, K.; Zhao, Y.; Wang, X.; Shu, H.; Yu, J.; Tang, W.; Sun, M. Electronic and optical properties of van der Waals heterostructures of g-GaN and transition metal dichalcogenides. Appl. Surf. Sci. 2019, 492, 513–519. [Google Scholar] [CrossRef]
- Ren, K.; Wang, S.; Luo, Y.; Xu, Y.; Sun, M.; Yu, J.; Tang, W. Strain-enhanced properties of van der Waals heterostructure based on blue phosphorus and g-GaN as a visible-light-driven photocatalyst for water splitting. RSC Adv. 2019, 9, 4816–4823. [Google Scholar] [CrossRef]
- Nie, K.; Wang, X.; Mi, W. Electronic structure and magnetic properties of 3d transition-metal atom adsorbed SnO monolayers. Appl. Surf. Sci. 2019, 493, 404–410. [Google Scholar] [CrossRef]
- Guo, J.-X.; Wu, S.-Y.; Zhong, S.-Y.; Zhang, G.-J.; Shen, G.-Q.; Yu, X.-Y. Janus WSSe monolayer adsorbed with transition-metal atoms (Fe, Co and Ni): Excellent performance for gas sensing and CO catalytic oxidation. Appl. Surf. Sci. 2021, 565, 150558. [Google Scholar] [CrossRef]
- Cui, Z.; Zhang, S.; Wang, L.; Yang, K. Optoelectronic and magnetic properties of transition metals adsorbed Pd2Se3 monolayer. Micro Nanostruct. 2022, 167, 207260. [Google Scholar] [CrossRef]
- Xu, B.; Chen, C.; Liu, X.; Ma, S.; Zhang, J.; Wang, Y.; Li, J.; Gu, Z.; Yi, L. Magnetic properties and electronic structure of 3d transition-metal atom adsorbed two-dimensional SnSe2. J. Magn. Magn. Mater. 2022, 562, 169817. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Hafner, J. Ab-initio simulations of materials using VASP: Density-functional theory and beyond. J. Comput. Chem. 2008, 29, 2044–2078. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Hybertsen, M.S.; Louie, S.G. Electron correlation in semiconductors and insulators: Band gaps and quasiparticle energies. Phys. Rev. B 1986, 34, 5390. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yong, Y.; Zhou, Q.; Kuang, Y.; Li, X. Gas-sensing properties of the SiC monolayer and bilayer: A density functional theory study. ACS Omega 2020, 5, 12364–12373. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Arnaldsson, A.; Jónsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comput. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Ehrenreich, H.; Cohen, M.H. Self-consistent field approach to the many-electron problem. Phys. Rev. 1959, 115, 786. [Google Scholar] [CrossRef]
- Toll, J.S. Causality and the dispersion relation: Logical foundations. Phys. Rev. 1956, 104, 1760. [Google Scholar] [CrossRef]
- Fox, M.; Bertsch, G.F. Optical properties of solids. Am. Assoc. Phys. Teach. 2002, 70, 1269–1270. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Style | Sx | Ead (eV) | D (Å) | Eg (eV) | M (μB) | C (e) |
|---|---|---|---|---|---|---|
| Ag | SC2 | −0.193 | 2.36 | 0.521 | 1.00 | +0.446 |
| Al | SC1 | −1.683 | 2.44 | 0.659 | 1.00 | −0.588 |
| Au | SC1 | −1.706 | 2.04 | 0.837 | 1.00 | +0.319 |
| Bi | SC1 | −1.135 | 2.45 | 0 | 1.01 | −0.109 |
| Ca | SC1 | −0.111 | 2.87 | 0 | 1.95 | −0.766 |
| Co | SH | −2.785 | 1.30 | 0 | 1.00 | −0.110 |
| Cr | SH | −2.545 | 1.42 | 0.199 | 4.26 | −0.560 |
| Cu | SC1 | −1.388 | 2.06 | 0.705 | 1.00 | +0.023 |
| Fe | SC1 | −2.104 | 1.36 | 0.734 | 2.00 | −0.280 |
| Ga | SC1 | −1.493 | 2.54 | 0.640 | 1.00 | −0.292 |
| K | SH | −5.357 | 3.07 | 0 | 0.99 | +1.455 |
| Li | SH | −1.026 | 1.61 | 0 | 1.00 | −0.867 |
| Mn | SH | −0.956 | 1.46 | 0.494 | 3.00 | −0.468 |
| Na | SH | −0.301 | 2.84 | 0.442 | 1.00 | −0.391 |
| Ni | SH | −3.845 | 1.61 | 1.754 | 0 | +0.045 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Dong, Y.; Cui, Z. Adsorption of Metal Atoms on SiC Monolayer. Inorganics 2023, 11, 240. https://doi.org/10.3390/inorganics11060240
Jiang L, Dong Y, Cui Z. Adsorption of Metal Atoms on SiC Monolayer. Inorganics. 2023; 11(6):240. https://doi.org/10.3390/inorganics11060240
Chicago/Turabian StyleJiang, Lei, Yanbo Dong, and Zhen Cui. 2023. "Adsorption of Metal Atoms on SiC Monolayer" Inorganics 11, no. 6: 240. https://doi.org/10.3390/inorganics11060240