
Covalent Grafting of Eosin Y to the Giant Keplerate {Mo132} through an Organosilicon Linker in Homogeneous Regime
 , ,
, ,  ,
,
Abstract
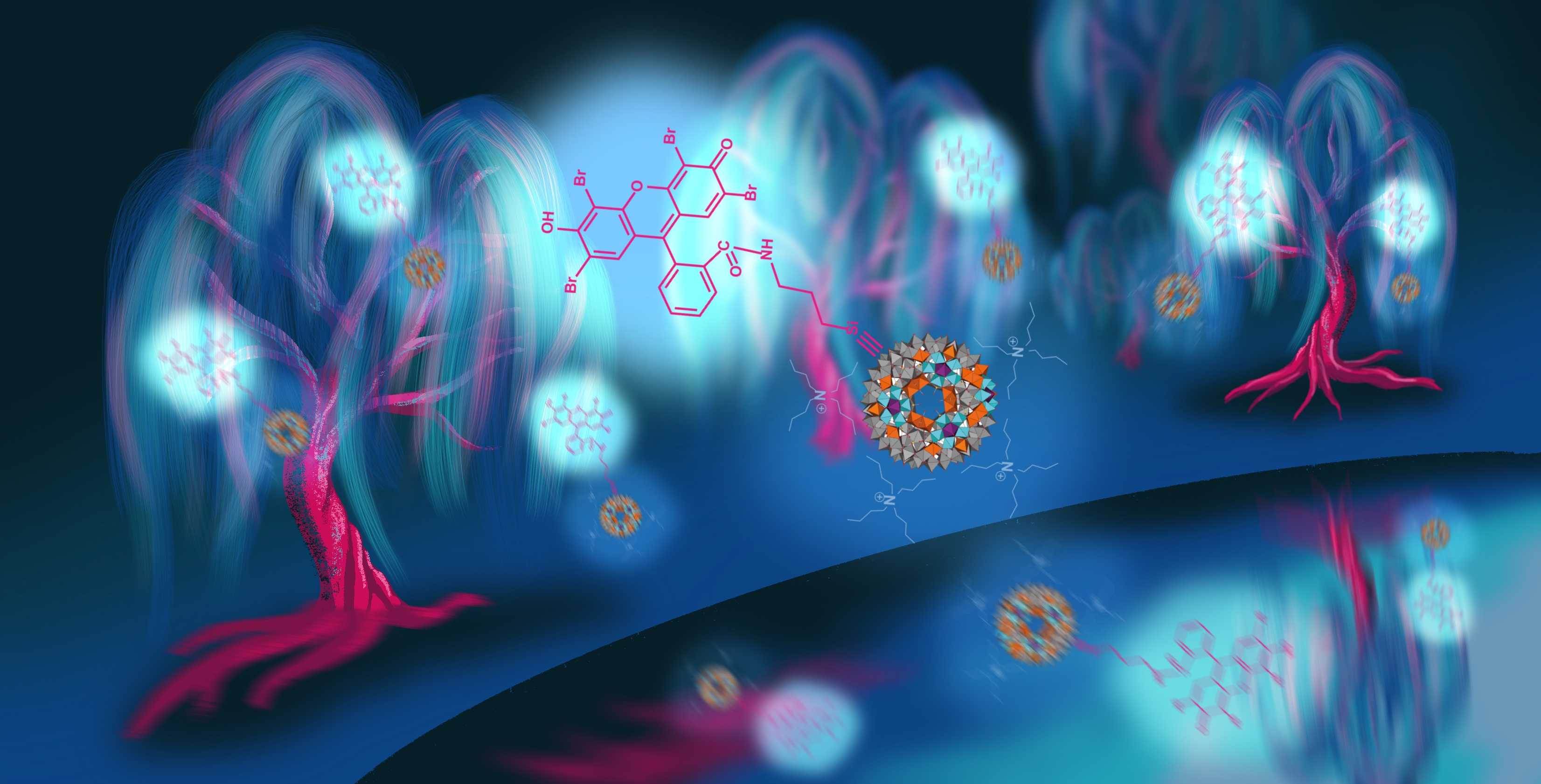
:
1. Introduction
2. Results and Discussion
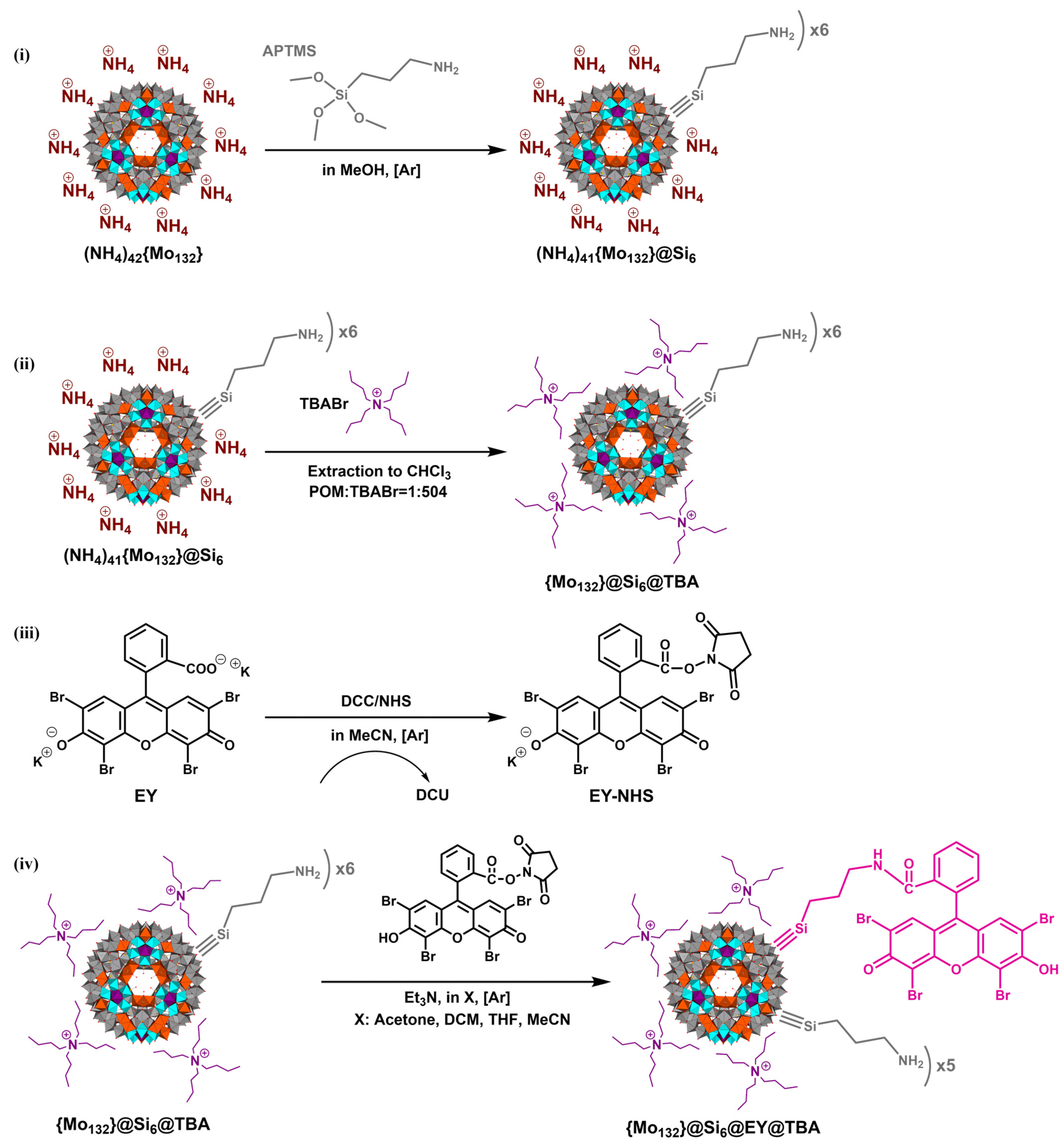
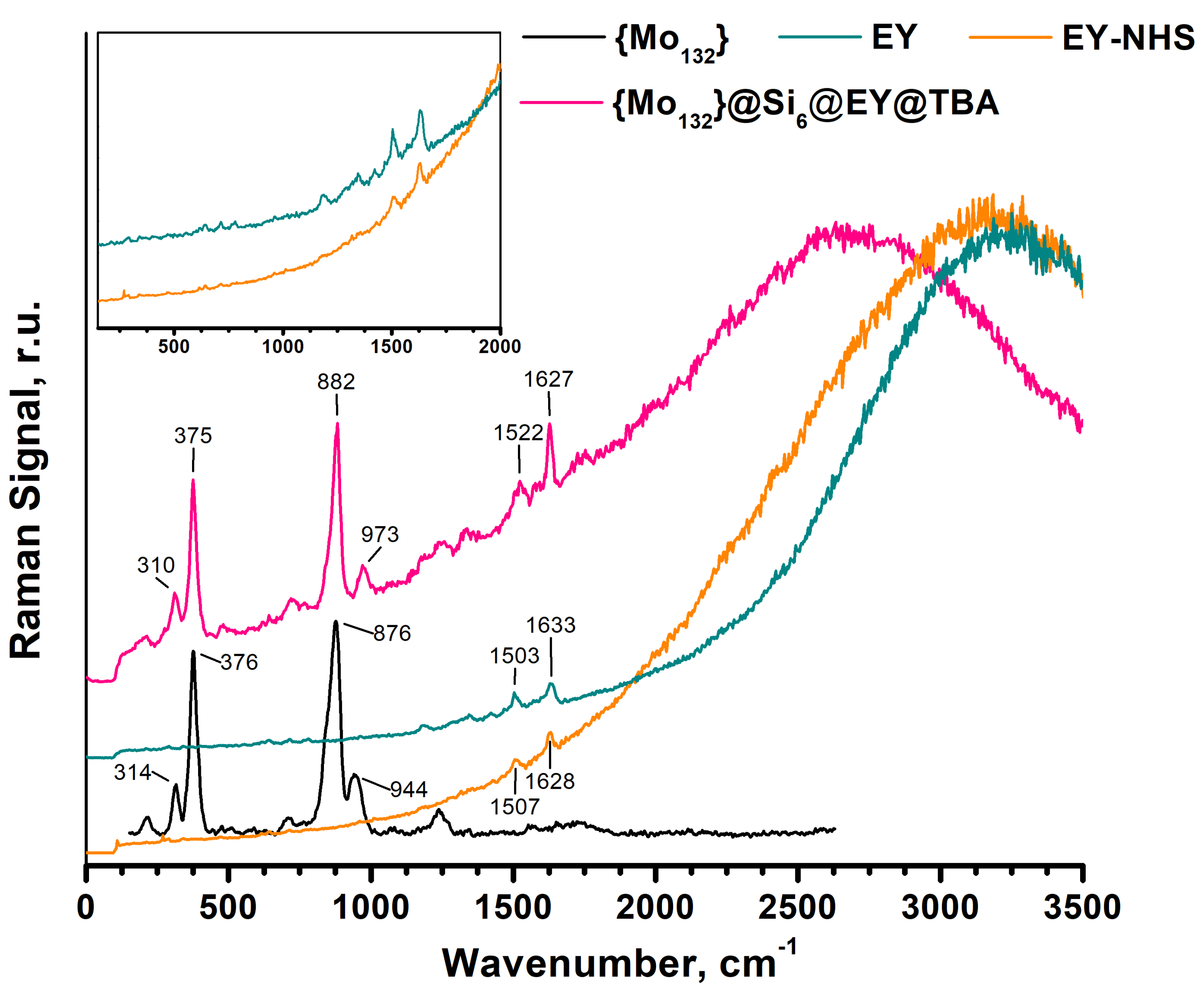
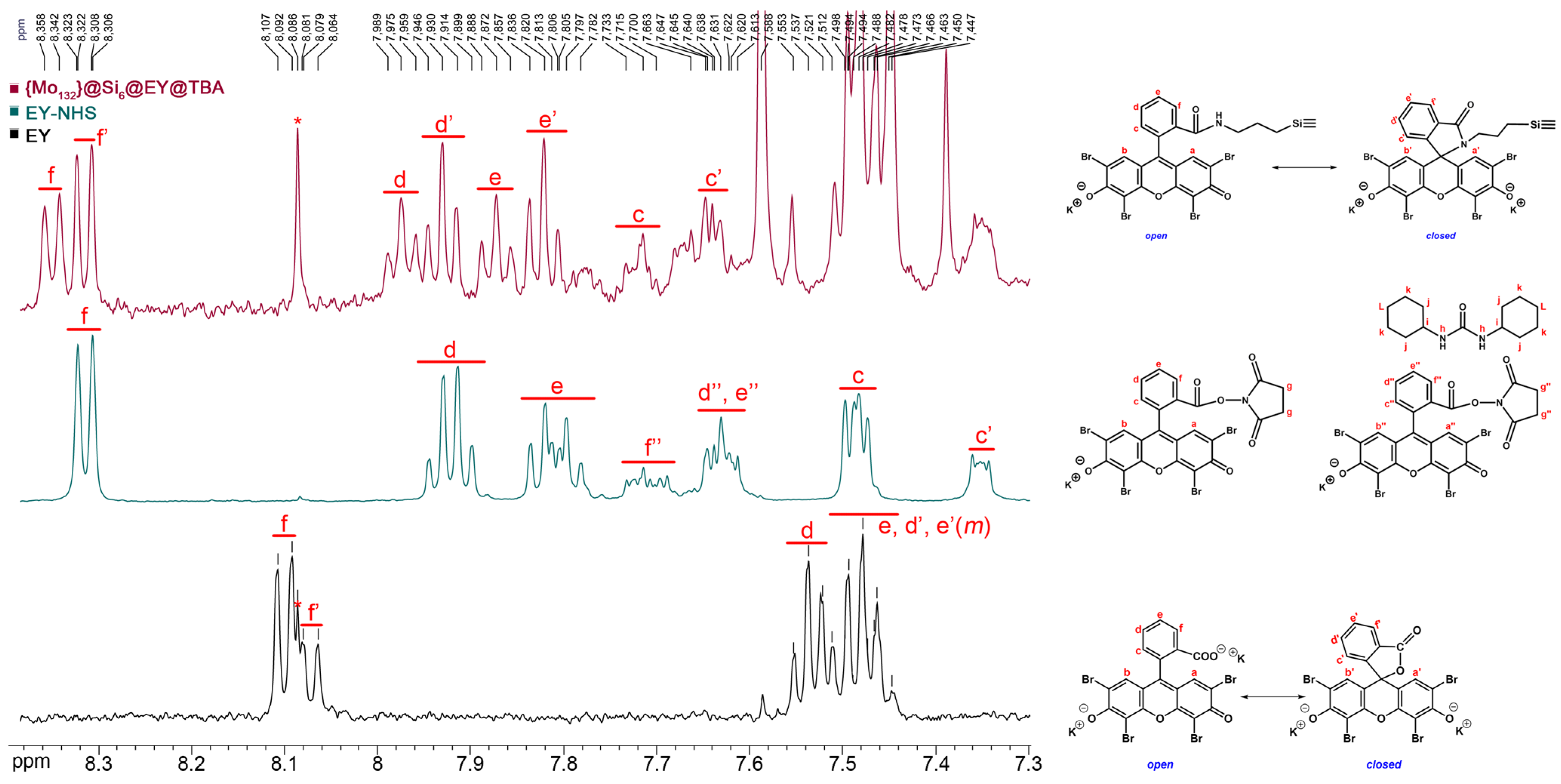
2.1. Conjugation of Esoin Y with {Mo132}
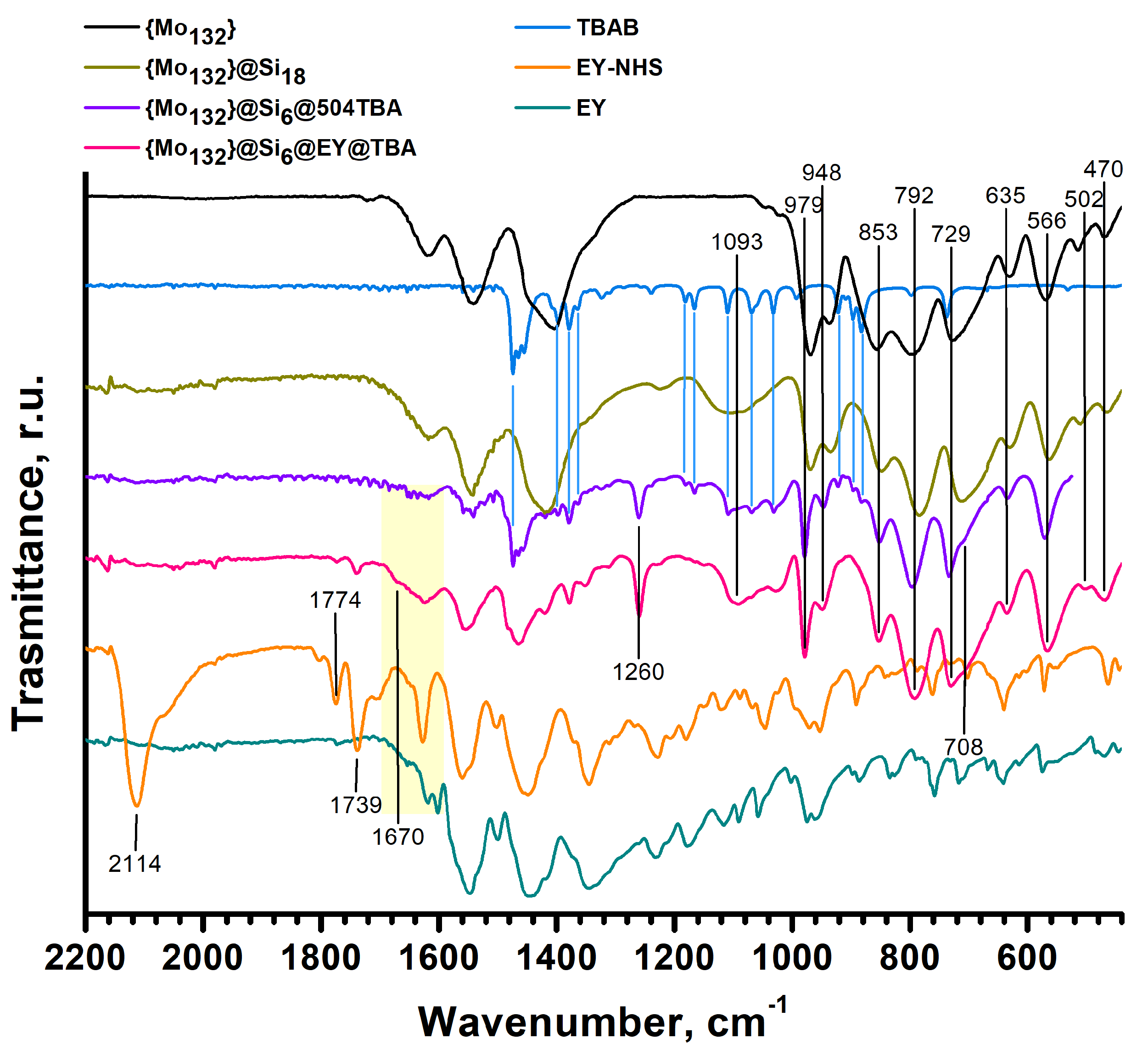
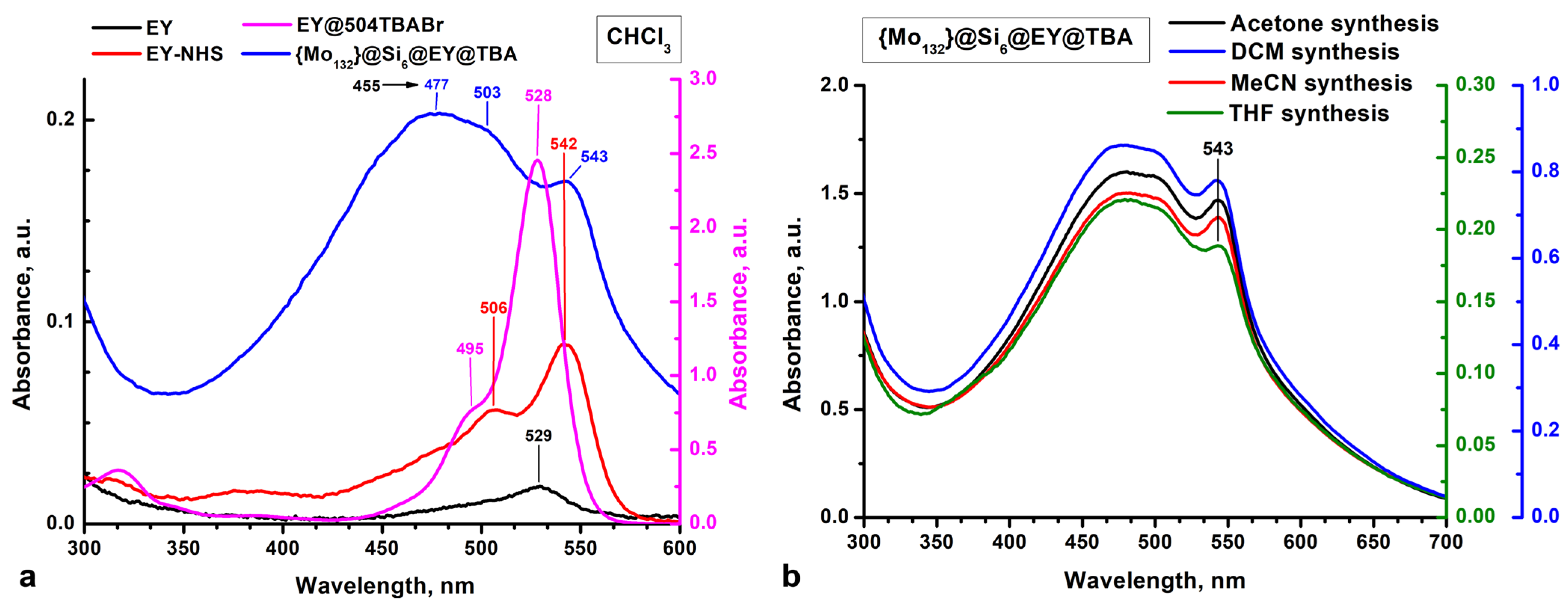
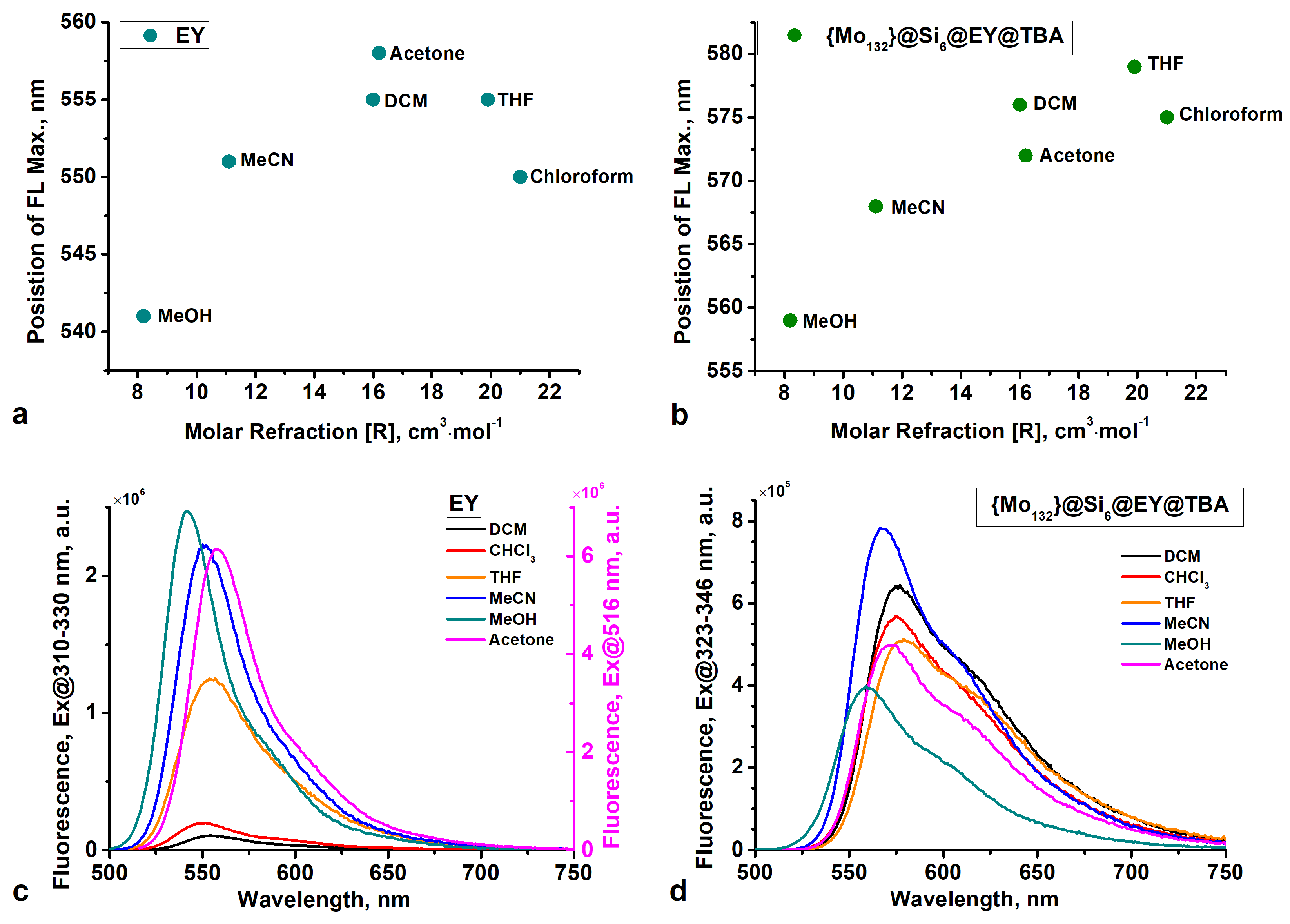
2.2. Analysis of Photophysical Properties of {Mo132}@Si6@EY@TBA
3. Experimental Section
3.1. Materials and Methods
3.2. Synthesis of {Mo132}@Si6@504TBA
3.3. Synthesis of {Mo132}@Si6@EY@TBA
3.3.1. Synthesis of EY-NHS
3.3.2. Grafting of EY-NHS to the {Mo132}@Si6@504TBA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grzhegorzhevskii, K.; Ostroushko, A.; Koriakova, O.; Ovchinnikova, I.; Kim, G. Photoinduced Charge Transfer in the Supramolecular Structure Based on Toroid Polyoxomolibdate Mo138 and Xanthene Dye—Rhodamine-B. Inorg. Chim. Acta 2015, 436, 205–213. [Google Scholar] [CrossRef]
- Fazylova, V.; Shevtsev, N.; Mikhailov, S.; Kim, G.; Ostroushko, A.; Grzhegorzhevskii, K. Fundamental Aspects of Xanthene Dye Aggregation on the Surfaces of Nanocluster Polyoxometalates: H- to J-Aggregate Switching. Chem. A Eur. J. 2020, 26, 5685–5693. [Google Scholar] [CrossRef] [PubMed]
- Grzhegorzhevskii, K.; Haouas, M.; Lion, M.; Vashurin, A.; Denikaev, A.; Marfin, Y.; Kim, G.; Falaise, C.; Cadot, E. Gigantic Supramolecular Assemblies Built from Dynamic Hierarchical Organization between Inorganic Nanospheres and Porphyrins. Chem. Commun. 2023, 59, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Lamare, R.; Ruppert, R.; Boudon, C.; Ruhlmann, L.; Weiss, J. Porphyrins and Polyoxometalate Scaffolds. Chem. A Eur. J. 2021, 27, 16071–16081. [Google Scholar] [CrossRef]
- Ahmed, I.; Farha, R.; Goldmann, M.; Ruhlmann, L. A Molecular Photovoltaic System Based on Dawson Type Polyoxometalate and Porphyrin Formed by Layer-by-Layer Self Assembly. Chem. Commun. 2013, 49, 496–498. [Google Scholar] [CrossRef] [PubMed]
- Black, F.A.; Jacquart, A.; Toupalas, G.; Alves, S.; Proust, A.; Clark, I.P.; Gibson, E.A.; Izzet, G. Rapid Photoinduced Charge Injection into Covalent Polyoxometalate–Bodipy Conjugates. Chem. Sci. 2018, 9, 5578–5584. [Google Scholar] [CrossRef]
- Luo, Y.; Maloul, S.; Wächtler, M.; Winter, A.; Schubert, U.S.; Streb, C.; Dietzek, B. Is Electron Ping-Pong Limiting the Catalytic Hydrogen Evolution Activity in Covalent Photosensitizer–Polyoxometalate Dyads? Chem. Commun. 2020, 56, 10485–10488. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Maloul, S.; Endres, P.; Schönweiz, S.; Ritchie, C.; Wächtler, M.; Winter, A.; Schubert, U.S.; Streb, C.; Dietzek, B. Organic Linkage Controls the Photophysical Properties of Covalent Photosensitizer–Polyoxometalate Hydrogen Evolution Dyads. Sustain. Energy Fuels 2020, 4, 4688–4693. [Google Scholar] [CrossRef]
- Toupalas, G.; Karlsson, J.; Black, F.A.; Masip-Sánchez, A.; López, X.; Ben M’Barek, Y.; Blanchard, S.; Proust, A.; Alves, S.; Chabera, P.; et al. Tuning Photoinduced Electron Transfer in POM-Bodipy Hybrids by Controlling the Environment: Experiment and Theory. Angew. Chem. Int. Ed. 2021, 60, 6518–6525. [Google Scholar] [CrossRef] [PubMed]
- Ben M’Barek, Y.; Rosser, T.; Sum, J.; Blanchard, S.; Volatron, F.; Izzet, G.; Salles, R.; Fize, J.; Koepf, M.; Chavarot-Kerlidou, M.; et al. Dye-Sensitized Photocathodes: Boosting Photoelectrochemical Performances with Polyoxometalate Electron Transfer Mediators. ACS Appl. Energy Mater. 2020, 3, 163–169. [Google Scholar] [CrossRef]
- Huo, Z.; Liang, Y.; Yang, S.; Zang, D.; Farha, R.; Goldmann, M.; Xu, H.; Antoine, B.; Matricardi, E.; Izzet, G.; et al. Photocurrent Generation from Visible Light Irradiation of Covalent Polyoxometalate–Porphyrin Copolymers. Electrochim. Acta 2021, 368, 137635. [Google Scholar] [CrossRef]
- Saad, A.; Oms, O.; Dolbecq, A.; Menet, C.; Dessapt, R.; Serier-Brault, H.; Allard, E.; Baczko, K.; Mialane, P. A High Fatigue Resistant, Photoswitchable Fluorescent Spiropyran–Polyoxometalate–BODIPY Single-Molecule. Chem. Commun. 2015, 51, 16088–16091. [Google Scholar] [CrossRef] [PubMed]
- Parrot, A.; Bernard, A.; Jacquart, A.; Serapian, S.A.; Bo, C.; Derat, E.; Oms, O.; Dolbecq, A.; Proust, A.; Métivier, R.; et al. Photochromism and Dual-Color Fluorescence in a Polyoxometalate-Benzospiropyran Molecular Switch. Angew. Chem. Int. Ed. 2017, 56, 4872–4876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, Y.; Li, G.; Wei, Y. Recent Advances in Alkoxylation Chemistry of Polyoxometalates: From Synthetic Strategies, Structural Overviews to Functional Applications. Coord. Chem. Rev. 2019, 378, 395–414. [Google Scholar] [CrossRef]
- Anyushin, A.V.; Kondinski, A.; Parac-Vogt, T.N. Hybrid Polyoxometalates as Post-Functionalization Platforms: From Fundamentals to Emerging Applications. Chem. Soc. Rev. 2020, 49, 382–432. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Lin, C.; Yang, Y.; Wang, Y.; Wu, Z.; Song, Y.; Russell, T.P.; Shi, S. Polyoxometalate-Surfactant Assemblies: Responsiveness to Orthogonal Stimuli. Angew. Chem. Int. Ed. 2022, 61, e202203741. [Google Scholar] [CrossRef]
- Joo, N.; Renaudineau, S.; Delapierre, G.; Bidan, G.; Chamoreau, L.-M.; Thouvenot, R.; Gouzerh, P.; Proust, A. Organosilyl/-Germyl Polyoxotungstate Hybrids for Covalent Grafting onto Silicon Surfaces: Towards Molecular Memories. Chem. A Eur. J. 2010, 16, 5043–5051. [Google Scholar] [CrossRef]
- Liu, T.; Diemann, E.; Li, H.; Dress, A.W.M.; Müller, A. Self-Assembly in Aqueous Solution of Wheel-Shaped Mo154 Oxide Clusters into Vesicles. Nature 2003, 426, 59–62. [Google Scholar] [CrossRef]
- Zhang, J.; Li, D.; Liu, G.; Glover, K.J.; Liu, T. Lag Periods during the Self-Assembly of {Mo72Fe30} Macroions: Connection to the Virus Capsid Formation Process. J. Am. Chem. Soc. 2009, 131, 15152–15159. [Google Scholar] [CrossRef]
- Fan, D.; Hao, J. Phase Stability of Keplerate-Type Polyoxomolybdates Controlled by Added Cationic Surfactant. J. Colloid Interface Sci. 2009, 333, 757–763. [Google Scholar] [CrossRef]
- Liu, T.; Imber, B.; Diemann, E.; Liu, G.; Cokleski, K.; Li, H.; Chen, Z.; Müller, A. Deprotonations and Charges of Well-Defined {Mo72Fe30} Nanoacids Simply Stepwise Tuned by PH Allow Control/Variation of Related Self-Assembly Processes. J. Am. Chem. Soc. 2006, 128, 15914–15920. [Google Scholar] [CrossRef] [PubMed]
- Kistler, M.L.; Bhatt, A.; Liu, G.; Casa, D.; Liu, T. A Complete Macroion−“Blackberry” Assembly−Macroion Transition with Continuously Adjustable Assembly Sizes in {Mo132} Water/Acetone Systems. J. Am. Chem. Soc. 2007, 129, 6453–6460. [Google Scholar] [CrossRef] [PubMed]
- Kistler, M.L.; Patel, K.G.; Liu, T. Accurately Tuning the Charge on Giant Polyoxometalate Type Keplerates through Stoichiometric Interaction with Cationic Surfactants. Langmuir 2009, 25, 7328–7334. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.M.; Guillemot, G.; Galambos, T.; Amin, S.S.; Hampson, E.; Mall Haidaraly, K.; Newton, G.N.; Izzet, G. Supramolecular Assemblies of Organo-Functionalised Hybrid Polyoxometalates: From Functional Building Blocks to Hierarchical Nanomaterials. Chem. Soc. Rev. 2022, 51, 293–328. [Google Scholar] [CrossRef] [PubMed]
- Izzet, G.; Abécassis, B.; Brouri, D.; Piot, M.; Matt, B.; Serapian, S.A.; Bo, C.; Proust, A. Hierarchical Self-Assembly of Polyoxometalate-Based Hybrids Driven by Metal Coordination and Electrostatic Interactions: From Discrete Supramolecular Species to Dense Monodisperse Nanoparticles. J. Am. Chem. Soc. 2016, 138, 5093–5099. [Google Scholar] [CrossRef]
- Di, A.; Schmitt, J.; da Silva, M.A.; Hossain, K.M.Z.; Mahmoudi, N.; Errington, R.J.; Edler, K.J. Self-Assembly of Amphiphilic Polyoxometalates for the Preparation of Mesoporous Polyoxometalate-Titania Catalysts. Nanoscale 2020, 12, 22245–22257. [Google Scholar] [CrossRef]
- Grzhegorzhevskii, K.V.; Shevtsev, N.S.; Abushaeva, A.R.; Chezganov, D.S.; Ostroushko, A.A. Prerequisites and Prospects for the Development of Novel Systems Based on the Keplerate Type Polyoxomolybdates for the Controlled Release of Drugs and Fluorescent Molecules. Russ. Chem. Bull. 2020, 69, 804–814. [Google Scholar] [CrossRef]
- Grzhegorzhevskii, K.V.; Denikaev, A.D.; Morozova, M.V.; Pryakhina, V.; Khairullina, E.; Tumkin, I.; Taniya, O.; Ostroushko, A.A. The Precise Modification of a Nanoscaled Keplerate-Type Polyoxometalate with NH2-Groups: Reactive Sites, Mechanisms and Dye Conjugation. Inorg. Chem. Front. 2022, 9, 1541–1555. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infra-Red Spectra of Complex Molecules; Springer: Dordrecht, The Netherlands, 1975; ISBN 978-94-011-6017-9. [Google Scholar]
- Grzhegorzhevskii, K.V.; Zelenovskiy, P.S.; Koryakova, O.V.; Ostroushko, A.A. Thermal Destruction of Giant Polyoxometalate Nanoclusters: A Vibrational Spectroscopy Study. Inorg. Chim. Acta 2019, 489, 287–300. [Google Scholar] [CrossRef]
- Bo, C.; Miró, P. On the Electronic Structure of Giant Polyoxometalates: Mo132vs. W72Mo60. Dalton Trans. 2012, 41, 9984. [Google Scholar] [CrossRef]
- Vosgröne, T.; Meixner, A.J. Surface- and Resonance-Enhanced Micro-Raman Spectroscopy of Xanthene Dyes: From the Ensemble to Single Molecules. ChemPhysChem 2005, 6, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Mchedlov-Petrossyan, N.O.; Kukhtik, V.I.; Bezugliy, V.D. Dissociation, Tautomerism and Electroreduction of Xanthene and Sulfonephthalein Dyes in N,N-Dimethylformamide and Other Solvents. J. Phys. Org. Chem. 2003, 16, 380–397. [Google Scholar] [CrossRef]
- Kopilevich, S.; Gil, A.; Garcia-Ratés, M.; Bonet-Ávalos, J.; Bo, C.; Müller, A.; Weinstock, I.A. Catalysis in a Porous Molecular Capsule: Activation by Regulated Access to Sixty Metal Centers Spanning a Truncated Icosahedron. J. Am. Chem. Soc. 2012, 134, 13082–13088. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wang, Y.; Zhao, Y.; Chen, W.; Wang, J.; He, L.; Su, Z.; Wang, E.; Kang, Z. Keplerate-Type Polyoxometalate/Semiconductor Composite Electrodes with Light-Enhanced Conductivity towards Highly Efficient Photoelectronic Devices. J. Mater. Chem. A 2016, 4, 14025–14032. [Google Scholar] [CrossRef]
- Roth, H.; Romero, N.; Nicewicz, D. Experimental and Calculated Electrochemical Potentials of Common Organic Molecules for Applications to Single-Electron Redox Chemistry. Synlett 2015, 27, 714–723. [Google Scholar] [CrossRef]
- De, S.; Das, S.; Girigoswami, A. Environmental Effects on the Aggregation of Some Xanthene Dyes Used in Lasers. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2005, 61, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006; ISBN 0-387-31278-1. [Google Scholar]
- Müller, A.; Krickemeyer, E.; Bögge, H.; Schmidtmann, M.; Peters, F. Organizational Forms of Matter: An Inorganic Super Fullerene and Keplerate Based on Molybdenum Oxide. Angew. Chemie Int. Ed. 1998, 37, 3359–3363. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EY | {Mo132}@Si6@EY@TBA | |||
|---|---|---|---|---|
| Solvent | ΦF, % | τ, ns | ΦF, % | τ, ns |
| DCM * | 7.3 | 3.58 | 2.5 | 4.27 |
| CHCl3 * | 4.7 | 3.3 | 2.4 | 3.91 |
| THF | 68.8 | 3.88 | 2.6 | 4.42 |
| MeCN | 77.6 | 4 | 2.6 | 4.47 |
| Acetone | - | 4.29 | 2.6 | 4.52 |
| MeOH | 54.9 | 2.95 | 1.3 | 3.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Denikaev, A.; Kim, G.; Greshnyakov, E.; Moskalenko, N.; Grzhegorzhevskii, K. Covalent Grafting of Eosin Y to the Giant Keplerate {Mo132} through an Organosilicon Linker in Homogeneous Regime. Inorganics 2023, 11, 239. https://doi.org/10.3390/inorganics11060239
Denikaev A, Kim G, Greshnyakov E, Moskalenko N, Grzhegorzhevskii K. Covalent Grafting of Eosin Y to the Giant Keplerate {Mo132} through an Organosilicon Linker in Homogeneous Regime. Inorganics. 2023; 11(6):239. https://doi.org/10.3390/inorganics11060239
Chicago/Turabian StyleDenikaev, Andrey, Grigory Kim, Evgeny Greshnyakov, Nikolai Moskalenko, and Kirill Grzhegorzhevskii. 2023. "Covalent Grafting of Eosin Y to the Giant Keplerate {Mo132} through an Organosilicon Linker in Homogeneous Regime" Inorganics 11, no. 6: 239. https://doi.org/10.3390/inorganics11060239