
An LC–MS/MS Analytical Method for Quantifying Tepotinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation

Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. In Silico TEP Metabolic Vulnerability Prediction
2.3. LC-MS/MS Method
2.4. TEP Stock Solutions
2.5. TEP Calibration Standards
2.6. Method Validation
2.7. TEP Metabolic Stability
3. Results and Discussions
3.1. In Silico TEP Metabolic Lability
3.2. Quantification of TEP with the Developed LC-MS/MS Method
3.3. Validation Parameters
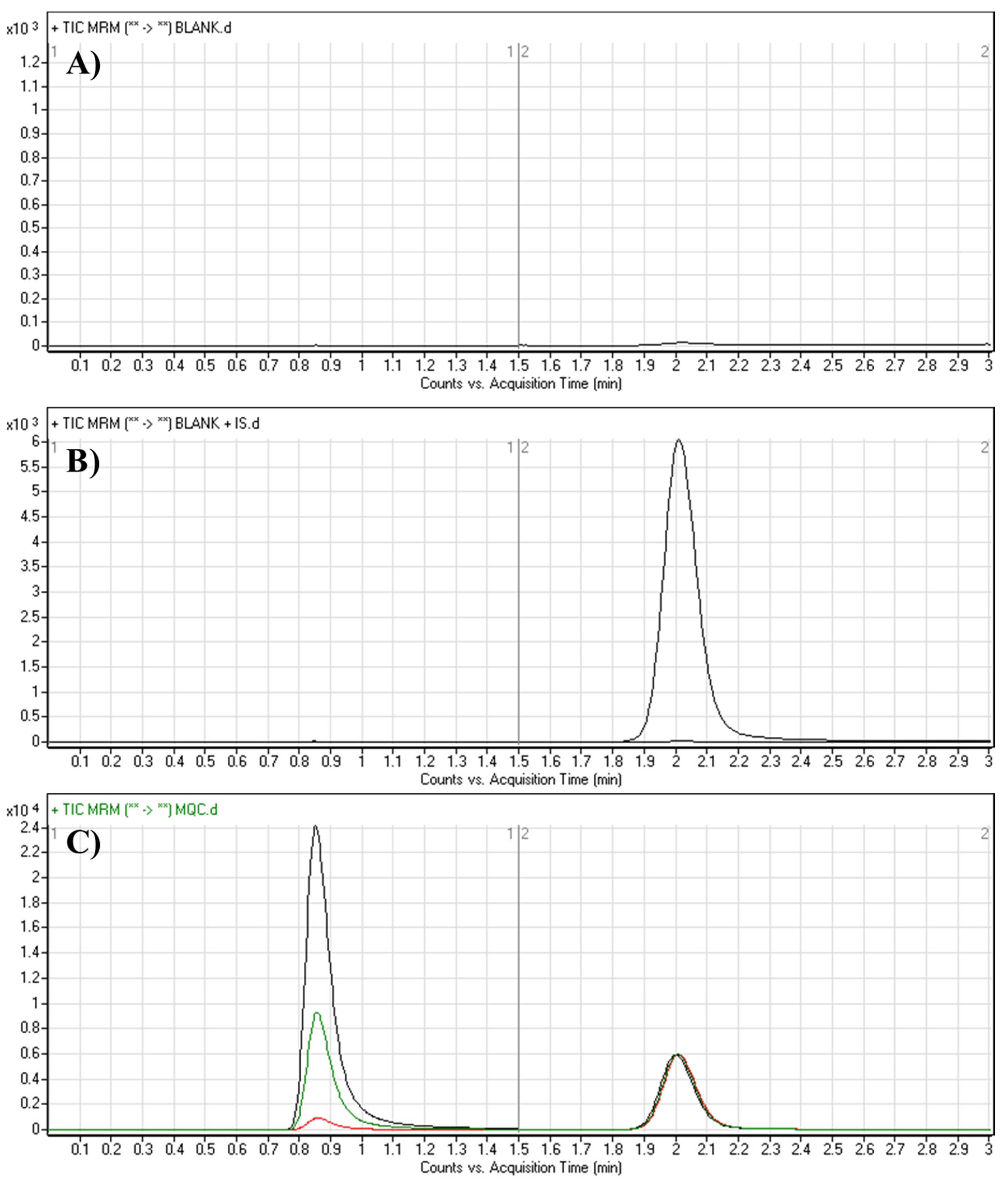
3.3.1. Specificity
3.3.2. Precision and Accuracy
3.3.3. Extraction Recovery and Matrix Effects
3.4. Metabolic Stability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA A Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung cancer statistics. In Lung Cancer and Personalized Medicine; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1–19. [Google Scholar]
- Chmielecki, J.; Foo, J.; Oxnard, G.R.; Hutchinson, K.; Ohashi, K.; Somwar, R.; Wang, L.; Amato, K.R.; Arcila, M.; Sos, M.L. Optimization of dosing for EGFR-mutant non–small cell lung cancer with evolutionary cancer modeling. Sci. Transl. Med. 2011, 3, 90ra59. [Google Scholar] [CrossRef]
- Katayama, R.; Khan, T.M.; Benes, C.; Lifshits, E.; Ebi, H.; Rivera, V.M.; Shakespeare, W.C.; Iafrate, A.J.; Engelman, J.A.; Shaw, A.T. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 7535–7540. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Yu, X.; Wu, S.; Wu, X.; Wang, Q.; Cheng, W.; Hu, W.; Kang, C.; Yang, W.; Li, Y.; et al. Instability Mechanism of Osimertinib in Plasma and a Solving Strategy in the Pharmacokinetics Study. Front. Pharmacol. 2022, 13, 928983. [Google Scholar] [CrossRef]
- Shenouda, S.K.; Alahari, S.K. MicroRNA function in cancer: Oncogene or a tumor suppressor? Cancer Metastasis Rev. 2009, 28, 369. [Google Scholar] [CrossRef]
- Sechler, M.; Cizmic, A.D.; Avasarala, S.; Van Scoyk, M.; Brzezinski, C.; Kelley, N.; Bikkavilli, R.K.; Winn, R.A. Non-small-cell lung cancer: Molecular targeted therapy and personalized medicine–drug resistance, mechanisms, and strategies. Pharm. Pers. Med. 2013, 6, 25. [Google Scholar]
- Cheng, L.; Alexander, R.E.; MacLennan, G.T.; Cummings, O.W.; Montironi, R.; Lopez-Beltran, A.; Cramer, H.M.; Davidson, D.D.; Zhang, S. Molecular pathology of lung cancer: Key to personalized medicine. Mod. Pathol. 2012, 25, 347–369. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.-H.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef]
- Kunii, K.; Davis, L.; Gorenstein, J.; Hatch, H.; Yashiro, M.; Di Bacco, A.; Elbi, C.; Lutterbach, B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008, 68, 2340–2348. [Google Scholar] [CrossRef]
- Abdelhameed, A.S.; Attwa, M.W.; Kadi, A.A. Identification of Iminium Intermediates Generation in the Metabolism of Tepotinib Using LC-MS/MS: In Silico and Practical Approaches to Bioactivation Pathway Elucidation. Molecules 2020, 25, 5004. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Veillon, R.; Cortot, A.B.; Felip, E.; Sakai, H.; Mazieres, J.; Griesinger, F.; Horn, L.; Senellart, H.; Meerbeeck, J.P.V.; et al. Phase II study of tepotinib in NSCLC patients with METex14 mutations. J. Clin. Oncol. 2019, 37, 9005. [Google Scholar] [CrossRef]
- Ruiz-Cordero, R.; Devine, W.P. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg. Pathol. Clin. 2020, 13, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Houston, J.B. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem. Pharm. 1994, 47, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar]
- Nichols, J.W.; Schultz, I.R.; Fitzsimmons, P.N. In vitro–in vivo extrapolation of quantitative hepatic biotransformation data for fish: I. A review of methods, and strategies for incorporating intrinsic clearance estimates into chemical kinetic models. Aquat. Toxicol. 2006, 78, 74–90. [Google Scholar] [CrossRef]
- Pelkonen, O.; Turpeinen, M. In vitro–in vivo extrapolation of hepatic clearance: Biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 2007, 37, 1066–1089. [Google Scholar] [CrossRef]
- Malapelle, U.; Muscarella, L.A.; Pisapia, P.; Rossi, A. Targeting emerging molecular alterations in the treatment of non-small cell lung cancer: Current challenges and the way forward. Expert Opin. Investig. Drugs 2020, 29, 363–372. [Google Scholar] [CrossRef]
- Baranczewski, P.; Stańczak, A.; Sundberg, K.; Svensson, R.; Wallin, A.; Jansson, J.; Garberg, P.; Postlind, H. Introduction to in vitro estimation of metabolic stability and drug interactions of new chemical entities in drug discovery and development. Pharmacol. Rep. 2006, 58, 453–472. [Google Scholar]
- Li, F.; MacKenzie, K.R.; Nyshadham, P.; Kerlec, K.A.; Matzuk, M.M. Identifying Metabolic Pathways of c-MET Tyrosine Kinase Inhibitor Tepotinib in Human and Mouse Liver Microsomes. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S. Detection and characterization of olmutinib reactive metabolites by LC–MS/MS: Elucidation of bioactivation pathways. J. Sep. Sci. 2020, 43, 708–718. [Google Scholar] [CrossRef]
- Attwa, M.W.; Kadi, A.A.; Abdelhameed, A.S.; Alhazmi, H.A. Metabolic Stability Assessment of New PARP Inhibitor Talazoparib Using Validated LC-MS/MS Methodology: In silico Metabolic Vulnerability and Toxicity Studies. Drug Des. Dev. Ther. 2020, 14, 783–793. [Google Scholar] [CrossRef]
- Vansteenkiste, J.F.; Van De Kerkhove, C.; Wauters, E.; Van Mol, P. Capmatinib for the treatment of non-small cell lung cancer. Expert Rev. Anticancer. Ther. 2019, 19, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Alrabiah, H.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S. A simple liquid chromatography-tandem mass spectrometry method to accurately determine the novel third-generation EGFR-TKI naquotinib with its applicability to metabolic stability assessment. RSC Adv. 2019, 9, 4862–4869. [Google Scholar] [CrossRef]
- Kadi, A.A.; Darwish, H.W.; Abuelizz, H.A.; Alsubi, T.A.; Attwa, M.W. Identification of reactive intermediate formation and bioactivation pathways in Abemaciclib metabolism by LC-MS/MS: In vitro metabolic investigation. R. Soc. Open Sci. 2019, 6, 181714. [Google Scholar] [CrossRef]
- Attwa, M.W.; AlRabiah, H.; Alsibaee, A.M.; Abdelhameed, A.S.; Kadi, A.A. An UPLC–ESI–MS/MS Bioanalytical Methodology for the Quantification of Gilteritinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation. Separations 2023, 10, 278. [Google Scholar] [CrossRef]
- Korzekwa, K.R.; Trager, W.F.; Gillette, J.R. Theory for the observed isotope effects from enzymatic systems that form multiple products via branched reaction pathways: Cytochrome P-450. Biochemistry 1989, 28, 9012–9018. [Google Scholar] [CrossRef] [PubMed]
- Busby, W.F.; Ackermann, J.M.; Crespi, C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. 1999, 27, 246–249. [Google Scholar]
- Taniguchi-Takizawa, T.; Shimizu, M.; Kume, T.; Yamazaki, H. Benzydamine N-oxygenation as an index for flavin-containing monooxygenase activity and benzydamine N-demethylation by cytochrome P450 enzymes in liver microsomes from rats, dogs, monkeys, and humans. Drug Metab. Pharmacokinet. 2015, 30, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Fouin-Fortunet, H.; Tinel, M.; Descatoire, V.; Letteron, P.; Larrey, D.; Geneve, J.; Pessayre, D. Inactivation of cytochrome P-450 by the drug methoxsalen. J. Pharmacol. Exp. Ther. 1986, 236, 237–247. [Google Scholar]
- Attwa, M.W.; Kadi, A.A.; Darwish, H.W.; Amer, S.M.; Al-Shakliah, N.S. Identification and characterization of in vivo, in vitro and reactive metabolites of vandetanib using LC-ESI-MS/MS. Chem. Cent. J. 2018, 12, 99. [Google Scholar] [CrossRef]
- Alrabiah, H.; Kadi, A.A.; Aljohar, H.I.; Attwa, M.W.; Al-Shakliah, N.S.; Attia, S.M.; Mostafa, G.A.E. A new validated HPLC-MS/MS method for quantification and pharmacokinetic evaluation of dovitinib, a multi-kinase inhibitor, in mouse Plasma. Drug Des. Dev. Ther. 2020, 14, 407–415. [Google Scholar] [CrossRef]
- Attwa, M.W.; Darwish, H.W.; Alhazmi, H.A.; Kadi, A.A. Investigation of metabolic degradation of new ALK inhibitor: Entrectinib by LC-MS/MS. Clin. Chim. Acta 2018, 485, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, M.M.; Alkahtani, H.M.; Almehizia, A.A.; Attwa, M.W.; Bakheit, A.H.; Darwish, H.W. Validated liquid chromatography tandem mass spectrometry for simultaneous quantification of foretinib and lapatinib, and application to metabolic stability investigation. RSC Adv. 2019, 9, 19325–19332. [Google Scholar] [CrossRef] [PubMed]
- Attwa, M.; Kadi, A.A.; Darwish, H.W.; Abdelhameed, A.S. Investigation of the metabolic stability of olmutinib by validated LC-MS/MS: Quantification in human plasma. RSC Adv. 2018, 8, 40387–40394. [Google Scholar] [CrossRef] [PubMed]
- McNaney, C.A.; Drexler, D.M.; Hnatyshyn, S.Y.; Zvyaga, T.A.; Knipe, J.O.; Belcastro, J.V.; Sanders, M. An automated liquid chromatography-mass spectrometry process to determine metabolic stability half-life and intrinsic clearance of drug candidates by substrate depletion. Assay Drug Dev. Technol. 2008, 6, 121–129. [Google Scholar] [CrossRef]
- Słoczyńska, K.; Gunia-Krzyżak, A.; Koczurkiewicz, P.; Wójcik-Pszczoła, K.; Żelaszczyk, D.; Popiół, J.; Pękala, E. Metabolic stability and its role in the discovery of new chemical entities. Acta Pharm. 2019, 69, 345–361. [Google Scholar] [CrossRef]
- Manzo, A.; Montanino, A.; Costanzo, R.; Sandomenico, C.; Palumbo, G.; Schettino, C.; Daniele, G.; Morabito, A.; Perrone, F.; Piccirillo, M.C. Chapter 33-EGFR Mutations: Best Results from Second- and Third-Generation Tyrosine Kinase Inhibitors. In Oncogenomics; Dammacco, F., Silvestris, F., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 477–486. [Google Scholar] [CrossRef]
- Kadian, N.; Raju, K.S.R.; Rashid, M.; Malik, M.Y.; Taneja, I.; Wahajuddin, M. Comparative assessment of bioanalytical method validation guidelines for pharmaceutical industry. J. Pharm. Biomed. Anal. 2016, 126, 83–97. [Google Scholar] [CrossRef]
- Leahy, D.E. Integrating in vitro ADMET data through generic physiologically based pharmacokinetic models. Expert Opin. Drug Metab. Toxicol. 2006, 2, 619–628. [Google Scholar] [CrossRef]
- Attwa, M.W.; Abdelhameed, A.S.; Alsibaee, A.M.; Kadi, A.A. A Rapid and Sensitive UPLC-MS/MS Method for Quantifying Capmatinib in Human Liver Microsomes: Evaluation of Metabolic Stability by In Silico and In Vitro Analysis. Separations 2023, 10, 247. [Google Scholar] [CrossRef]






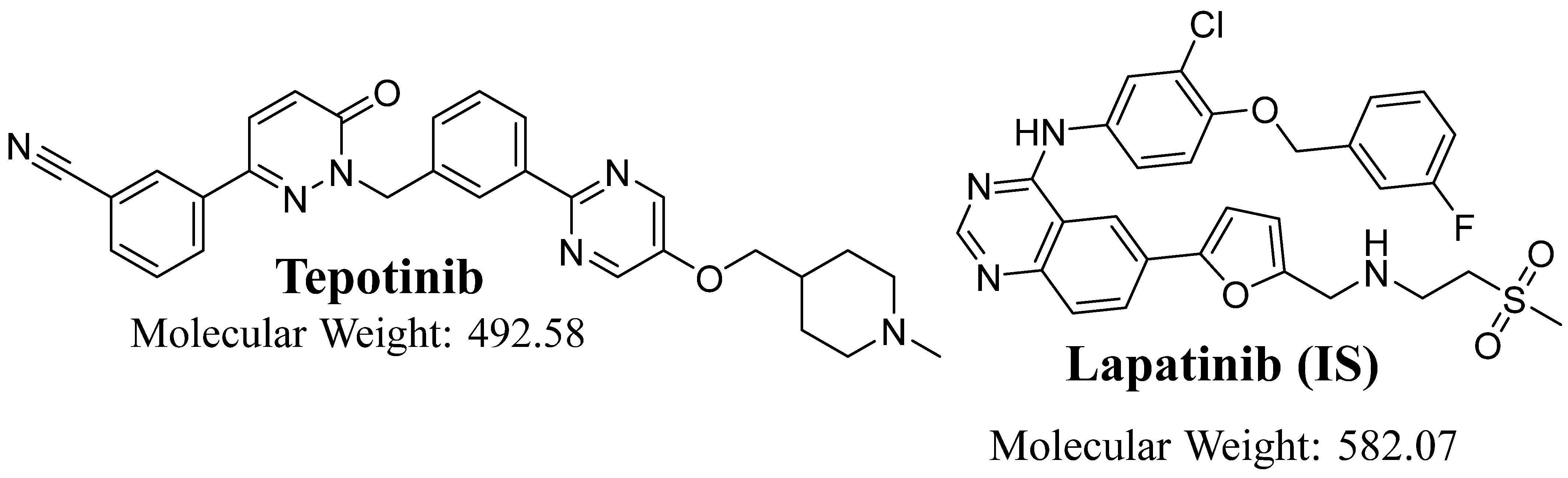{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LC (Agilent 1200) | MS/MS (Agilent 6410 QqQ) | |||
|---|---|---|---|---|
| Mobile phase (isocratic) | 70% acetonitrile | ESI | ESI (positive mode) | |
| 10 mM NH4COOH | 30% | Drying gas: nitrogen of high purity at 11 L/min flow rate and pressure of 60 psi | ||
| pH 3.5 | ||||
| 0.15 mL/min. | ||||
| 5 μL | ||||
| Agilent eclipse plus C18 Column | T: 20 ± 2 °C. | Capillary voltage: 4000 V | ||
| 1.8 μm particle size | Source T: 350 °C | |||
| 2.1 mm i.d. | Collision cell | Nitrogen gas (high purity) | ||
| 50 mm long | Mode | MRM | ||
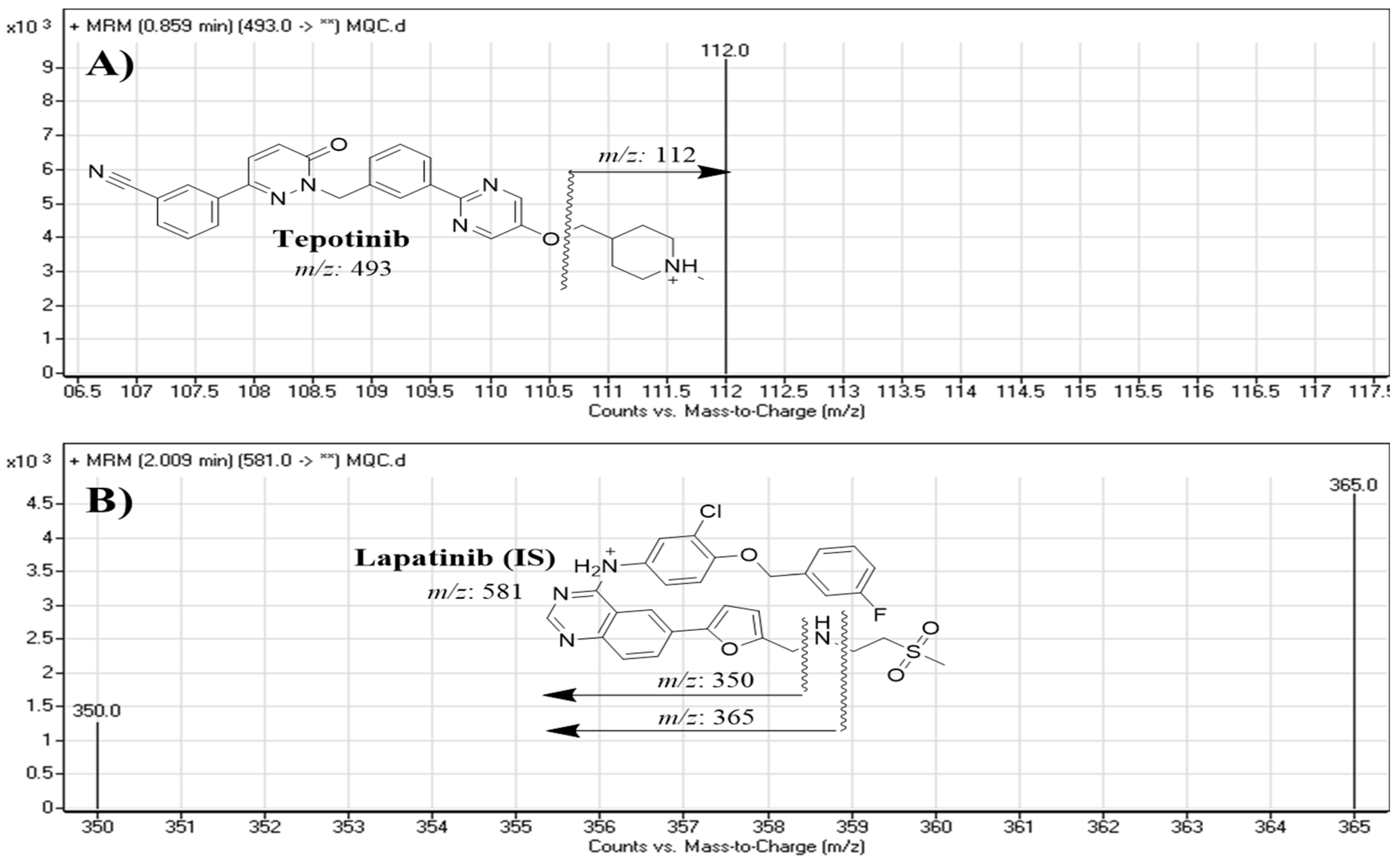
| Mass spectra segment | 0.0 to 1.5 min | TEP (0.86 min) | Analyte: Tepotinib (TEP) | m/z 493→ m/z 112, FV a: 140 V CE b: of 18 eV |
| 1.5 to 3.0 min | LTP (2.0 min) | Lapatinib (LTP, IS) | m/z 581.1→ m/z 350, FV a: 145 V, CE b: 32 eV | |
| m/z 581.1→ m/z 365, FV a: 140 V, CE b: 30 eV | ||||
| Parameter | Value |
|---|---|
| Slope | 0.5635714 |
| Intercept | 0.4066 |
| Standard deviation of the intercept | 0.1123472 |
| Standard deviation of the slope | 0.002105 |
| Correlation coefficient (R2) | 0.999 |
| Standard deviation of the residuals (Sy.x) | 0.0812733 |
| Limit of detection (LOD), ng/mL | 0.4759 |
| Retention time of TEP, min | 0.86 |
| Retention time of IS, min | 2.0 |
| Lower limit of quantification (LOQ), ng/mL | 1.4421 |
| Linearity range, ng/ml | 5–500 |
| HLM Matrix | Intra-Day Assay * | |||
|---|---|---|---|---|
| 5 ng/mL (LLQC) | 15 ng/mL (LQC) | 150 ng/mL (MQC) | 400 ng/mL (HQC) | |
| Mean | 5.09 | 14.92 | 151.04 | 401.36 |
| SD | 0.21 | 0.19 | 2.12 | 4.22 |
| Relative error | 1.84 | −0.55 | 0.69 | 0.34 |
| Precision (RSD), % | 4.19 | 1.24 | 1.40 | 1.05 |
| Inter-day assay ** | ||||
| Mean | 5.17 | 15.02 | 146.73 | 397.46 |
| SD | 0.23 | 0.31 | 2.67 | 4.94 |
| Relative error | 3.48 | 0.11 | −2.18 | −0.63 |
| Precision (RSD), % | 4.39 | 2.05 | 1.82 | 1.24 |
| TEP Nominal Concentrations (ng/mL) | Mean * | SD | RSD % | Relative ERROR % | Recovery % |
|---|---|---|---|---|---|
| 5 | 5.09 | 0.21 | 4.19 | 1.84 | 100.8 |
| 10 | 10.03 | 0.19 | 1.92 | 0.28 | 100.3 |
| 30 | 29.92 | 0.72 | 2.41 | −0.28 | 99.73 |
| 50 | 50.38 | 2.23 | 4.43 | 0.75 | 100.76 |
| 100 | 98.90 | 3.18 | 3.21 | −1.10 | 98.9 |
| 200 | 201.31 | 4.26 | 2.12 | 0.66 | 100.65 |
| 300 | 297.44 | 7.07 | 2.38 | −0.85 | 99.15 |
| 500 | 500.81 | 3.61 | 0.72 | 0.16 | 100.16 |
| TEP (ng/mL) | Mean ± SD | RSD, % | Recovery % |
|---|---|---|---|
| 5 (LLOQ) | 5.09 ± 0.21 | 4.19 | 101.80 |
| 15 (LQC) | 14.92 ± 0.19 | 1.24 | 99.47 |
| 150 (MQC) | 151.04 ± 2.12 | 1.40 | 100.69 |
| 400 (HQC) | 401.36 ± 4.22 | 1.05 | 100.34 |
| TEP extraction recovery± SD | 100.58 ± 0.97 | ||
| Time (min) | Mean (ng/mL) a | X b | ln (X) | Analytical Features |
|---|---|---|---|---|
| 0.00 | 467.74 | 100.00 | 4.61 | Regression equation: y = −0.0306x + 4.5813 R2 = 0.9914 Slope: −0.0306 t1/2: 22.65 min Clint: 35.79 mL/min/kg |
| 0.50 | 455.73 | 95.96 | 4.56 | |
| 2.5 | 450.58 | 88.12 | 4.48 | |
| 7.5 | 439.12 | 77.65 | 4.35 | |
| 15.00 | 375.32 | 62.00 | 4.13 | |
| 30.00 | 347.06 | 54.30 | 3.99 | |
| 50.00 | 336.11 | 47.97 | 3.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Attwa, M.W.; Mostafa, G.A.E.; AlRabiah, H.; Kadi, A.A. An LC–MS/MS Analytical Method for Quantifying Tepotinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation. Separations 2023, 10, 330. https://doi.org/10.3390/separations10060330
Attwa MW, Mostafa GAE, AlRabiah H, Kadi AA. An LC–MS/MS Analytical Method for Quantifying Tepotinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation. Separations. 2023; 10(6):330. https://doi.org/10.3390/separations10060330
Chicago/Turabian StyleAttwa, Mohamed W., Gamal A. E. Mostafa, Haitham AlRabiah, and Adnan A. Kadi. 2023. "An LC–MS/MS Analytical Method for Quantifying Tepotinib in Human Liver Microsomes: Application to In Vitro and In Silico Metabolic Stability Estimation" Separations 10, no. 6: 330. https://doi.org/10.3390/separations10060330