

Quantitative Analysis of Eight Compounds in Traditional Korean Medicine, Gongjindan Using HPLC, UPLC–MS/MS, and GC–MS/MS Systems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical and Reagents
2.2. HPLC–PDA Analysis
2.3. UPLC–MS/MS Analytical Conditions and Sample Preparation
2.4. GC–MS/MS SIM Analysis
2.5. Validation of Developed Analytical Method
3. Results and Discussion
3.1. HPLC–PDA Analysis
3.1.1. Determination of Target Compounds in GJD for Analytical Method Development Using HPLC–PDA
3.1.2. HPLC Simultaneous Analysis Conditions
3.1.3. Validation of the Developed HPLC Analytical Method
3.1.4. Simultaneous Quantitation of the Seven Target Components in GJD Samples Using the HPLC System
3.2. UPLC–MS/MS Analysis
3.2.1. UPLC–MS/MS Multiple Reaction Monitoring (MRM) Method for Simultaneous Analysis
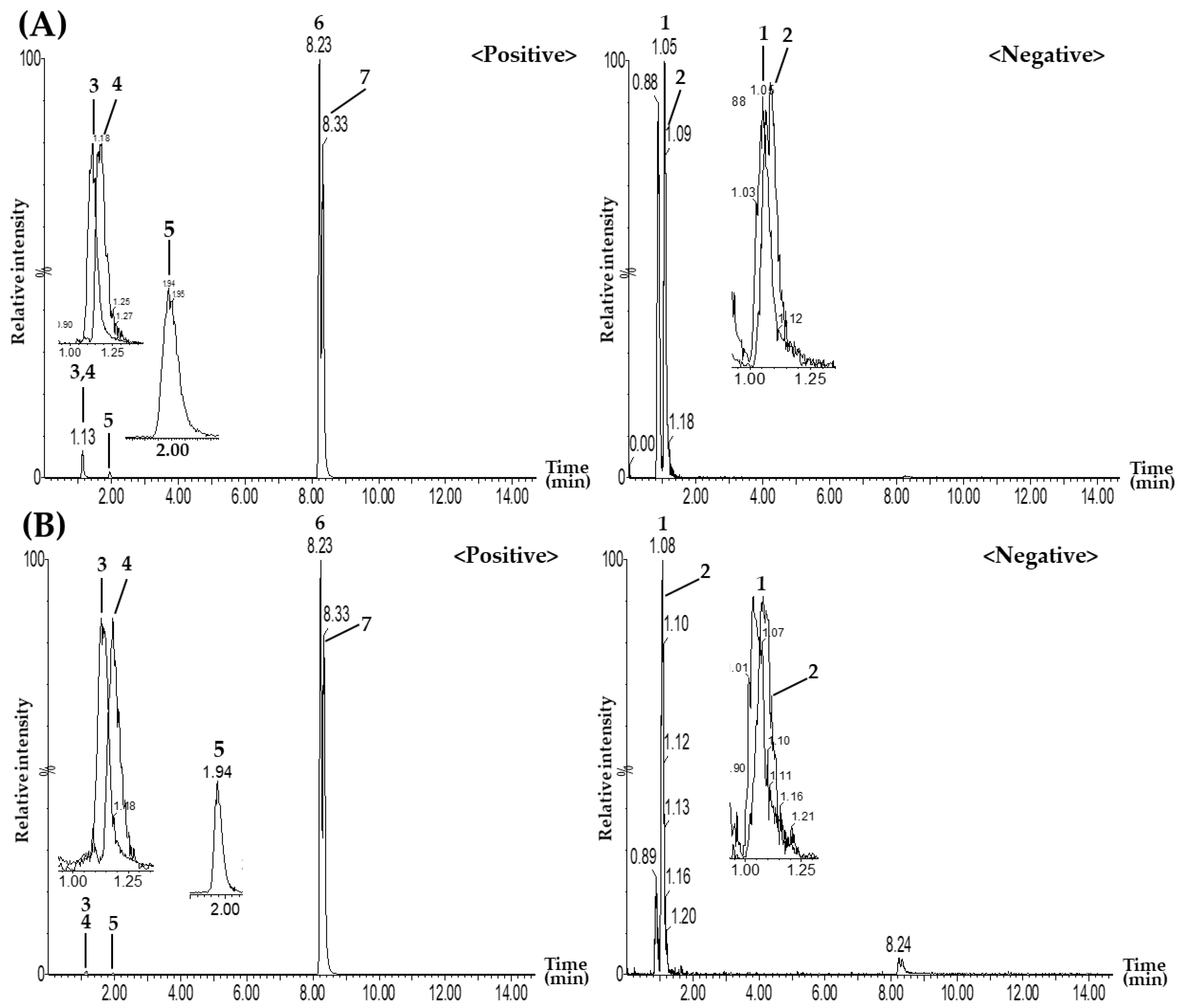
3.2.2. Quantitation of the Seven Target Components in GJD Samples
3.3. GC–MS/MS Analysis
3.3.1. Establishment of the GC–MS/MS Method
3.3.2. GC–MS/MS Method Validation for Analysis of Muscone
3.3.3. Quantitation of Muscone in GJD Samples Using the GC–MS/MS Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Han, M.G.; Kim, K.S.; Joo, J.H.; Choi, H.S.; Kim, S.M. Favorable hepatoprotective effects of Gongjin-dan on the acute ethanol—induced liver damaged C57BL/6 mice. J. Physiol. Pathol. Korean Med. 2016, 30, 279–288. [Google Scholar] [CrossRef]
- Choi, K.H.; Park, C.S. An analysis of the Gongjindan’s ingredients and its efficacy on anti-oxidation. Kor. J. Herbol. 2007, 22, 51–63. [Google Scholar]
- Moon, E.; Her, Y.; Lee, J.B.; Park, J.H.; Lee, E.H.; Kim, S.H.; Oh, M.S.; Jang, C.G.; Kim, S.Y. The multi-herbal medicine Gongjin-dan enhances memory and learning tasks via NGF regulation. Neurosci. Lett. 2009, 466, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.S.; Lee, J.; Lee, J.S.; Lee, H.W.; Kim, H.G.; Lee, S.K.; Park, B.K.; Son, C.G. The traditional drug Gongjin-dan ameliorates chronic fatigue in a forced-stress mouse exercise model. J. Ethnopharmacol. 2015, 168, 268–278. [Google Scholar] [CrossRef]
- Hwang, S.M.; Chung, D.K. The effects of Kongjin-dan (KJD) on the Alzheimer’s disease model induced by CT105. J. Orient. Neuropsychiatry 2004, 15, 103–118. [Google Scholar]
- Jeong, S.Y.; Kim, H.M.; Lee, K.H.; Kim, K.Y.; Huang, D.S.; Kim, J.H.; Seong, R.S. Quantitative analysis of marker compounds in Angelica gigas, Angelica sinensis, and Angelica acutiloba by HPLC/DAD. Chem. Pharm. Bull. 2015, 63, 504–511. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Chen, H.; Wang, L.; Zou, J.; Zheng, X.; Liu, Z. Quality evaluation of polar and active components in crude and processed Fructus corni by quantitative analysis of multicomponents with single marker. J. Anal. Methods Chem. 2016, 2016, 6496840. [Google Scholar] [CrossRef] [Green Version]
- Shan, S.M.; Luo, J.G.; Huang, F.; Kong, L.Y. Chemical characteristics combined with bioactivity for comprehensive evaluation of Panax ginseng C.A. Meyer in different ages and seasons based on HPLC-DAD and chemometric methods. J. Pharm. Biomed. Anal. 2014, 89, 76–82. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, E.J.; Kim, J.S.; Lee, J.H.; Kang, S.S. Phytochemical studies on Rehmanniae Radix Preparata. Kor. J. Pharmacogn. 2011, 42, 117–126. [Google Scholar]
- Fujimoto, S.; Yoshikawa, K.; Itoh, M.; Kitahara, T. Synthesis of (R)- and (S)-muscone. Biosci. Biotechnol. Biochem. 2002, 66, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.H. The Korean Herbal Pharmacopoeia, 5th ed.; Shinilbooks: Seoul, Republic of Korea, 2016; pp. 97–99. [Google Scholar]
- Liu, K.; Xie, L.; Deng, M.; Zhang, X.; Luo, J.; Li, X. Zoology, chemical composition, pharmacology, quality control and future perspective of Musk (Moschus): A review. Chin. Med. 2021, 16, 46. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Yin, Y.; Zhou, Y.; He, G.; Qi, Y. Isolation and characterization of peptidoglycan recognition protein 1 from antler base of silka deer (Cervus nippon). Int. J. Biol. Macromol. 2014, 64, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.O.; Kim, J.W.; Jo, S.J.; Kim, J.H.; Han, B.H. A new HPLC-analytical method for total sphingosine contents as an indirect index for the ganglioside contents of deer antlers. Nat. Prod. Sci. 2011, 17, 315–320. [Google Scholar]
- Ahn, M.J.; Lee, M.K.; Kim, Y.C.; Sung, S.H. The simultaneous determination of coumarins in Angelica gigas root by high performance liquid chromatography–diode array detector coupled with electrospray ionization/mass spectrometry. J. Pharm. Biomed. Anal. 2008, 46, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.Y.; Hwang, H.Y.; Choi, W.H.; Jung, H.J.; Huh, J.D.; Lee, K.S.; Ro, J.S. Quantitative determination of 5-hydroxymethyl-2-furaldehyde in the Rehmanniae Radix Preparata samples at various processing stages. Kor. J. Pharmacogn. 2001, 32, 116–120. [Google Scholar]
- Jin, C.; Yan, C.; Luo, Y.; Li, B.; He, J.; Xiao, X. Fast and direct quantification of underivatized muscone by ultra performance liquid chromatography coupled with evaporative light scattering detection. J. Sep. Sci. 2013, 36, 1762–1767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Tao, Y.; Hong, X.; Wang, Z. Steroids in musk by gas chromatography/mass spectrometry. Chin. Tradit. Pat. Med. 2005, 27, 79–83. [Google Scholar]
- Lee, J.S.; Hong, S.S.; Kim, H.G.; Lee, H.W.; Kim, W.Y.; Lee, S.K.; Son, C.G. Gongjin-dan enhances hippocampal memory in a mouse model of scopolamine-induced amnesia. PLoS ONE 2016, 11, e0159823. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Jiang, C.; Hu, Y.; Zhao, X.; Shi, C.; Yu, Y.; Liu, C.; Tao, Y.; Pan, H.; Feng, Y.; et al. Immunoenhancement effect of Rehmannia glutinosa polysaccharide on lymphocyte proliferation and dendritic cell. Carbohydr. Polym 2013, 96, 516–521. [Google Scholar] [CrossRef]
- Liu, Y.F.; Liang, D.; Luo, H.; Hao, Z.Y.; Wang, Y.; Zhang, C.L.; Zhang, Q.J.; Chen, R.Y.; Yu, D.Q. Hepatoprotective iridoid glycosides from the roots of Rehmannia glutinosa. J. Nat. Prod 2012, 75, 1625–1631. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, A.; Jiang, B.; Bao, Y.; Wang, J.; An, L. Further pharmacological evidence of the neuroprotective effect of catalpol from Rehmannia glutinosa. Phytomedicine 2008, 15, 484–490. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonisation (ICH). Guidance for Industry, Q2B, Validation of Analytical Procedures: Methodology; Food and Drug Administration: Rockville, MD, USA, 1996. [Google Scholar]
- Jiang, M.; Zhou, M.; Han, Y.; Xing, L.; Zhao, H.; Dong, L.; Bai, G.; Luo, G. Identification of NF-κB inhibitors in Xuebijing injection for sepsis treatment based on bioactivity-integrated UPLC-Q/TOF. J. Ethnopharmacol. 2013, 147, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, X.; Dai, W.; Yan, S.; Huang, H.; Liang, X.; Li, Y.; Zhang, W. Chemical fingerprinting of Liuwei Dihuang Pill and simultaneous determination of its major bioactive constituents by HPLC coupled with multiple detections of DAD, ELSD and ESI-MS. J. Pharm. Biomed. Anal. 2009, 49, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Dan, G.; Cho, C.W.; Vinh, L.B.; Kim, J.H.; Cho, K.W.; Kim, Y.H.; Kang, J.S. Simultaneous qualitative and quantitative analysis of morroniside and hederacoside D in extract mixture of Cornus officinalis and Stauntonia hexaphylla leaves to improve benign prostatic hyperplasia by HPLC–UV. Anal. Sci. Technol. 2020, 33, 224–231. [Google Scholar]
- Song, Y.; Li, S.L.; Wu, M.H.; Li, H.J.; Li, P. Qualitative and quantitative analysis of iridoid glycosides in the flower buds of Lonicera species by capillary high performance liquid chromatography coupled with mass spectrometric detector. Anal. Chim. Acta 2006, 564, 211–218. [Google Scholar] [CrossRef]
- Kim, M.K.; Yang, D.H.; Jung, M.; Jung, E.H.; Eom, H.Y.; Suh, J.H.; Min, J.W.; Kim, U.; Min, H.; Kim, J.; et al. Simultaneous determination of chromones and coumarins in Radix Saposhnikoviae by high performance liquid chromatography with diode array and tandem mass detectors. J. Chromatogr. A 2011, 1218, 6319–6330. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Analyte 1 | Ion Mode | Molecular Weight | MRM Conditions | Cone Voltage (V) | Collision Energy (eV) |
|---|---|---|---|---|---|
| 1 | negative | 170.02 | 169.0 → 125.0 | 25 | 15 |
| 2 | negative | 406.15 | 405.4 → 243.2 | 30 | 13 |
| 3 | positive | 126.03 | 127.0 → 109.0 | 20 | 10 |
| 4 | positive | 390.15 | 391.4 → 229.2 | 20 | 10 |
| 5 | positive | 408.14 | 409.4 → 247.2 | 30 | 15 |
| 6 | positive | 328.13 | 329.2 → 229.0 | 35 | 20 |
| 7 | positive | 328.13 | 329.2 → 229.0 | 35 | 20 |
| Analyte 1 | Measured Wavelength (nm) | Linear Range (µg/mL) | Regression Equation 2 | r2 | LOD (µg/mL) | LOQ (µg/mL) |
|---|---|---|---|---|---|---|
| 1 | 270 | 0.31–20.00 | y = 66,489.40x + 3119.59 | 1.0000 | 0.03 | 0.10 |
| 2 | 280 | 0.47–30.00 | y = 92,324.67x + 666.45 | 1.0000 | 0.03 | 0.09 |
| 3 | 240 | 0.31–20.00 | y = 47,649.17x + 2644.87 | 1.0000 | 0.02 | 0.06 |
| 4 | 235 | 0.47–30.00 | y = 22,917.18x + 1539.24 | 1.0000 | 0.04 | 0.13 |
| 5 | 335 | 0.31–20.00 | y = 45,241.57x + 2789.69 | 1.0000 | 0.02 | 0.06 |
| 6 | 330 | 1.56–100.00 | y = 48,629.16x + 25,391.82 | 1.0000 | 0.05 | 0.16 |
| 7 | 330 | 1.56–100.00 | y = 32,740.22x + 12,709.59 | 1.0000 | 0.04 | 0.11 |
| Analyte 1 | Spiked Amount (µg/mL) | Found Amount (µg/mL) | Recovery (%) | SD 2 | RSD (%) |
|---|---|---|---|---|---|
| 1 | 1.00 | 1.04 | 104.00 | 1.69 | 1.62 |
| 2.00 | 2.11 | 105.65 | 1.05 | 0.99 | |
| 4.00 | 4.17 | 104.14 | 0.56 | 0.54 | |
| 2 | 2.00 | 1.94 | 97.19 | 1.74 | 1.79 |
| 4.00 | 3.97 | 99.37 | 0.74 | 0.74 | |
| 8.00 | 7.74 | 96.73 | 0.53 | 0.55 | |
| 3 | 1.00 | 1.03 | 102.81 | 1.38 | 1.34 |
| 2.00 | 2.13 | 106.41 | 0.56 | 0.53 | |
| 4.00 | 4.28 | 107.04 | 0.72 | 0.68 | |
| 4 | 1.00 | 0.97 | 97.16 | 1.75 | 1.80 |
| 2.00 | 2.01 | 100.30 | 0.15 | 0.15 | |
| 4.00 | 4.03 | 100.75 | 1.30 | 1.29 | |
| 5 | 1.00 | 1.02 | 102.07 | 1.62 | 1.59 |
| 2.00 | 2.05 | 102.54 | 2.11 | 2.06 | |
| 4.00 | 4.21 | 105.31 | 1.20 | 1.14 | |
| 6 | 6.00 | 6.03 | 100.29 | 0.83 | 0.83 |
| 15.00 | 15.77 | 105.16 | 0.31 | 0.29 | |
| 30.00 | 32.44 | 108.17 | 0.47 | 0.43 | |
| 7 | 8.00 | 7.50 | 93.70 | 0.62 | 0.67 |
| 20.00 | 19.40 | 97.01 | 0.23 | 0.24 | |
| 40.00 | 40.92 | 102.30 | 0.53 | 0.52 |
| Analyte 1 | Conc. (µg/mL) | Intraday (n = 5) | Interday (n = 5) | ||||
|---|---|---|---|---|---|---|---|
| Observed Conc. (µg/mL) | Precision (RSD, %) | Accuracy (%) | Observed Conc. (µg/mL) | Precision (RSD, %) | Accuracy (%) | ||
| 1 | 5.00 | 5.19 | 0.17 | 103.88 | 5.21 | 0.53 | 104.27 |
| 10.00 | 10.41 | 0.89 | 104.12 | 10.44 | 1.01 | 104.37 | |
| 20.00 | 20.38 | 0.32 | 101.88 | 20.52 | 0.94 | 102.59 | |
| 2 | 7.50 | 7.89 | 0.28 | 105.13 | 7.91 | 0.96 | 105.52 |
| 15.00 | 15.74 | 0.70 | 104.94 | 15.77 | 1.22 | 105.12 | |
| 30.00 | 30.93 | 0.63 | 103.10 | 31.08 | 1.08 | 103.60 | |
| 3 | 5.00 | 5.18 | 0.17 | 103.62 | 5.21 | 0.52 | 104.17 |
| 10.00 | 10.39 | 0.68 | 103.87 | 10.42 | 0.96 | 104.20 | |
| 20.00 | 20.40 | 0.40 | 101.98 | 20.54 | 0.91 | 102.71 | |
| 4 | 7.50 | 7.78 | 0.18 | 103.80 | 7.81 | 0.48 | 104.11 |
| 15.00 | 15.56 | 0.65 | 103.73 | 15.62 | 1.00 | 104.13 | |
| 30.00 | 30.55 | 0.47 | 101.84 | 30.79 | 0.90 | 102.62 | |
| 5 | 5.00 | 5.19 | 0.06 | 103.82 | 5.22 | 0.52 | 104.30 |
| 10.00 | 10.40 | 0.86 | 104.01 | 10.43 | 1.00 | 104.29 | |
| 20.00 | 20.33 | 0.32 | 101.65 | 20.48 | 0.88 | 102.42 | |
| 6 | 25.00 | 26.01 | 0.08 | 104.03 | 26.13 | 0.48 | 104.52 |
| 50.00 | 52.09 | 0.69 | 104.19 | 52.22 | 0.94 | 104.44 | |
| 100.00 | 101.91 | 0.38 | 101.91 | 102.53 | 0.84 | 102.53 | |
| 7 | 25.00 | 26.06 | 0.08 | 104.24 | 26.18 | 0.46 | 104.71 |
| 50.00 | 52.06 | 0.77 | 104.13 | 52.19 | 0.92 | 104.37 | |
| 100.00 | 102.10 | 0.38 | 102.10 | 102.65 | 0.85 | 102.65 | |
| Analyte 2 | GJD–1 | GJD–2 | GJD–3 | GJD–4 | GJD–5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | |
| 1 | 0.19 | 0.85 | 0.09 | 0.90 | 0.13 | 0.61 | 0.06 | 0.89 | 0.23 | 0.42 |
| 2 | 0.77 | 0.29 | 0.15 | 0.23 | 0.01 | 1.66 | 0.08 | 1.69 | 1.99 | 0.16 |
| 3 | 0.39 | 0.36 | 0.12 | 0.30 | 0.43 | 0.40 | 0.44 | 0.21 | 0.15 | 0.15 |
| 4 | 0.58 | 0.41 | 0.42 | 0.27 | 0.71 | 0.29 | 0.65 | 0.03 | 0.38 | 0.19 |
| 5 | 0.28 | 0.35 | 0.22 | 0.32 | 0.91 | 0.70 | 0.22 | 0.20 | 0.30 | 0.62 |
| 6 | 2.78 | 0.18 | 3.24 | 0.10 | 7.06 | 0.11 | 2.76 | 0.18 | 2.57 | 0.03 |
| 7 | 4.03 | 0.28 | 3.87 | 0.21 | 6.54 | 0.03 | 2.35 | 0.20 | 3.24 | 0.06 |
| Analyte 1 | Retention Time (min) | Linear Range (µg/mL) | Regression Equation 2 | r2 | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|---|---|---|---|
| 1 | 1.06 | 0.10–5.00 | y = 0.61x − 14.81 | 0.9967 | 3.27 | 9.82 |
| 2 | 1.08 | 0.10–5.00 | y = 0.47x − 6.36 | 0.9917 | 3.50 | 10.49 |
| 3 | 1.13 | 0.10–5.00 | y = 23.77x + 425.4 | 0.9929 | 1.61 | 4.82 |
| 4 | 1.18 | 0.10–5.00 | y = 1.01x + 12.56 | 0.9921 | 9.92 | 29.75 |
| 5 | 1.94 | 0.10–5.00 | y = 5.33x + 114.23 | 0.9943 | 3.56 | 10.68 |
| 6 | 8.23 | 0.10–5.00 | y = 189.03x + 1684.86 | 0.9945 | 0.15 | 0.46 |
| 7 | 8.33 | 0.10–5.00 | y = 168.94x + 2700.37 | 0.9959 | 0.16 | 0.48 |
| Analyte 2 | GJD–1 | GJD–2 | GJD–3 | GJD–4 | GJD–5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | |
| 1 | 0.30 | 3.97 | 0.11 | 2.31 | 0.19 | 1.16 | 0.06 | 1.57 | 0.37 | 2.62 |
| 2 | 0.67 | 2.45 | 0.29 | 1.84 | 0.73 | 0.69 | 0.78 | 4.79 | 0.24 | 3.79 |
| 3 | 0.44 | 0.93 | 0.11 | 4.57 | ND 3 | – | 0.02 | 3.58 | 1.34 | 0.85 |
| 4 | 0.64 | 3.20 | 0.57 | 2.41 | 0.91 | 3.44 | 0.78 | 1.95 | 0.46 | 2.62 |
| 5 | 0.75 | 2.47 | 0.64 | 1.30 | 2.41 | 4.65 | 0.77 | 1.71 | 0.87 | 2.80 |
| 6 | 4.03 | 0.67 | 4.89 | 2.23 | 10.75 | 1.20 | 4.44 | 1.53 | 3.86 | 0.81 |
| 7 | 4.49 | 0.49 | 4.45 | 3.08 | 7.81 | 2.16 | 2.95 | 0.24 | 3.62 | 0.33 |
| Analyte | Spiked Amount (µg/mL) | Found Amount (µg/mL) | Recovery (%) | SD | RSD (%) |
|---|---|---|---|---|---|
| Muscone | 110.00 | 109.00 | 99.13 | 2.53 | 2.55 |
| 275.00 | 277.30 | 100.84 | 1.49 | 1.48 | |
| 550.00 | 590.30 | 107.33 | 0.83 | 0.77 |
| Analyte | Conc. (µg/mL) | Intraday (n = 5) | Interday (n = 5) | ||||
|---|---|---|---|---|---|---|---|
| Observed Conc. (µg/mL) | Precision (RSD, %) | Accuracy (%) | Observed Conc. (µg/mL) | Precision (RSD, %) | Accuracy (%) | ||
| Muscone | 2.50 | 2.49 | 0.67 | 99.72 | 2.52 | 1.30 | 100.68 |
| 5.00 | 4.91 | 0.97 | 98.19 | 4.97 | 0.95 | 99.40 | |
| 10.00 | 10.26 | 0.66 | 102.56 | 10.22 | 1.44 | 102.19 | |
| Analyte | GJD–1 | GJD–2 | GJD–3 | GJD–4 | GJD–5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | Mean (mg/g) | RSD (%) | |
| Muscone | 0.48 | 3.43 | 0.51 | 14.31 | 0.52 | 4.13 | 0.49 | 4.61 | 0.44 | 2.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, C.-S.; Shin, H.-K. Quantitative Analysis of Eight Compounds in Traditional Korean Medicine, Gongjindan Using HPLC, UPLC–MS/MS, and GC–MS/MS Systems. Separations 2023, 10, 231. https://doi.org/10.3390/separations10040231
Seo C-S, Shin H-K. Quantitative Analysis of Eight Compounds in Traditional Korean Medicine, Gongjindan Using HPLC, UPLC–MS/MS, and GC–MS/MS Systems. Separations. 2023; 10(4):231. https://doi.org/10.3390/separations10040231
Chicago/Turabian StyleSeo, Chang-Seob, and Hyeun-Kyoo Shin. 2023. "Quantitative Analysis of Eight Compounds in Traditional Korean Medicine, Gongjindan Using HPLC, UPLC–MS/MS, and GC–MS/MS Systems" Separations 10, no. 4: 231. https://doi.org/10.3390/separations10040231