

Development and Validation of a Confirmatory Method for the Determination of 12 Coccidiostat Residues in Eggs and Muscle by Means of Liquid Chromatography Coupled to Hybrid High Resolution Mass Spectrometry

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals, Reagents, Stock and Intermediates Solutions
2.2. Chromatographic Conditions
2.3. MS Conditions
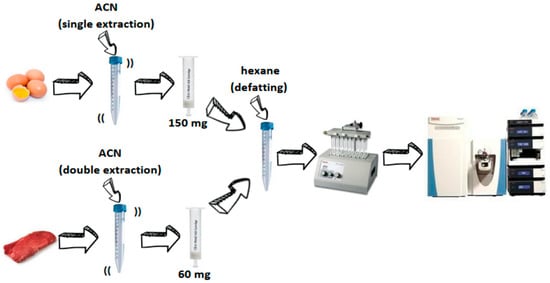
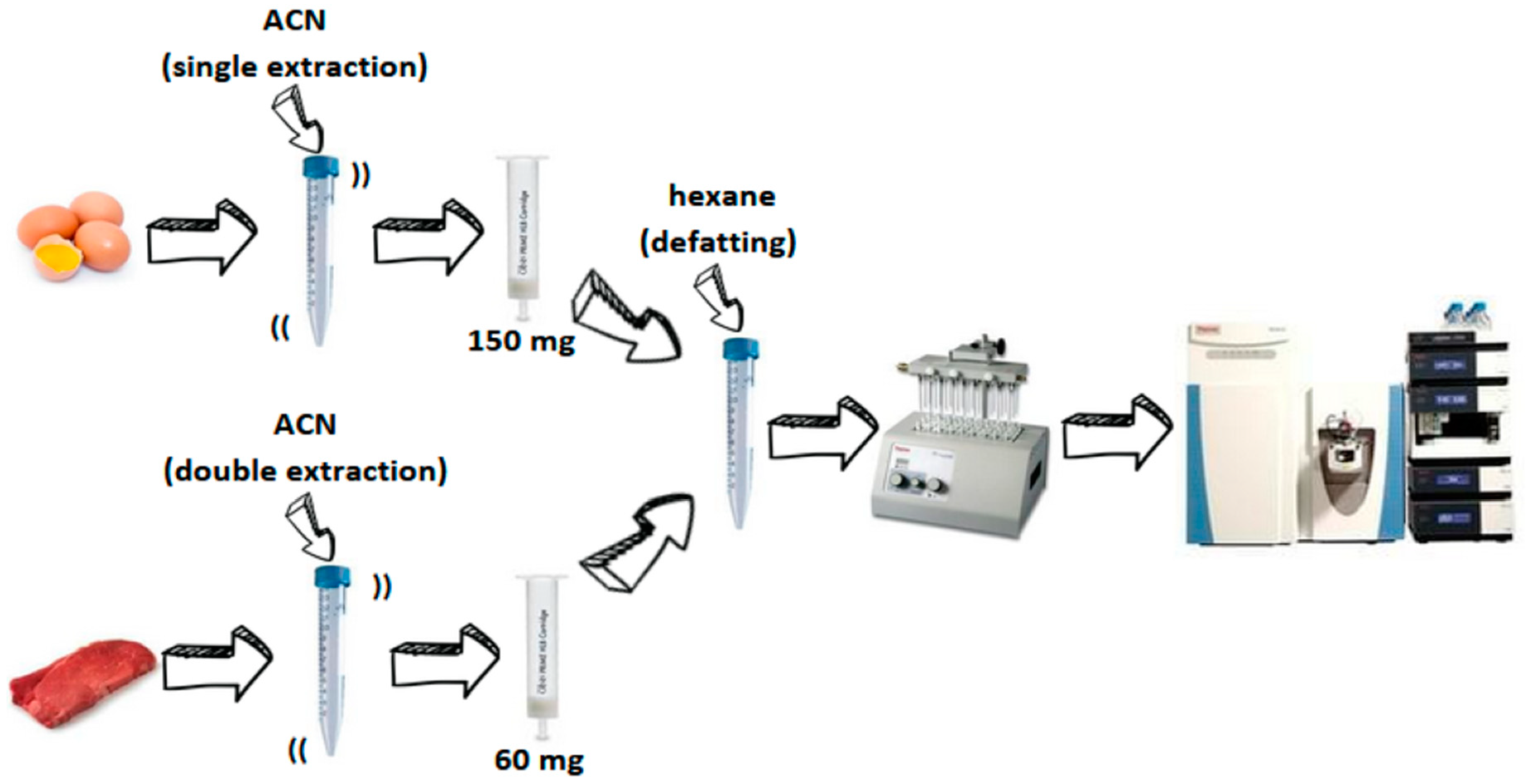
2.4. Sample Preparation
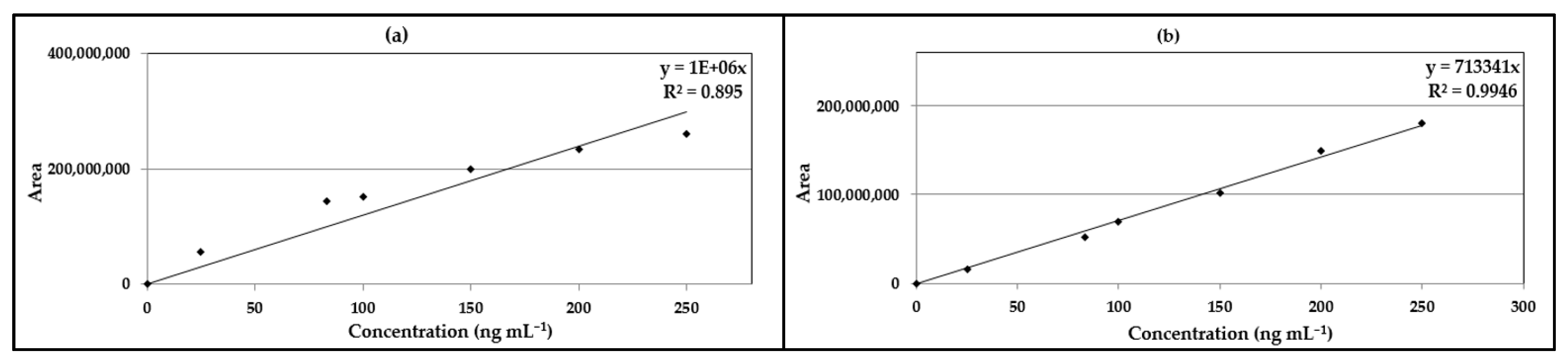
2.5. Method Validation
2.6. Real Samples Analysis
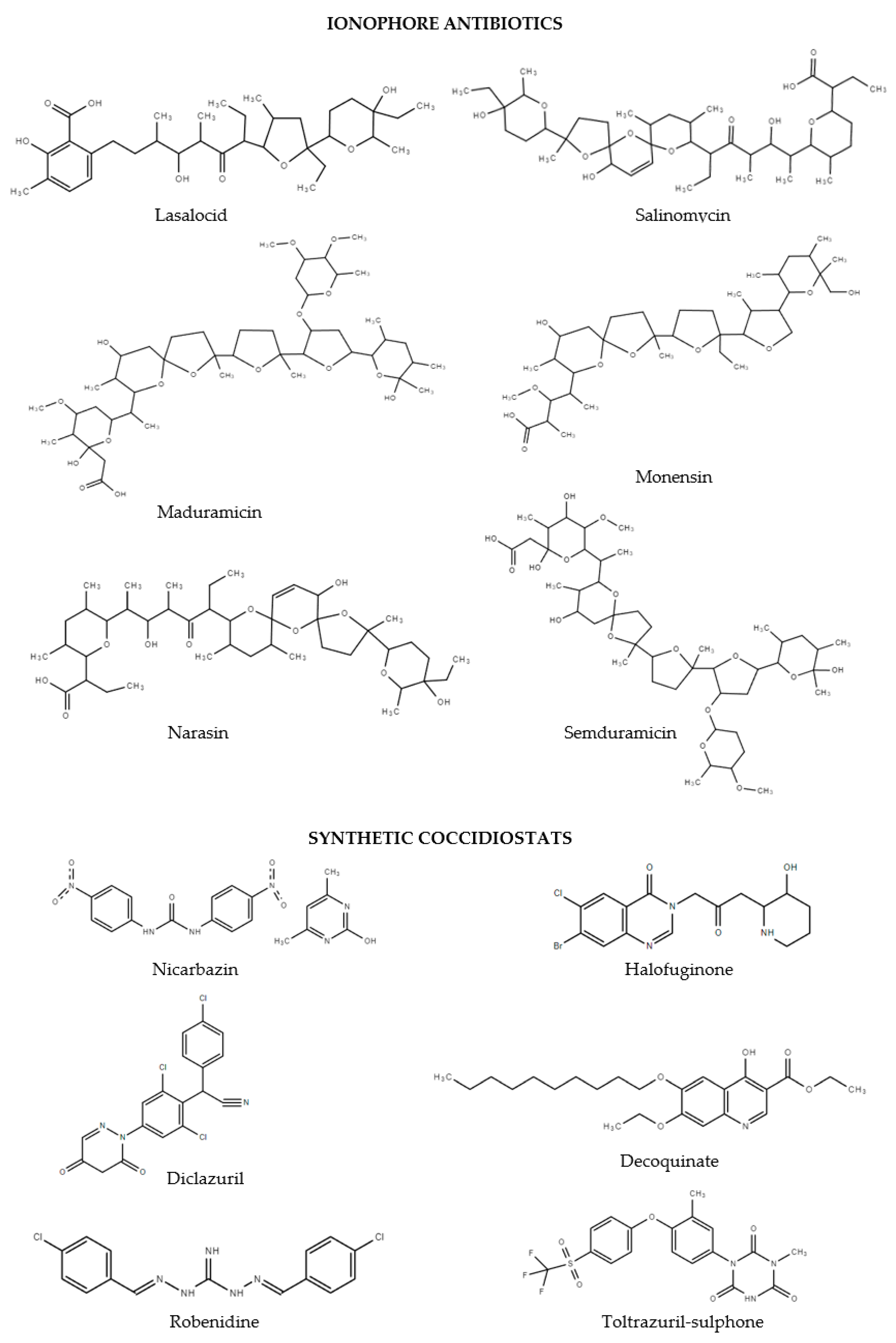
3. Results and Discussion
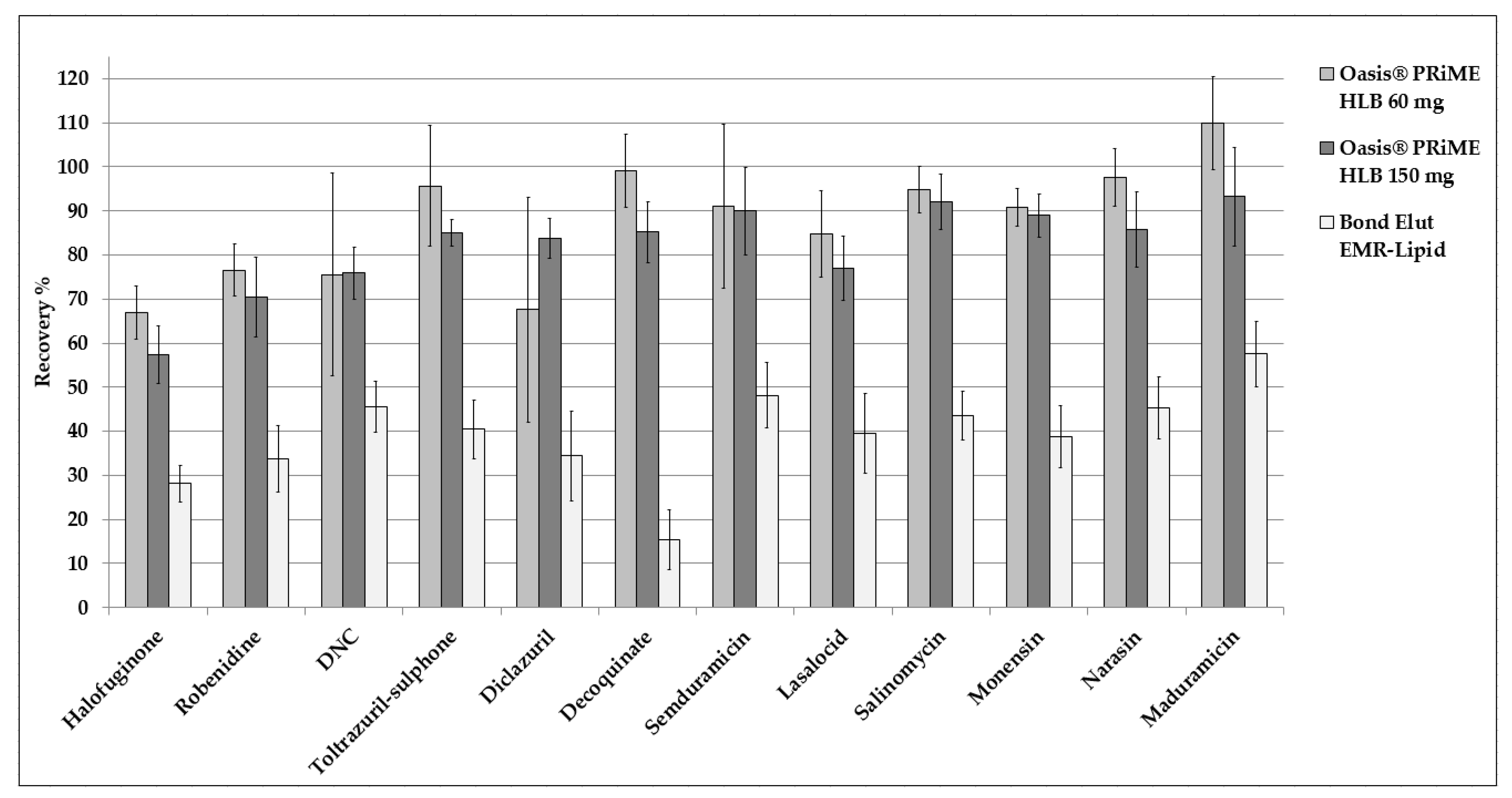
3.1. Sample Preparation
3.2. Optimization of LC-HRMS/MS Conditions
3.3. Method Validation
3.4. Real Samples Analysis and QC Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Clarke, L.; Fodey, T.L.; Crooks, S.R.H.; Moloney, M.; O’Mahony, J.; Delahaut, P.; O’Kennedy, R.; Danaher, M. A Review of Coccidiostats and the Analysis of Their Residues in Meat and Other Food. Meat Sci. 2014, 97, 358–374. [Google Scholar] [CrossRef] [PubMed]
- The Commission of the European Communities. Commission Regulation (EU) No 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin. Off. J. Eur. Communities 2009. Available online: http://data.europa.eu/eli/reg/2010/37(1)/2022-05-09 (accessed on 9 March 2023).
- Commission Regulation. Reg 388/2011. 2006, Volume 1881, pp. 1–5. Available online: http://data.europa.eu/eli/reg_impl/2011/388/2013-11-12 (accessed on 9 March 2023).
- Union, T.; Journal, O.; Union, E. Reg 124/2009. Ec 2074/2005 2017. Volume 10, pp. 1–21. Available online: http://data.europa.eu/eli/reg/2009/124/2020-04-27 (accessed on 9 March 2023).
- The Commission of the European Communities. Reg 495/2011. 2011, Volume 9. Available online: http://data.europa.eu/eli/reg_impl/2011/495/oj (accessed on 9 March 2023).
- European Commission. Reg 2094/2021. 2018, Volume 2016, pp. 48–119. Available online: http://data.europa.eu/eli/reg_impl/2021/2094/oj (accessed on 9 March 2023).
- The Commission of the European Communities. Reg 1417/2015. 2015, Volume 13, pp. 20–22. Available online: http://data.europa.eu/eli/reg_impl/2015/1417/oj (accessed on 9 March 2023).
- The Commission of the European Communities. Reg 118/2012. 2012. No. 118. pp. 36–39. Available online: http://data.europa.eu/eli/reg_impl/2012/118/oj (accessed on 9 March 2023).
- Commission Regulation. Reg 532/2011. 2006, Volume 1881, pp. 1–5. Available online: http://data.europa.eu/eli/reg_impl/2011/532/2013-11-12 (accessed on 9 March 2023).
- The Commission of the European Communities. Reg 875/2010. 2010, Volume 8, pp. 4–6. Available online: http://data.europa.eu/eli/reg/2010/875/oj (accessed on 9 March 2023).
- Union, T.; Journal, O.; Union, E. Reg 885/2010. Ec 2074/2005 2017. Volume 10, pp. 1–21. Available online: http://data.europa.eu/eli/reg/2010/885/oj (accessed on 9 March 2023).
- The Commission of the European Communities. Scientific Opinion on the Safety and Efficacy of Koffogran (Nicarbazin) as a Feed Additive for Chickens for Fattening. EFSA J. 2010, 8, 2011–2013. [Google Scholar] [CrossRef]
- Barreto, F.; Ribeiro, C.; Hoff, R.B.; Costa, T.D. A Simple and High-Throughput Method for Determination and Confirmation of 14 Coccidiostats in Poultry Muscle and Eggs Using Liquid Chromatography–Quadrupole Linear Ion Trap-Tandem Mass Spectrometry (HPLC–QqLIT-MS/MS): Validation According to European. Talanta 2017, 168, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Buiarelli, F.; Di Filippo, P.; Riccardi, C.; Pomata, D.; Giannetti, L.; Neri, B.; Rago, D. Liquid Chromatography Tandem Mass Spectrometry Analysis of Synthetic Coccidiostats in Eggs. Separations 2017, 4, 15. [Google Scholar] [CrossRef]
- Galarini, R.; Fioroni, L.; Moretti, S.; Pettinacci, L.; Dusi, G. Development and Validation of a Multi-Residue Liquid Chromatography-Tandem Mass Spectrometry Confirmatory Method for Eleven Coccidiostats in Eggs. Anal. Chim. Acta 2011, 700, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-Residue Methodology for the Determination of 16 Coccidiostats in Animal Tissues and Eggs by Hydrophilic Interaction Liquid Chromatography–Tandem Mass Spectrometry. Food Chem. 2019, 275, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Wu, X.; Zhang, J.; Duan, H.; Chu, X.; Wu, Y. Development of a Rapid LC-MS-MS Method for Multi-Class Determination of 14 Coccidiostat Residues in Eggs and Chicken. Chromatographia 2009, 69, 1083–1088. [Google Scholar] [CrossRef]
- Clarke, L.; Moloney, M.; O’Mahony, J.; O’Kennedy, R.; Danaher, M. Determination of 20 Coccidiostats in Milk, Duck Muscle and Non-Avian Muscle Tissue Using UHPLC-MS/MS. Food Addit. Contam.-Part A 2013, 30, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Lai, Y.H.; Huang, C.N.; Peng, G.J.; Liao, C.D.; Kao, Y.M.; Tseng, S.H.; Wang, D.Y. Multi-Residue Analysis Using Liquid Chromatography Tandem Mass Spectrometry for Detection of 20 Coccidiostats in Poultry, Livestock, and Aquatic Tissues. J. Food Drug Anal. 2019, 27, 703–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Liu, J.; Zhao, X.; Xie, K.; Diao, Z.; Zhang, G.; Zhang, T.; Dai, G. Determination of Eight Coccidiostats in Eggs by Liquid-Liquid Extraction-Solid-Phase Extraction and Liquid Chromatography-Tandem Mass Spectrometry. Molecules 2020, 25, 987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, K.; Vincent, U.; von Holst, C. Analysis of Antimicrobial Agents in Pig Feed by Liquid Chromatography Coupled to Orbitrap Mass Spectrometry. J. Chromatogr. A 2013, 1293, 60–74. [Google Scholar] [CrossRef] [PubMed]
- Matus, J.L.; Boison, J.O. A Multi-Residue Method for 17 Anticoccidial Drugs and Ractopamine in Animal Tissues by Liquid Chromatography-Tandem Mass Spectrometry and Time-of-Flight Mass Spectrometry. Drug Test. Anal. 2016, 8, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusko, J.; Jansons, M.; Pugajeva, I.; Zacs, D.; Bartkevics, V. Development and Optimization of Confirmatory Liquid Chromatography—Orbitrap Mass Spectrometry Method for the Determination of 17 Anticoccidials in Poultry and Eggs. J. Pharm. Biomed. Anal. 2019, 164, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Commission, E. Commission Implementing Regulation (EU) 2021/808 of 22 March 2021 on the Performance of Analytical Methods for Residues of Pharmacologically Active Substances Used in Food-Producing Animals and on the Interpretation of Results as Well as on the Methods To. Off. J. Eur. Union 2021, 180, 84–109. [Google Scholar]
- Ha, J.; Song, G.; Ai, L.F.; Li, J.C. Determination of Six Polyether Antibiotic Residues in Foods of Animal Origin by Solid Phase Extraction Combined with Liquid Chromatography-Tandem Mass Spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1017–1018, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.R.; Silva, L.J.G.; Pereira, A.M.P.T.; Esteves, A.; Duarte, S.C.; Pena, A. Coccidiostats and Poultry: A Comprehensive Review and Current Legislation. Foods 2022, 11, 2738. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, M.; Szprengier-Juszkiewicz, T.; Jedziniak, P.; Śledzińska, E.; Szymanek-Bany, I.; Korycińska, B.; Pietruk, K.; Zmudzki, J. Residue Control of Coccidiostats in Food of Animal Origin in Poland during 2007–2010. Food Addit. Contam. Part B Surveill. 2011, 4, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Rauniyar, N. Parallel Reaction Monitoring: A Targeted Experiment Performed Using High Resolution and High Mass Accuracy Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 28566–28581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, H. A Non-Covalent Dimer Formed in Electrospray Ionisation Mass Spectrometry Behaving as a Precursor for Fragmentations. Rapid Commun. Mass Spectrom. 2008. [Google Scholar] [CrossRef] [PubMed]
- European Comission. Document N0 SANTE 11312/2021. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. 2021, pp. 1–57. Available online: https://www.Eurl-Pesticides.Eu/Docs/Public/Tmplt_article.Asp?CntID=727 (accessed on 9 March 2023).
- Galarini, R.; Moretti, S.; Saluti, G. Quality Assurance and Validation General Considerations and Trends. In Chromatogr. Anal. Environ. Mass Spectrom. Based Approaches, 4th ed.; CRC Press: Boca Raton, FL, USA, 2017; p. 45. [Google Scholar]
- Magnusson, B.; Naikki, T.; Hovind, H.; Krysell, M. Handbook for Calculation of Measurement Uncertainty in Environmental Laboratories. NT TR 537, ed. 3.1. 2012. Available online: http://www.nordtest.info (accessed on 9 March 2023).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | RT (min) | RRT | Molecular Formula | Adduct | Monoisotopic Exact Mass (m/z) | CE (eV) | Fragment 1 Accurate Mass 1 (m/z) | Fragment 2 Accurate Mass (m/z) |
|---|---|---|---|---|---|---|---|---|
| Halofuginone-13C6 (IS) | 5.77 | - | 13C6C10H17BrClN3O3 | [M+H]+ | 420.0416 | 25 | 100.0757 | - |
| Halofuginone | 5.77 | 1.00 | C16H17BrClN3O3 | [M+H]+ | 414.0416 | 25 | 100.0757 | 120.0808 |
| Robenidine-d8 (IS) | 8.36 | - | C15H5D8Cl2N5 | [M+H]+ | 342.1123 | 25 | 159.0622 | - |
| Robenidine | 8.38 | 1.00 | C15H13Cl2N5 | [M+H]+ | 334.0621 | 25 | 138.0105 | 155.0372 |
| DNC-d8 (IS) | 12.09 | - | C13H2D8N4O5 | [M-H]− | 309.1080 | 45 | 141.0603 | - |
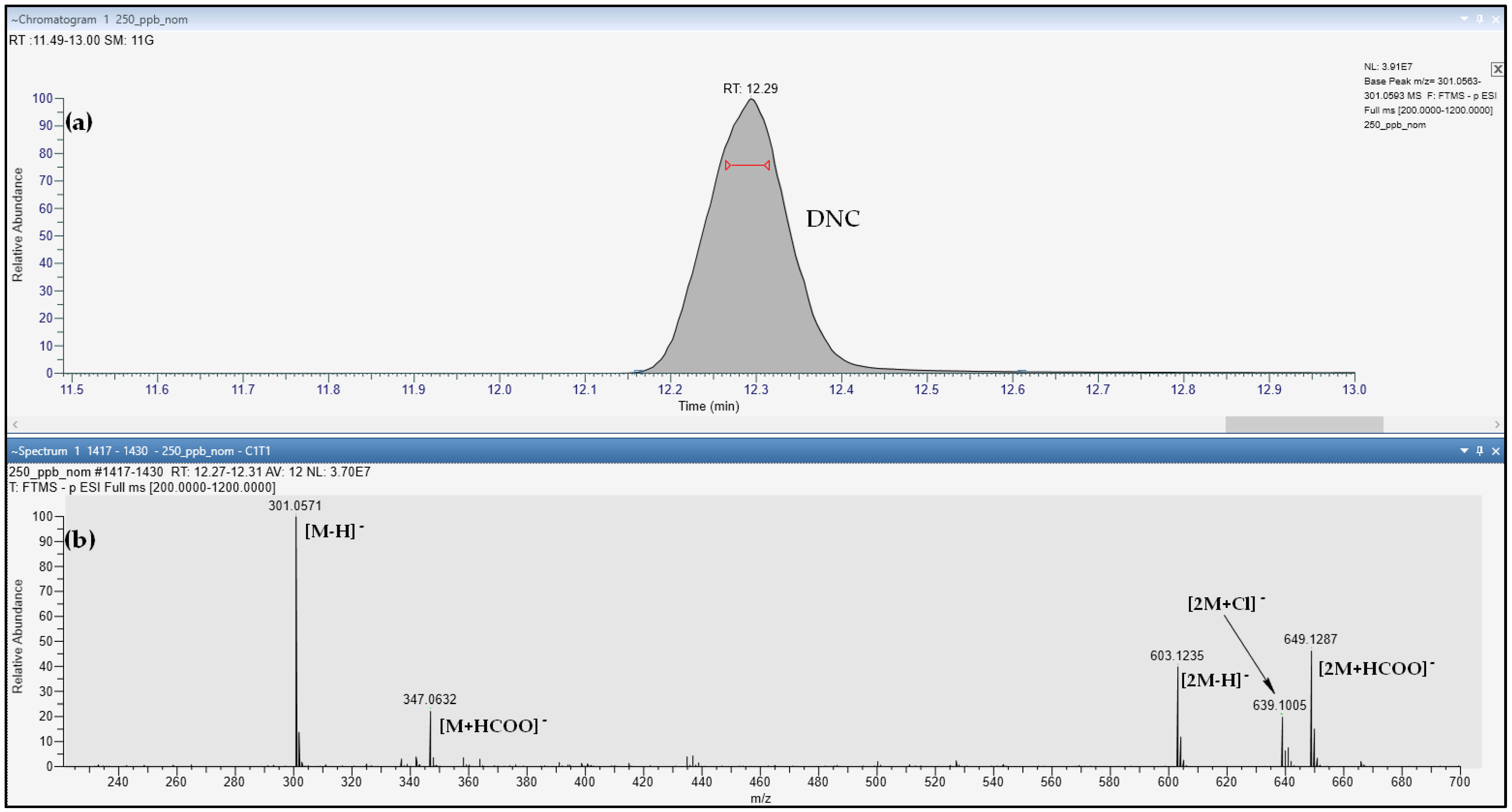
| DNC | 12.13 | 1.00 | C13H10N4O5 | [M-H]− | 301.0578 | 45 | 137.0352 | 107.0369 |
| Toltrazuril-sulphone | 12.18 | 0.92 | C18H14F3N3O6S | [M-H]− | 456.0484 | 10 | 456.0484 2 | - |
| Diclazuril | 12.72 | 1.02 | C17H9Cl3N4O2 | [M-H]− | 404.9718 | 35 | 333.9713 | 298.9785 |
| Diclazuril-methyl (IS) | 12.97 | - | C18H11Cl3N4O2 | [M-H]− | 418.9874 | 35 | 320.9760 | - |
| Toltrazuril-d3 (IS) | 13.19 | - | C18D3H11F3N3O4S | [M-H]− | 427.0772 | 10 | 427.0772 2 | - |
| Decoquinate-d5 (IS) | 14.90 | - | C24H30D5NO5 | [M+H]+ | 423.2902 | 40 | 255.1020 | - |
| Decoquinate | 14.99 | 1.01 | C24H35NO5 | [M+H]+ | 418.2588 | 40 | 250.0709 | 390.2275 |
| Semduramicin | 17.48 | 0.83 | C45H76O16 | [M+Na]+ | 895.5026 | 65 | 833.5019 | 705.4189 |
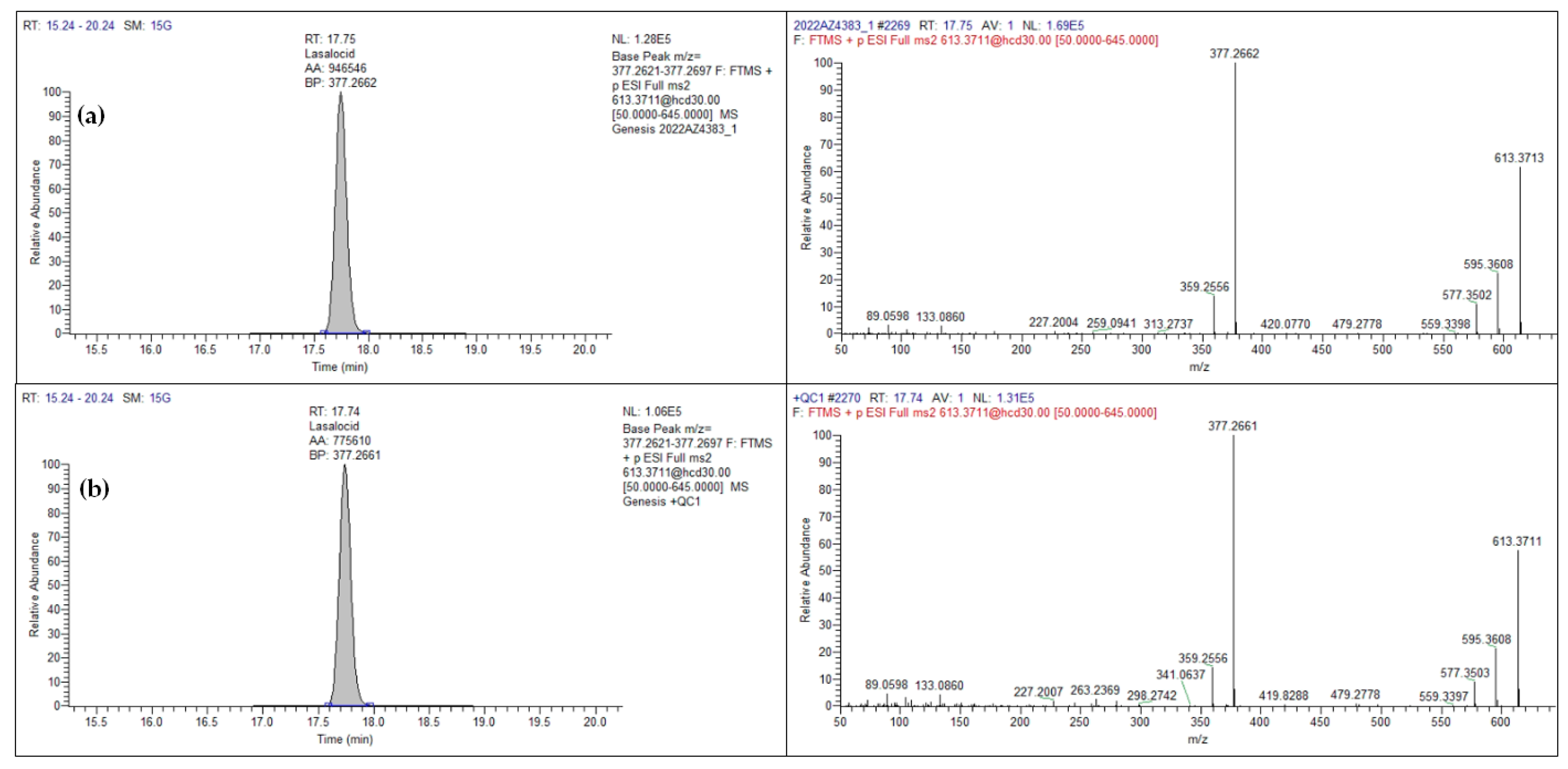
| Lasalocid | 17.65 | 0.84 | C34H54O8 | [M+Na]+ | 613.3711 | 30 | 377.2659 | 613.3711 |
| Salinomycin | 18.85 | 0.90 | C42H70O11 | [M+Na]+ | 773.4810 | 45 | 431.2401 | 531.3294 |
| Monensin | 19.01 | 0.91 | C36H62O11 | [M+Na]+ | 693.4184 | 70 | 461.2876 | 501.3186 |
| Narasin | 19.83 | 0.94 | C43H72O11 | [M+Na]+ | 787.4967 | 50 | 431.2409 | 531.3301 |
| Maduramicin | 20.16 | 0.96 | C47H80O17 | [M+Na]+ | 939.5288 | 75 | 877.5291 | 719.4343 |
| Nigericin (IS) | 21.00 | - | C40H67O11 | [M+H]+ | 747.4654 | 75 | 237.1093 | - |
| Eggs | |||||||
|---|---|---|---|---|---|---|---|
| Spiking Level (µg kg−1) | Number of Spiked Samples Day−1 | Concentration of Analyte Solution (µg mL−1) | Added Volume of Analyte Solution (µL) | IS Spiking Level (µg kg−1) | Concentration of IS Solution (µg mL−1) | Added Volume of IS Solution (µL) | Dilution Factor |
| 1 | 4 | 0.1 | 25 | 10 | 1 | 25 | 0.4 |
| 2 | 4 | 0.1 | 50 | 10 | 1 | 25 | 0.4 |
| 3.33 | 4 | 0.1 | 83.3 | 10 | 1 | 25 | 0.4 |
| 10 | 4 | 1 | 25 | 10 | 1 | 25 | 0.4 |
| 33.3 | 4 | 1 | 83.3 | 10 | 1 | 25 | 0.4 |
| 100 | 4 | 10 | 25 | 100 | 10 | 25 | 4 |
| 333 * | 4 | 10 | 83.3 | 100 | 10 | 25 | 4 |
| 1000 * | 4 | 10 | 250 | 1000 | 10 | 250 | 40 |
| Muscle | |||||||
| 1 | 4 | 0.1 | 25 | 10 | 1 | 25 | 0.4 |
| 2 | 4 | 0.1 | 50 | 10 | 1 | 25 | 0.4 |
| 3.33 | 4 | 0.1 | 83.3 | 10 | 1 | 25 | 0.4 |
| 10 | 4 | 1 | 25 | 10 | 1 | 25 | 0.4 |
| 33.3 | 4 | 1 | 83.3 | 10 | 1 | 25 | 0.4 |
| 100 | 4 | 10 | 25 | 100 | 10 | 25 | 4 |
| 333 * | 4 | 10 | 83.3 | 100 | 10 | 25 | 4 |
| 1000 * | 4 | 10 | 250 | 1000 | 10 | 250 | 40 |
| 3333 * | 4 | 100 | 83.3 | 1000 | 10 | 250 | 40 |
| 6000 * | 4 | 100 | 150 | 10000 | 100 | 250 | 200 |
| Time (min) | Polarity | Analytes in the Inclusion List |
|---|---|---|
| 3–11 | + | halofuginone, halofuginone-13C6, robenidine, robenidine-d8 |
| 11–14 | - | DNC, DNC-d8, toltrazuril-sulphone, toltrazuril-d3, diclazuril, diclazuril-methyl |
| 14–24 | + | decoquinate, decoquinate-d5, semduramicin, lasalocid, salinomycin, monensin, narasin, maduramicin, nigericin |
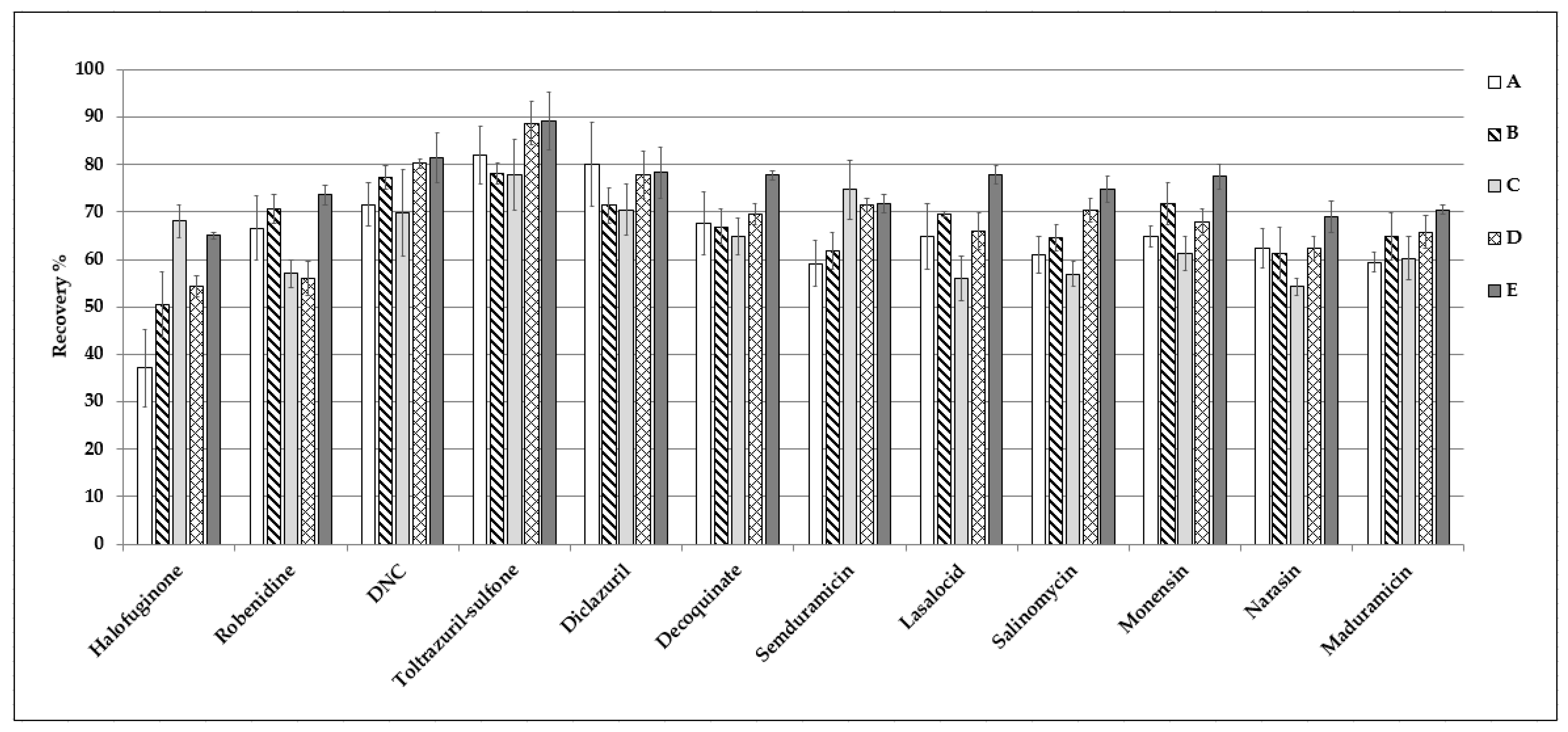
| Analyte | Mean Recovery (%) | CVr (%) | CVwR (%) | uc (%) | MRL or LL a (µg kg−1) | CCα (µg kg−1) | LOD (µg kg−1) | LOQ (µg kg−1) | ME (%) | CVME (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| Eggs | ||||||||||
| Halofuginone | 93 | 3.6 | 5.8 | 9.6 | 6 | 6.9 | 1 | 1 | 100 | 11 |
| Robenidine | 100 | 3.5 | 5.7 | 9.6 | 25 | 29 | 1 | 1 | 98 | 17 |
| DNC | 100 | 6.3 | 7.0 | 10 | 300 | 349 | 1 | 1 | 100 | 10 |
| Toltrazuril-sulphone b | 84 | 4.7 | 6.5 | 9.8 | Banned | - | 1 | - | 90 | 10 |
| Diclazuril | 102 | 5.1 | 6.4 | 9.8 | 2 | 2.3 | 1 | 1 | 111 | 11 |
| Decoquinate | 100 | 4.1 | 4.6 | 9.3 | 20 | 23 | 1 | 1 | 98 | 11 |
| Semduramicin | 83 | 5.6 | 8.9 | 11 | 2 | 2.4 | 1 | 1 | 96 | 15 |
| Lasalocid | 71 | 7.9 | 9.6 | 11 | 150 | 177 | 1 | 1 | 80 | 15 |
| Salinomycin | 70 | 5.4 | 13 | 13 | 3 | 3.6 | 1 | 1 | 63 | 15 |
| Monensin | 75 | 7.1 | 12 | 12 | 2 | 2.4 | 1 | 1 | 71 | 20 |
| Narasin | 68 | 5.5 | 7.7 | 10 | 2 | 2.3 | 1 | 1 | 81 | 19 |
| Maduramicin | 84 | 6.6 | 12 | 12 | 12 | 14 | 1 | 1 | 92 | 18 |
| Muscle | ||||||||||
| Halofuginone | 95 | 4.7 | 5.4 | 9.5 | Banned | 1 | 1 | 1 | 105 | 16 |
| Robenidine | 102 | 6.0 | 7.0 | 10 | 200 | 233 | 1 | 1 | 102 | 16 |
| DNC | 100 | 6.5 | 8.2 | 10 | 4000 | 4682 | 1 | 1 | 109 | 17 |
| Toltrazuril-sulphone | 89 | 6.3 | 10 | 11 | 100 | - | 1 | - | 84 | 12 |
| Diclazuril | 99 | 7.0 | 8.0 | 10 | 500 | 584 | 1 | 1 | 104 | 14 |
| Decoquinate | 102 | 6.6 | 8.2 | 10 | 500 | 585 | 1 | 1 | 99 | 3 |
| Semduramicin | 81 | 8.6 | 10 | 11 | 2 | 2.4 | 1 | 1 | 108 | 17 |
| Lasalocid | 76 | 7.0 | 8.8 | 11 | 60 | 70 | 1 | 1 | 106 | 14 |
| Salinomycin | 77 | 7.1 | 8.8 | 11 | 15 | 18 | 1 | 1 | 90 | 8 |
| Monensin | 80 | 7.1 | 11 | 12 | 8 | 10 | 1 | 1 | 95 | 17 |
| Narasin | 74 | 7.4 | 9.6 | 11 | 50 | 59 | 1 | 1 | 89 | 11 |
| Maduramicin | 84 | 9.3 | 13 | 13 | 30 | 36 | 1 | 1 | 90 | 13 |
| Analyte | Bovine R ± SD (%) | Swine R ± SD (%) |
|---|---|---|
| Halofuginone | 99 ± 1 | 110 ± 7 |
| Robenidine | 104 ± 1 | 101 ± 6 |
| DNC | 98 ± 9 | 94 ± 7 |
| Toltrazuril-sulphone | 79 ± 4 | 88 ± 7 |
| Diclazuril | 97 ± 4 | 101 ± 7 |
| Decoquinate | 98 ± 1 | 99 ± 7 |
| Semduramicin | 80 ± 3 | 83 ± 6 |
| Lasalocid | 74 ± 1 | 77 ± 6 |
| Salinomycin | 76 ± 2 | 82 ± 6 |
| Monensin | 79 ± 4 | 82 ± 5 |
| Narasin | 75 ± 4 | 77 ± 7 |
| Maduramicin | 77 ± 3 | 84 ± 10 |
| Analyte | Obtained Value ± SD (n = 2) (µg kg−1) | Assigned Value ± σ (µg kg−1) | Satisfactory Range (µg kg−1) | Accuracy (%) |
|---|---|---|---|---|
| Nicarbazin (as DNC fraction) a | 95.72 ± 7.32 a | 67.28 ± 17.43 | 32.41–102.14 | 142 |
| Salinomycin | 21.28 ± 0.64 | 20.19 ± 5.81 | 8.57–31.82 | 105 |
| Monensin | 14.35 ± 0.35 | 16.18 ± 4.82 | 6.55–25.81 | 89 |
| Diclazuril | 22.35 ± 1.12 | 20.54 ± 5.90 | 8.75–32.34 | 109 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellani, F.; Ricci, M.; Colagrande, M.N.; Scortichini, G.; Saluti, G. Development and Validation of a Confirmatory Method for the Determination of 12 Coccidiostat Residues in Eggs and Muscle by Means of Liquid Chromatography Coupled to Hybrid High Resolution Mass Spectrometry. Separations 2023, 10, 202. https://doi.org/10.3390/separations10030202
Castellani F, Ricci M, Colagrande MN, Scortichini G, Saluti G. Development and Validation of a Confirmatory Method for the Determination of 12 Coccidiostat Residues in Eggs and Muscle by Means of Liquid Chromatography Coupled to Hybrid High Resolution Mass Spectrometry. Separations. 2023; 10(3):202. https://doi.org/10.3390/separations10030202
Chicago/Turabian StyleCastellani, Federica, Matteo Ricci, Maria Novella Colagrande, Giampiero Scortichini, and Giorgio Saluti. 2023. "Development and Validation of a Confirmatory Method for the Determination of 12 Coccidiostat Residues in Eggs and Muscle by Means of Liquid Chromatography Coupled to Hybrid High Resolution Mass Spectrometry" Separations 10, no. 3: 202. https://doi.org/10.3390/separations10030202