
Histopathological Variants of Cutaneous Neurofibroma: A Compendious Review

Abstract
:1. Introduction
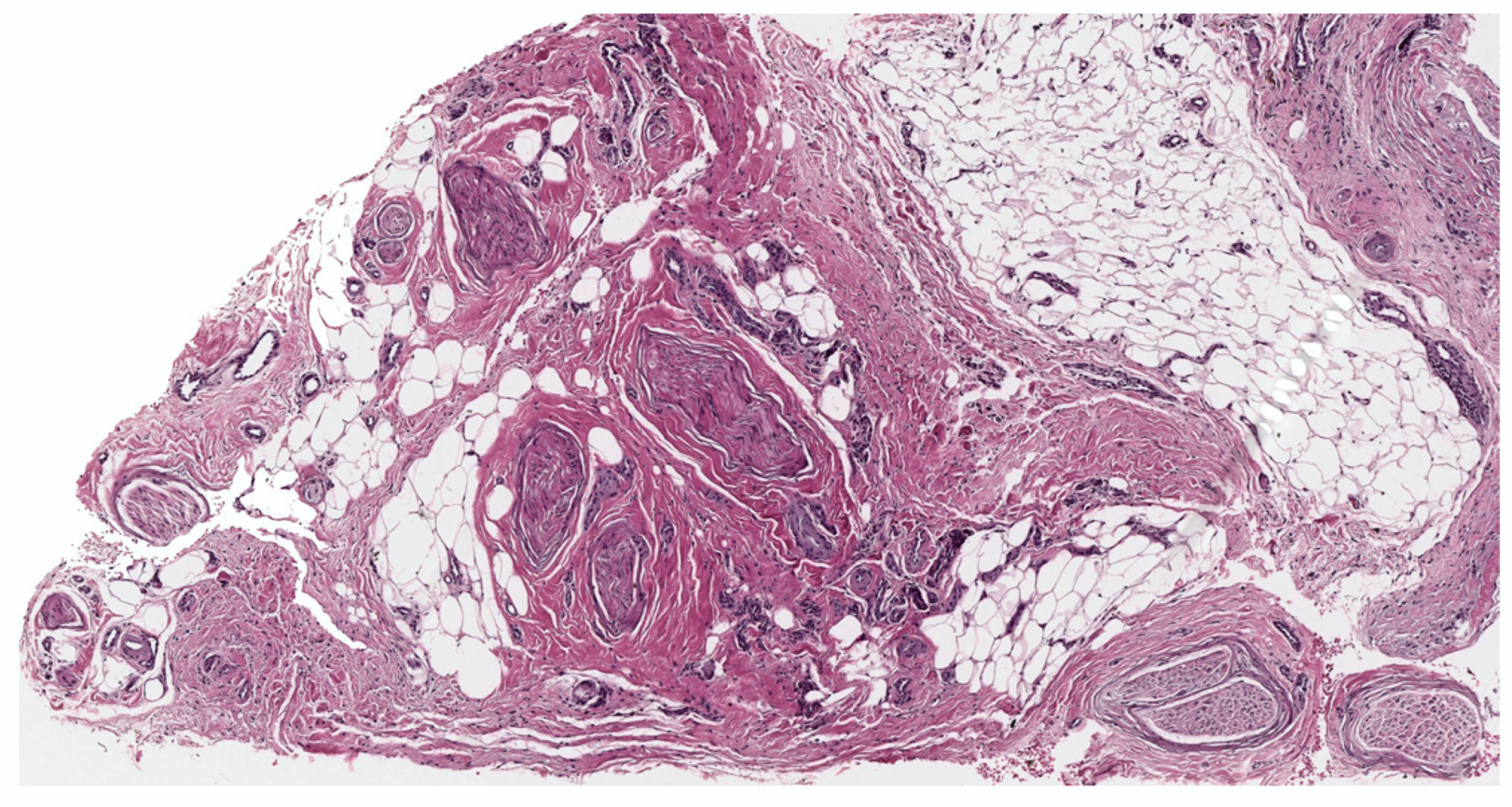
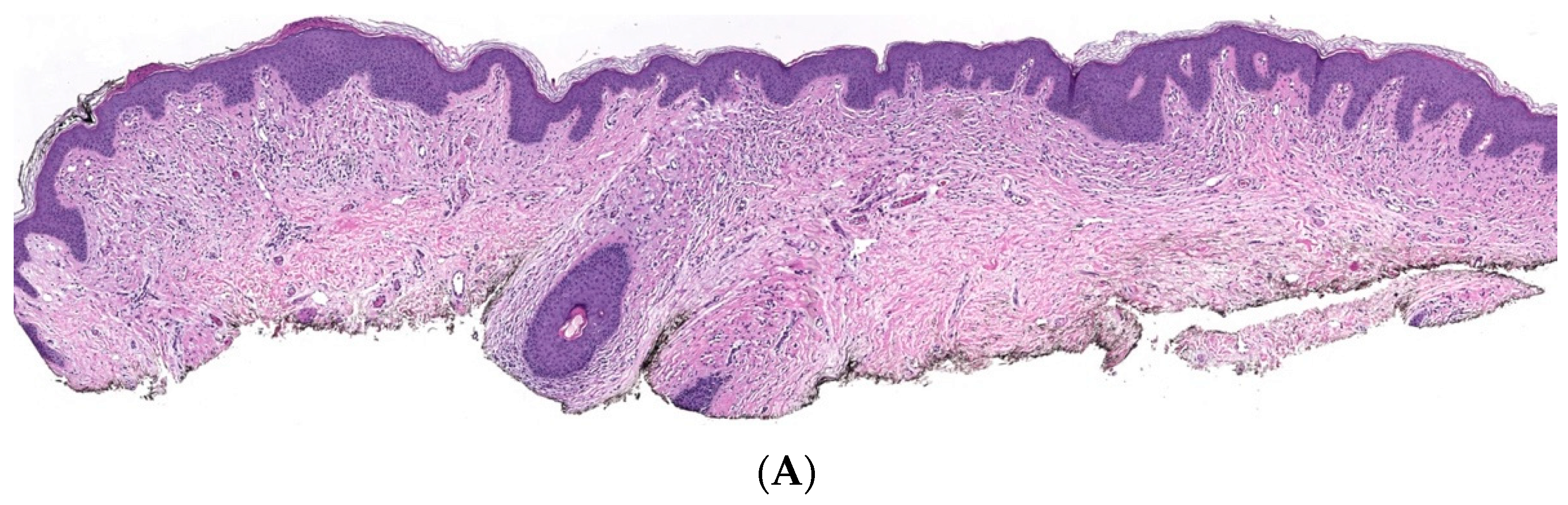
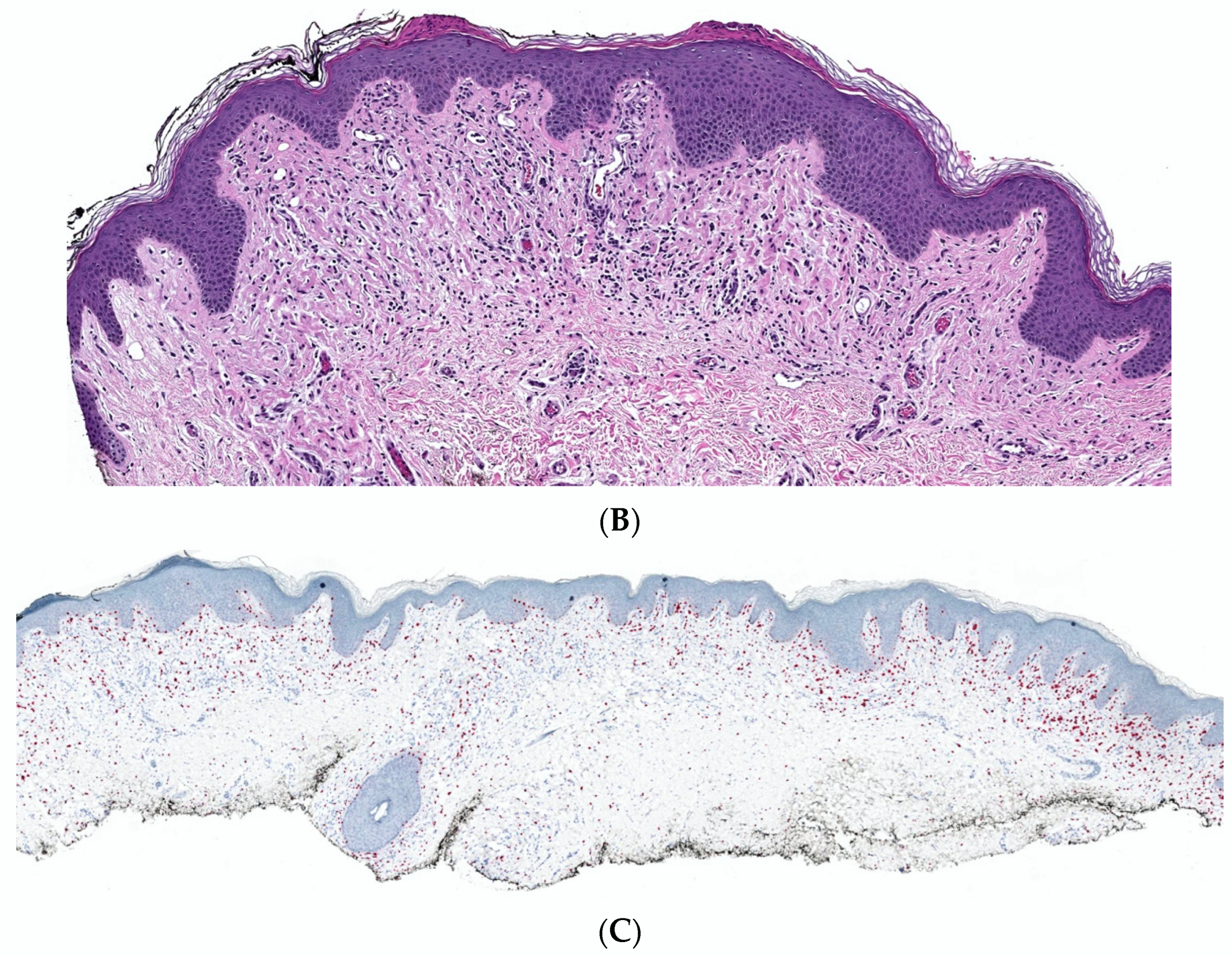
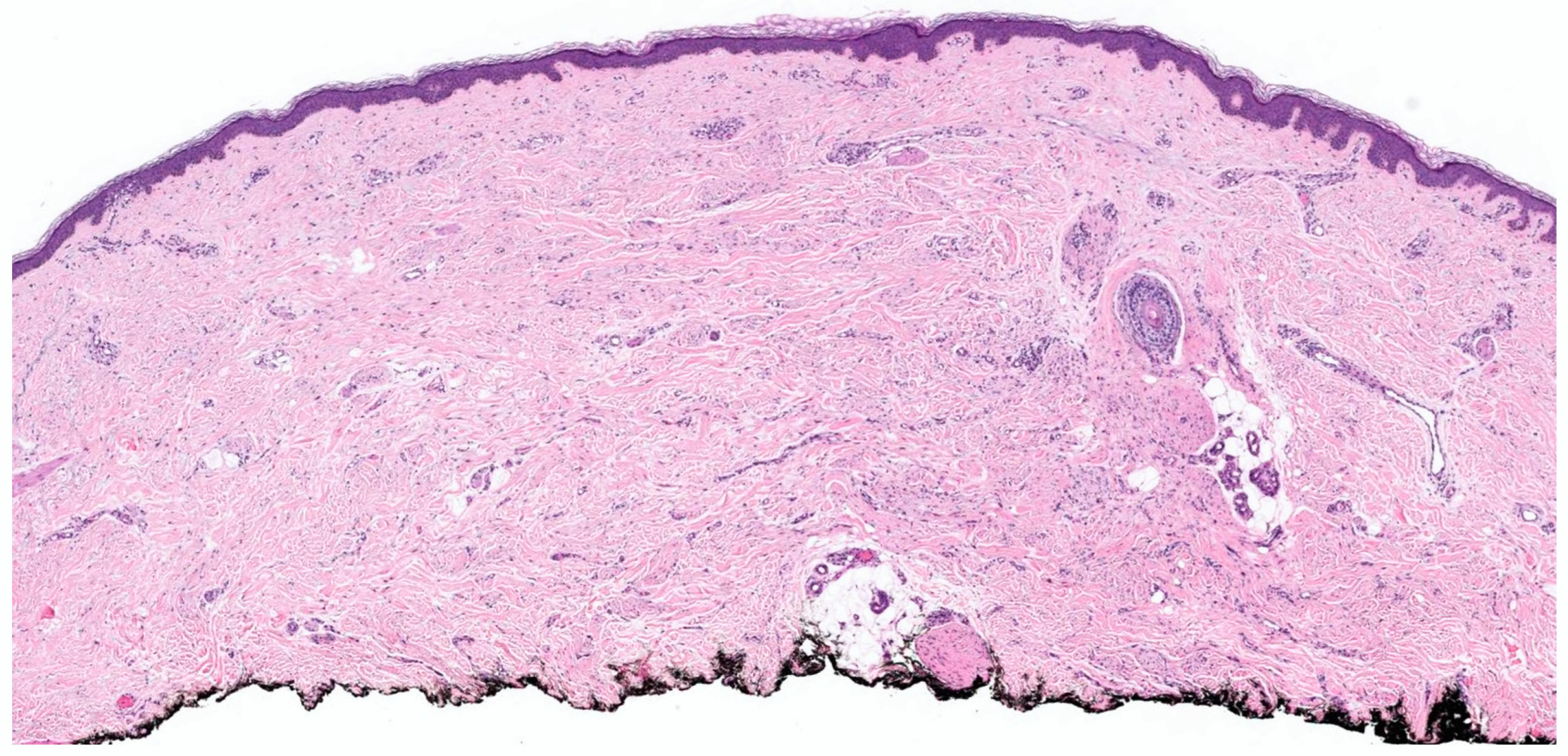
2. Conventional/Classic cNF
3. Histopathological Variants
3.1. According to the Variations in Cell Morphology
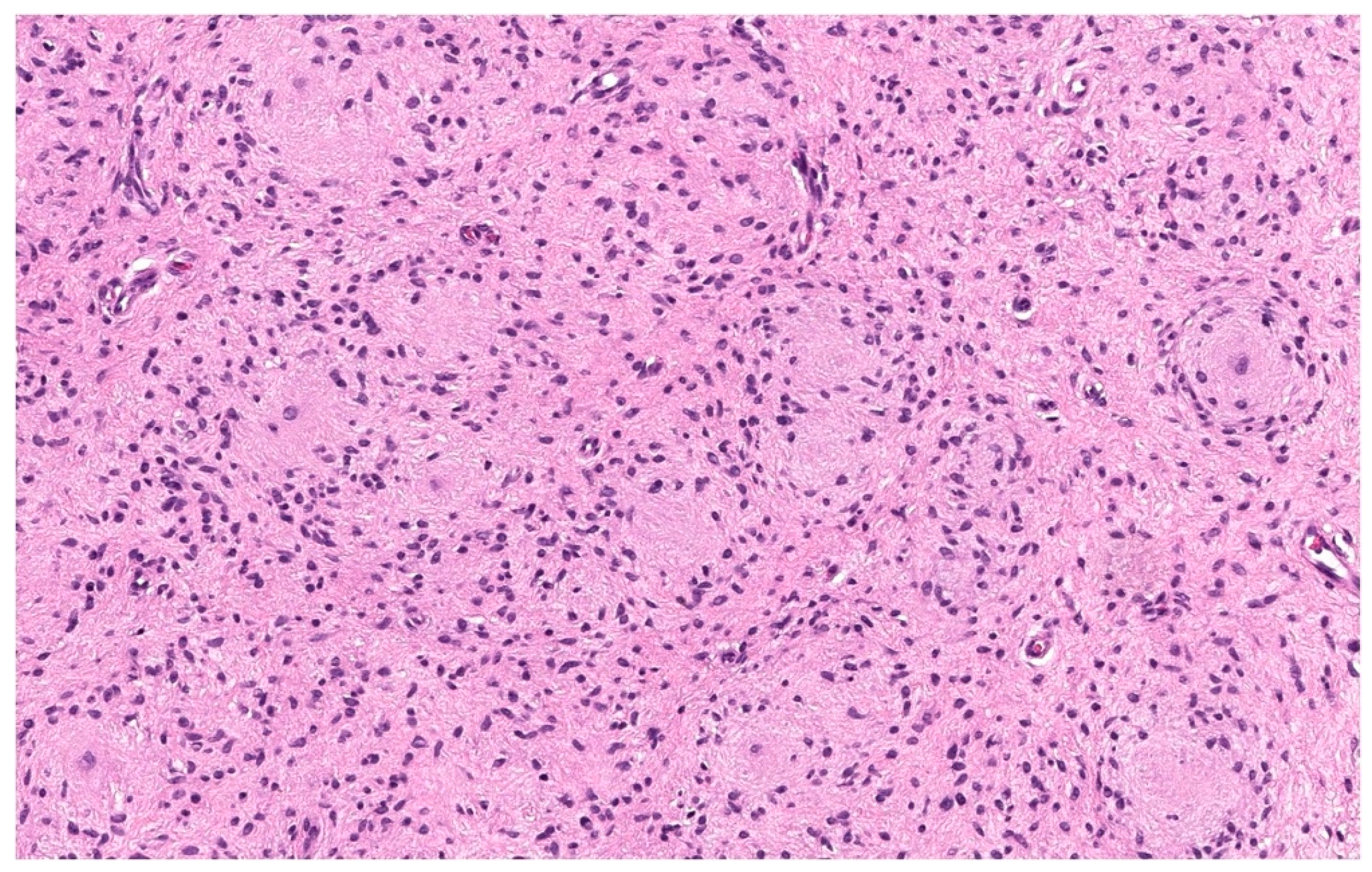
3.1.1. Epithelioid cNF
3.1.2. Granular Cell Cant
3.1.3. Balloon Cell/Clear Cell cNF
3.1.4. Dendritic Cell cNF with Pseudorosettes
3.1.5. Floret-Like Multinucleated Giant Cells
3.1.6. Meissnerian cNF
3.1.7. Pacinian cNF
3.1.8. Pigmented cNF
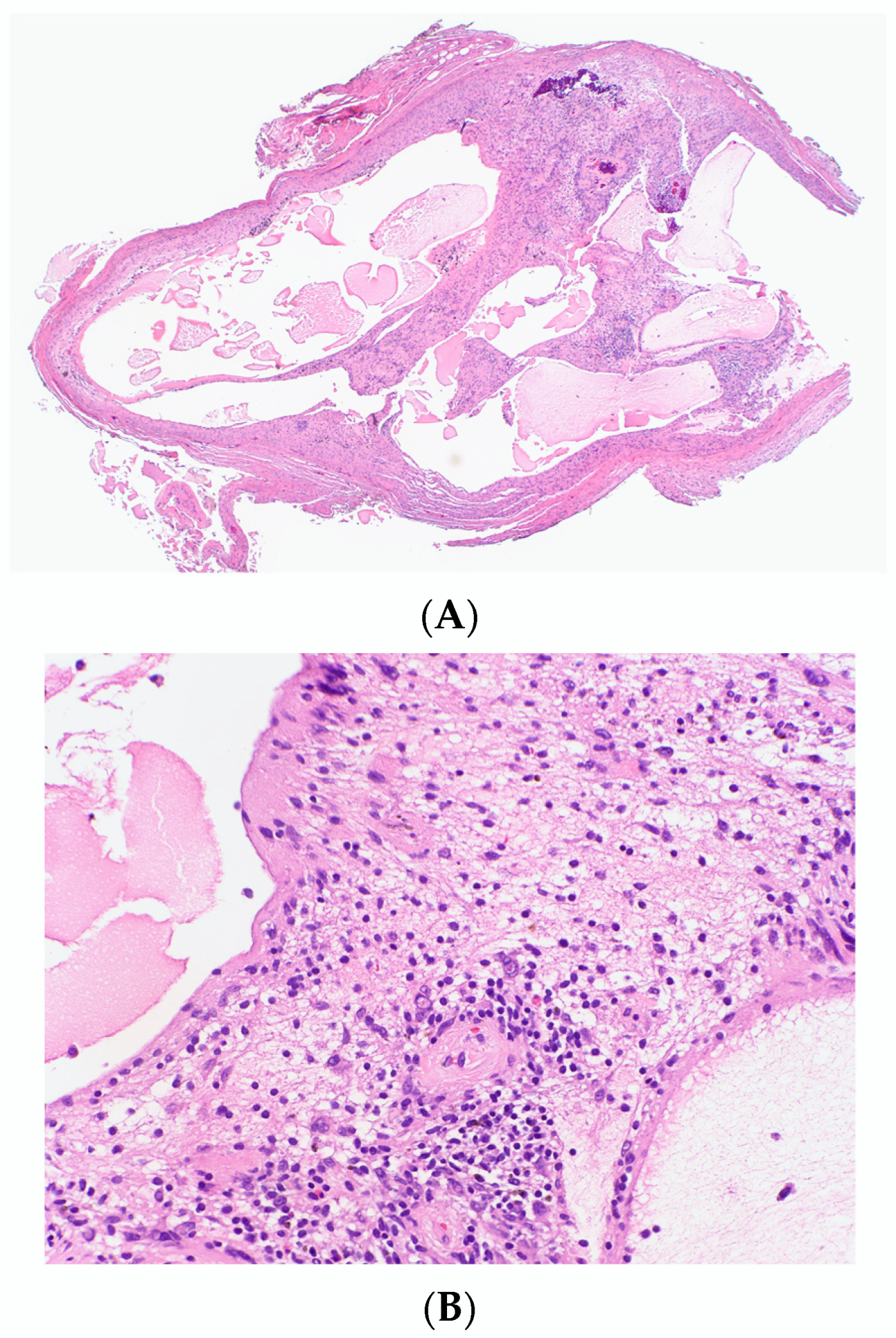
3.1.9. Atypical cNF
3.1.10. Cellular cNF
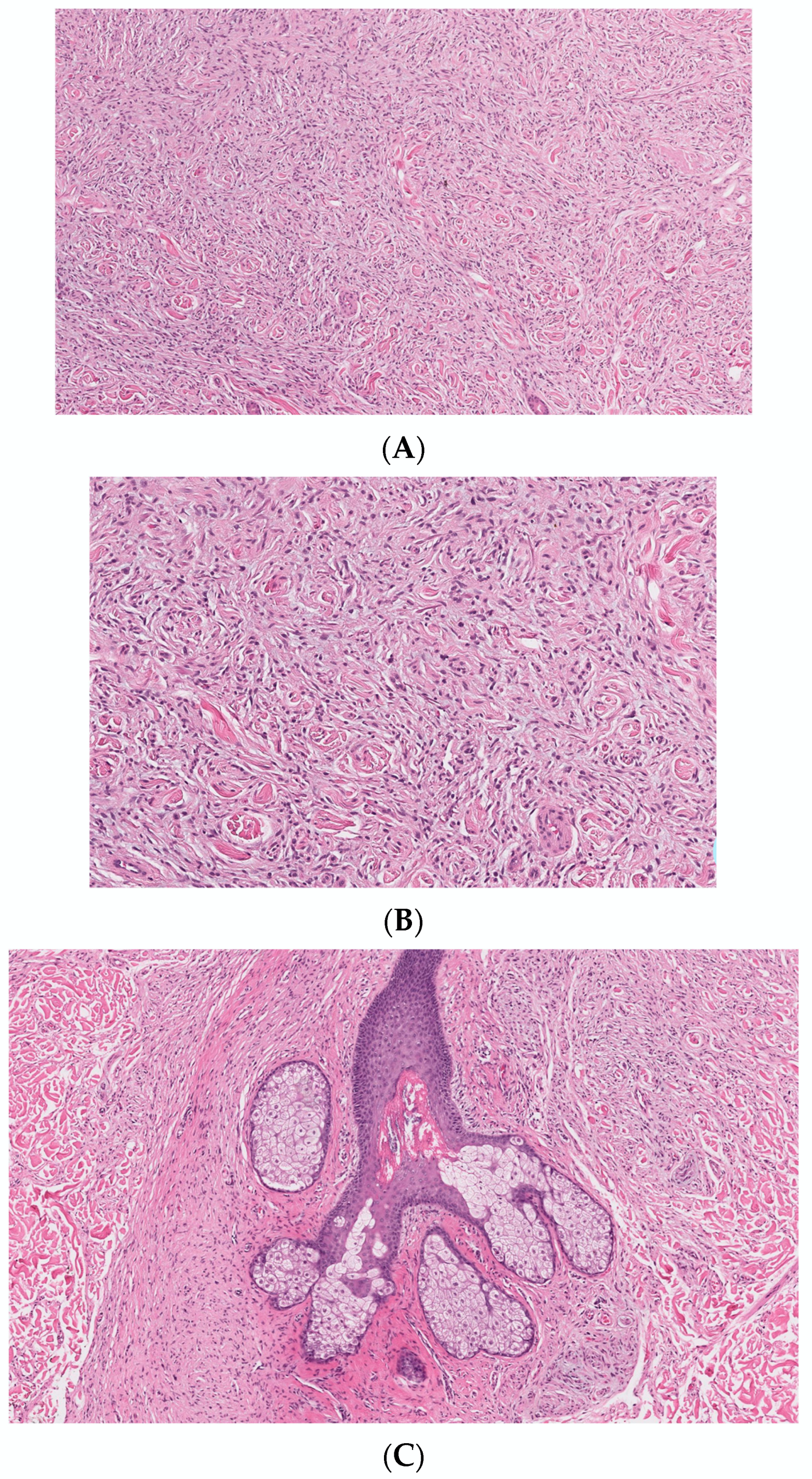
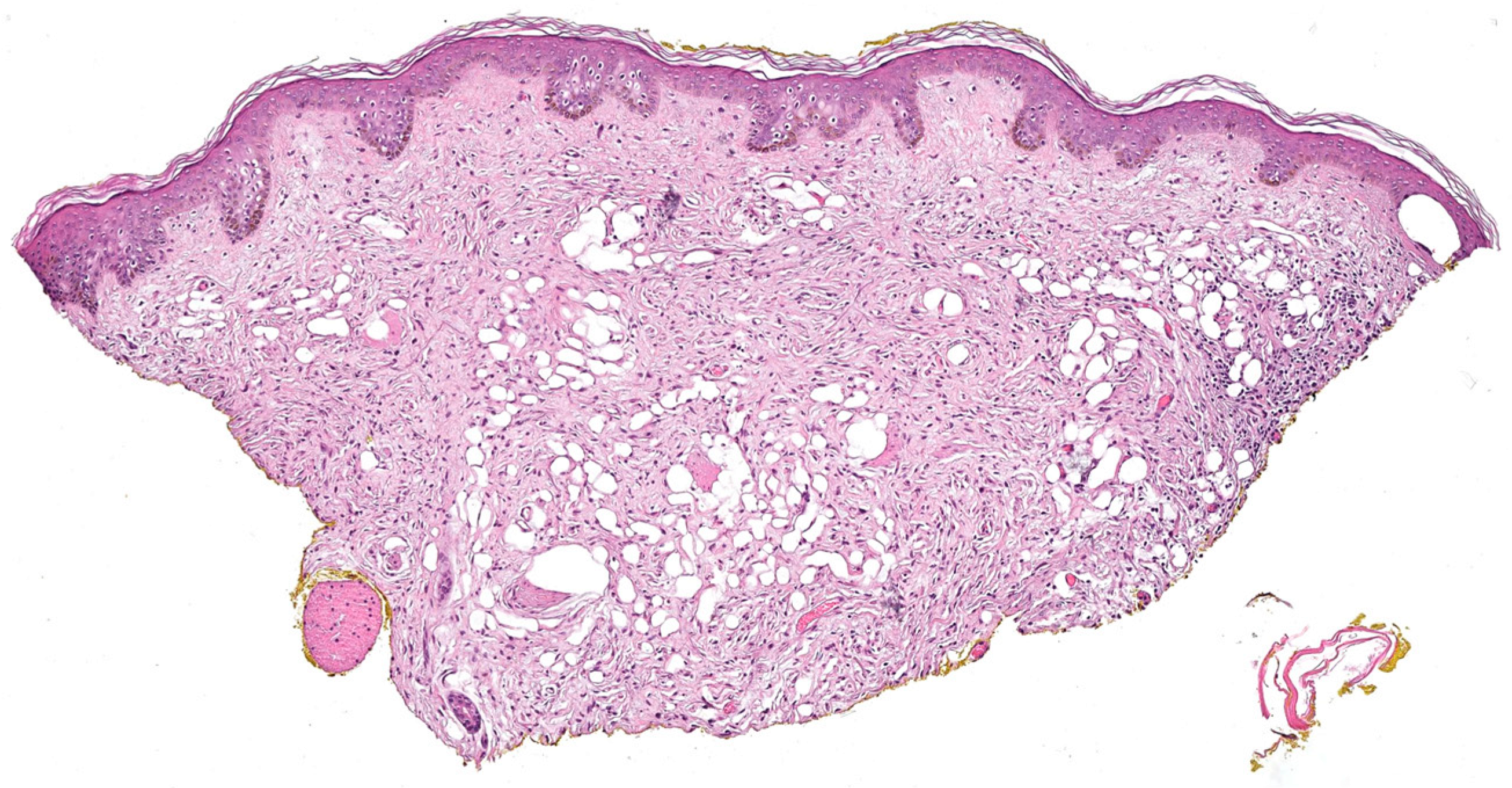
3.1.11. Microcystic (Pseudoglandular) cNF
3.2. According to Variation in Stroma
3.2.1. Myxoid cNF
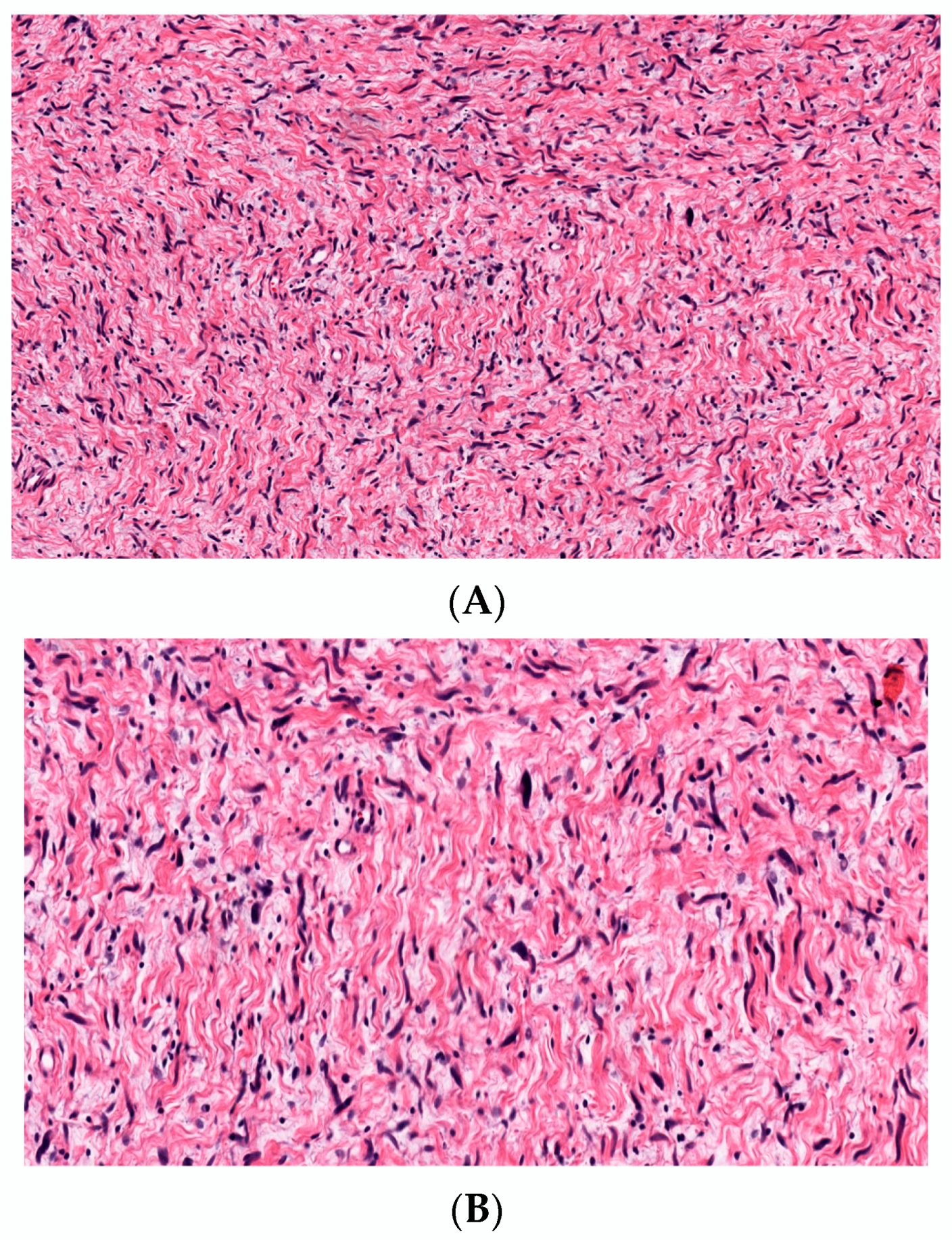
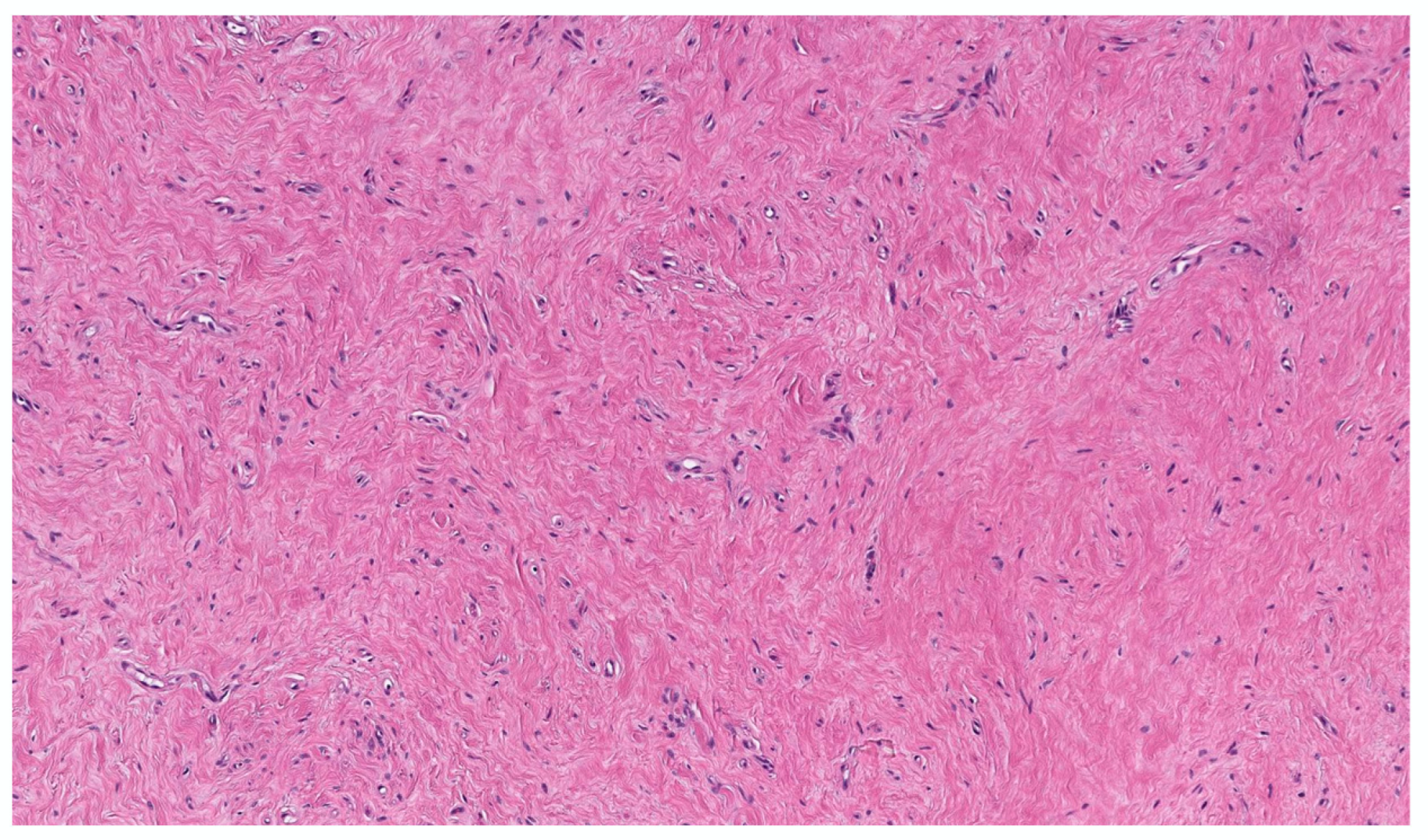
3.2.2. Hyalinized cNF
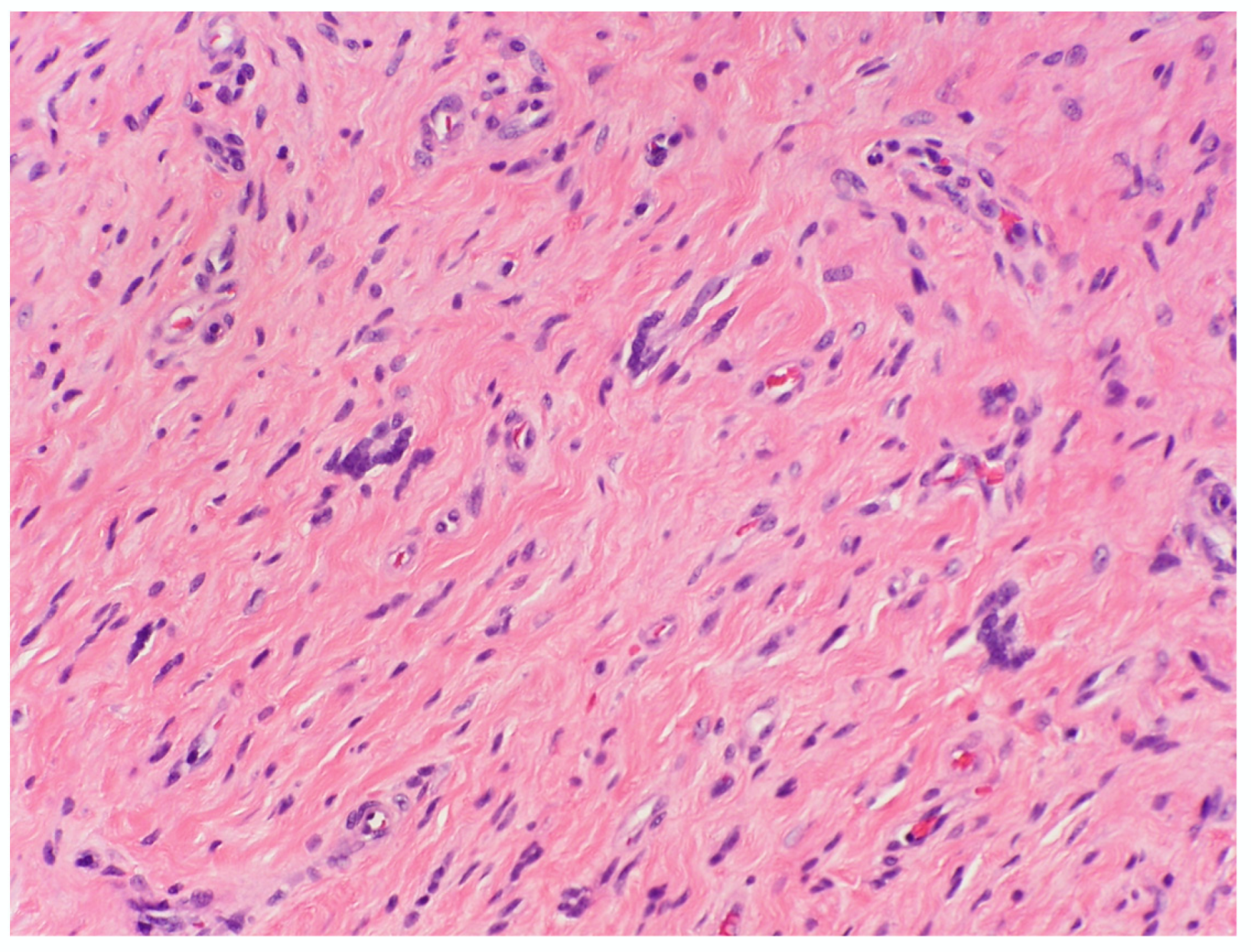
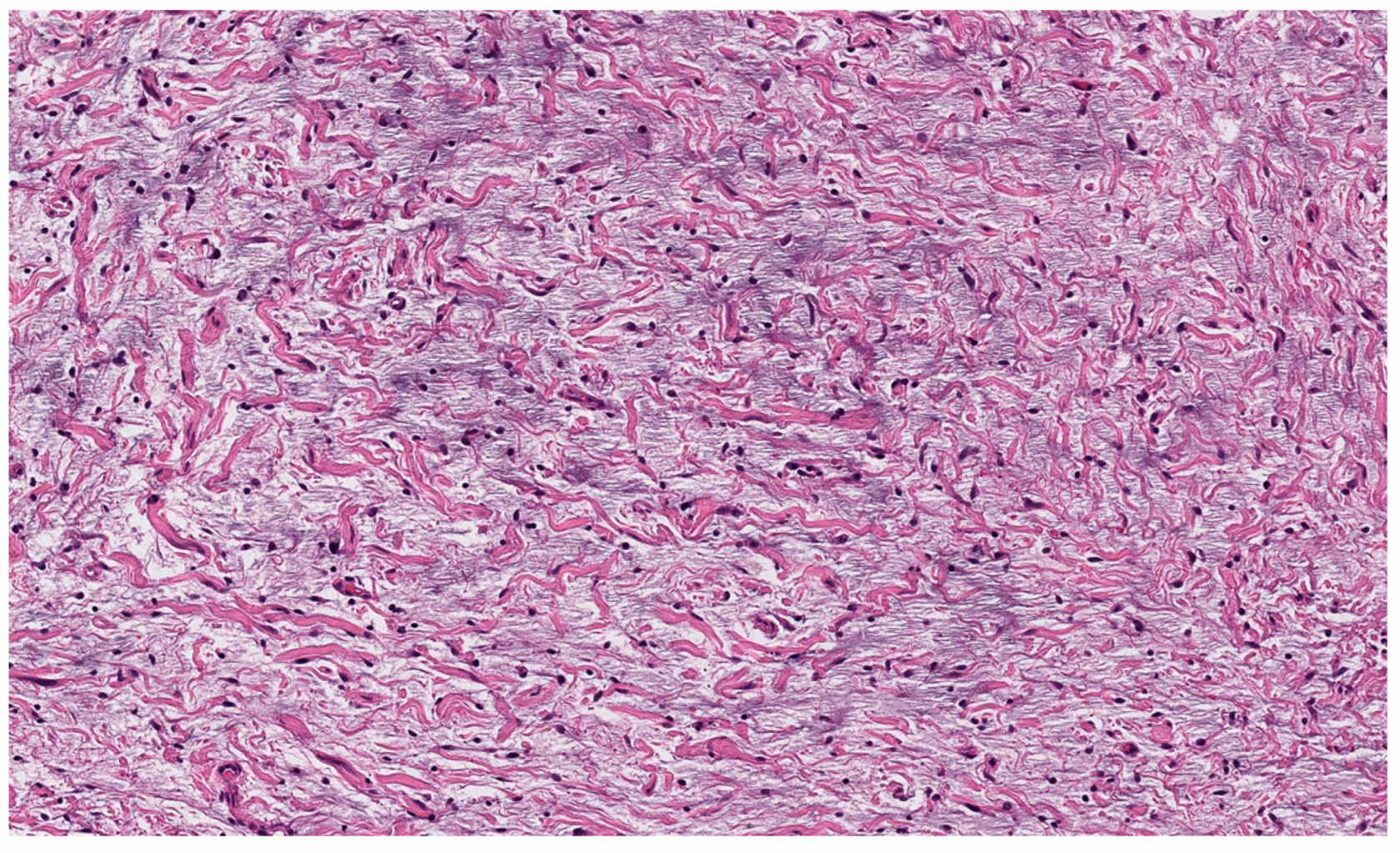
3.2.3. Sclerotic cNF
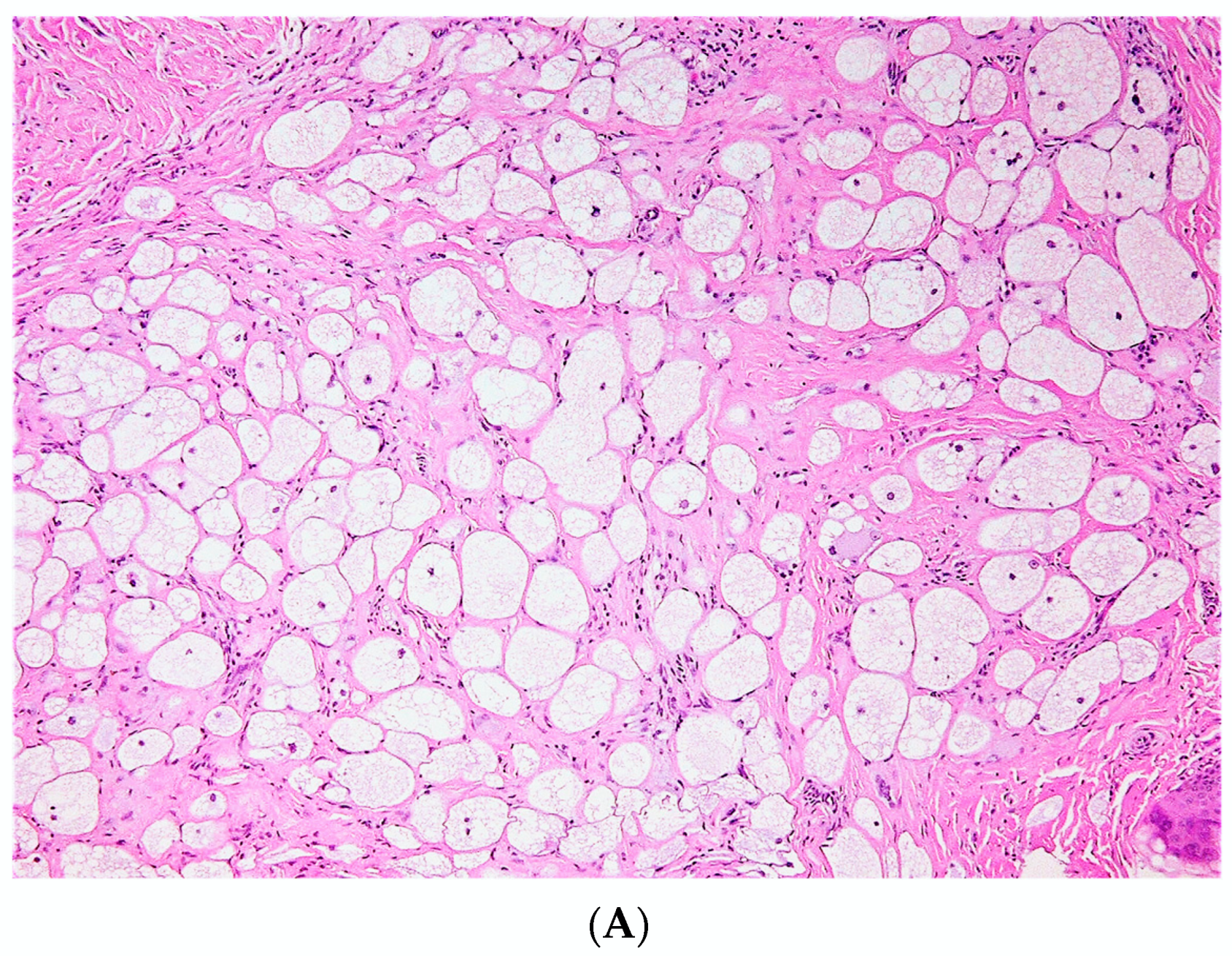
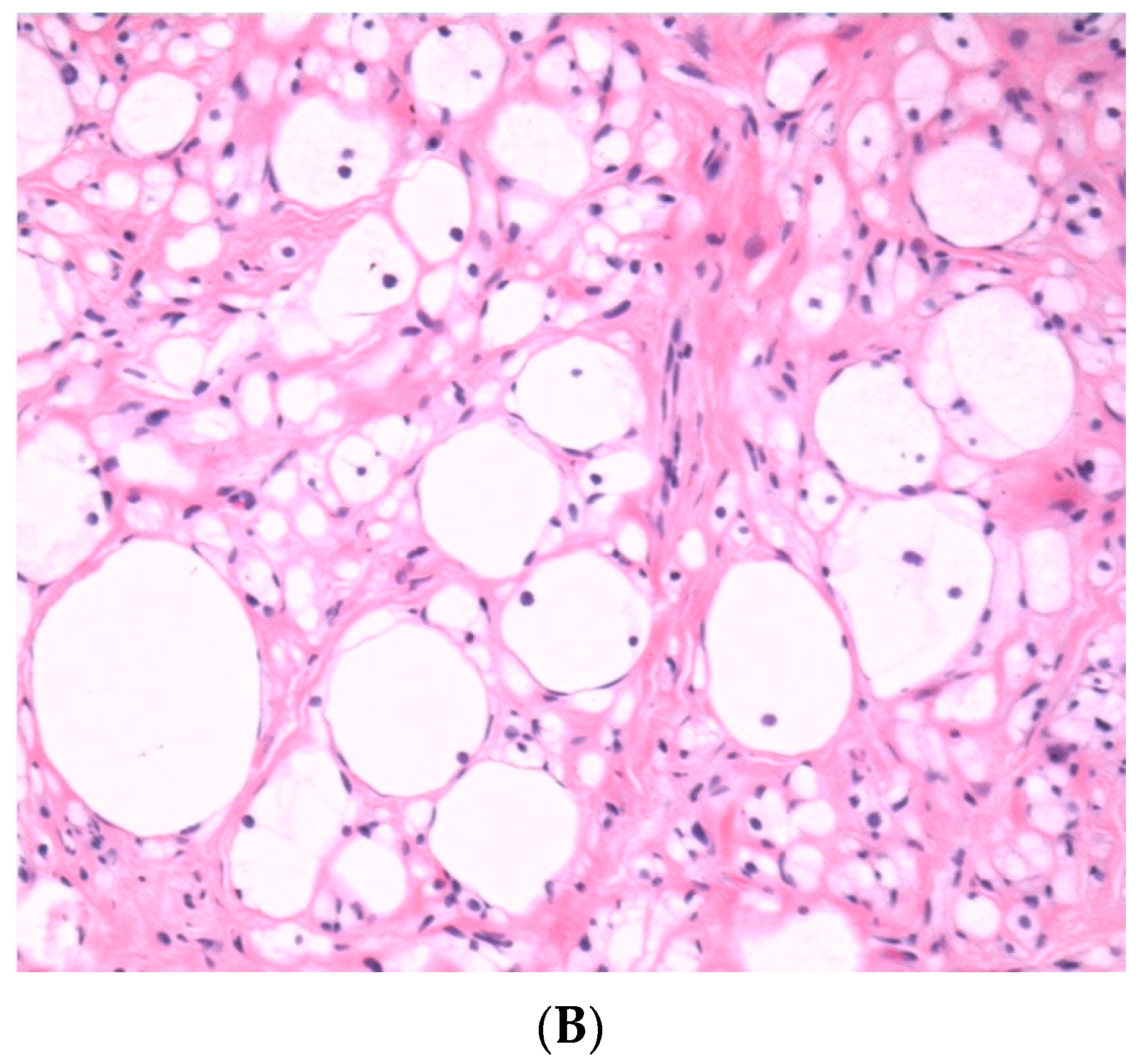
3.2.4. Lipomatous cNF
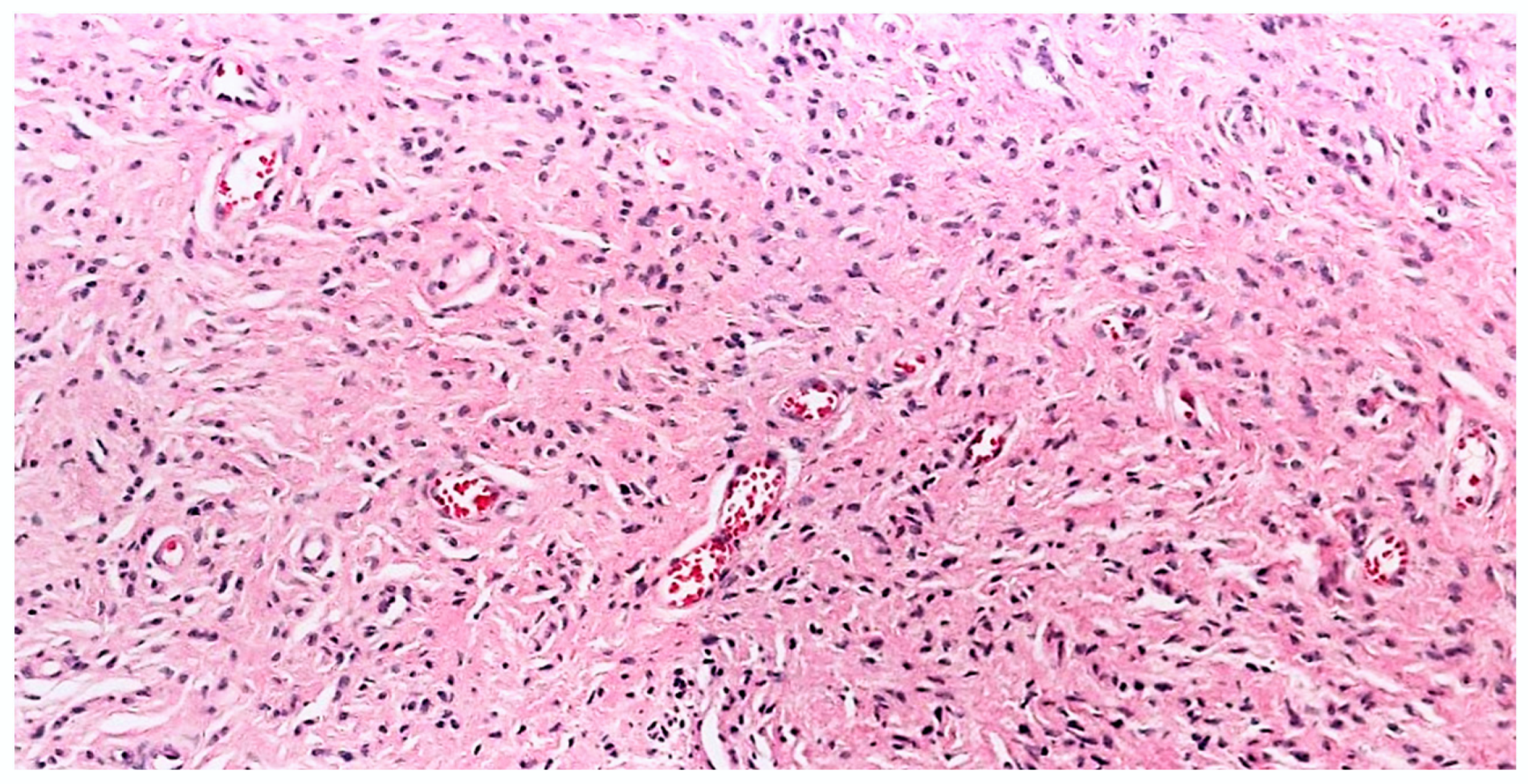
3.2.5. Angioneurofibroma
4. Other Potential Histopathological Variants Encountered in our Practice
4.1. Plaque
4.2. Dispersed
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ruggieri, M.; Praticò, A.D.; Caltabiano, R.; Polizzi, A. Early history of the different forms of neurofibromatosis from ancient Egypt to the British Empire and beyond: First descriptions, medical curiosities, misconceptions, landmarks, and the persons behind the syndromes. Am. J. Med. Genet. Part A 2018, 176, 515–550. [Google Scholar] [CrossRef] [PubMed]
- Wander, J.V.; Das Gupta, T.K. Neurofibromatosis. Curr. Probl. Surg. 1977, 14, 1–81. [Google Scholar] [CrossRef] [PubMed]
- Antônio, J.R.; Goloni-Bertollo, E.M.; Trídico, L.A. Neurofibromatosis: Chronological history and current issues. An. Bras. De Dermatol. 2013, 88, 329–343. [Google Scholar] [CrossRef] [PubMed]
- The WHO Classification of Tumours Editorial Board. WHO Classification of Tumours Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020. [Google Scholar]
- Li, S.; Chen, Z.; Le, L.Q. New insights into the neurofibroma tumor cells of origin. Neurooncol. Adv. 2019, 2 (Suppl. S1), i13–i22. [Google Scholar] [CrossRef] [Green Version]
- Ortonne, N.; Wolkenstein, P.; Blakeley, J.O.; Korf, B.; Plotkin, S.R.; Riccardi, V.M.; Miller, D.C.; Huson, S.; Peltonen, J.; Rosenberg, A.; et al. Cutaneous neurofibromas: Current clinical and pathologic issues. Neurology 2018, 91 (Suppl. S1), S5–S13. [Google Scholar] [CrossRef] [Green Version]
- Megahed, M. Histopathological variants of neurofibroma. A study of 114 lesions. Am. J. Dermatopathol. 1994, 16, 486–495. [Google Scholar] [CrossRef]
- Riccardi, V.M. The genetic predisposition to and histogenesis of neurofibromas and neurofibrosarcoma in neurofibromatosis type 1. Neurosurg. Focus 2007, 22, E3. [Google Scholar] [CrossRef]
- Argenyi, Z.; Jokinen, C.H. Cutaneous Neural Neoplasms: A Practical Guide (Current Clinical Pathology). 2016. Available online: www.springer.com/series/7632 (accessed on 1 October 2022).
- Hong, S.P.; Ahn, S.K. Abundant eosinophil infiltration in a neurofibroma. Am. J. Dermatopathol. 2007, 29, 187–189. [Google Scholar] [CrossRef]
- Hogan, S.R.; Speiser, J.J.; Lee, J.M.; Hutchens, K.A. Neurofibroma with Numerous Lymphoid Aggregates Simulating Spindle Cell Melanoma. Am. J. Dermatopathol. 2013, 35, 870–871. [Google Scholar] [CrossRef]
- Meyer, A.; Billings, S.D. What’s new in nerve sheath tumors. Virchows Arch. 2020, 476, 65–80. [Google Scholar] [CrossRef]
- Ryan, D.; Wright, G. 46. The fingerprint pattern of CD34 in neurofibroma. Pathology 2015, 47, S114. [Google Scholar] [CrossRef]
- Yeh, I.; McCalmont, T.H. Distinguishing neurofibroma from desmoplastic melanoma: The value of the CD34 fingerprint. J. Cutan. Pathol. 2011, 38, 625–630. [Google Scholar] [CrossRef]
- Weiss, S.W. Enzinger and Weiss’s Soft Tissue Tumors; Mosby: St. Louis, MO, USA, 2001. [Google Scholar]
- Calonje, E.; Brenn, T.; Lazar, A.; McKee, P.H. Mckee’s Pathology of the Skin: With Clinical Correlations; Elsevier: Philadelphia, PA, USA, 2020. [Google Scholar]
- Laskin, W.B.; Fetsch, J.F.; Lasota, J.; Miettinen, M. Benign epithelioid peripheral nerve sheath tumors of the soft tissues: Clinicopathologic spectrum of 33 cases. Am. J. Surg. Pathol. 2005, 29, 39–51. [Google Scholar] [CrossRef]
- Finkel, G.; Lane, B. Granular cell variant of neurofibromatosis: Ultrastructure of benign and malignant tumors. Hum. Pathol. 1982, 13, 959–963. [Google Scholar] [CrossRef]
- Puri, P.K.; Tyler, W.B.; Ferringer, T.C. Neurofibroma with clear cell change. Am. J. Dermatopathol. 2009, 31, 453–456. [Google Scholar] [CrossRef]
- Cazzato, G.; Cascardi, E.; Colagrande, A.; Cimmino, A.; Ingravallo, G.; Lospalluti, L.; Romita, P.; Demarco, A.; Arezzo, F.; Loizzi, V.; et al. Balloon Cell Melanoma: Presentation of Four Cases with a Comprehensive Review of the Literature. Dermatopathology 2022, 9, 100–110. [Google Scholar] [CrossRef]
- Michal, M.; Fanburg-Smith, J.C.; Mentzel, T.; Kutzner, H.; Requena, L.; Zamecnik, M.; Miettinen, M. Dendritic cell neurofibroma with pseudorosettes: A report of 18 cases of a distinct and hitherto unrecognized neurofibroma variant. Am. J. Surg. Pathol. 2001, 25, 587–594. [Google Scholar] [CrossRef]
- Vanessa, L.; Ramos, B.S.; Alice, R. Dendritic cell neurofibroma with pseudorosettes. J. Cutan. Pathol. 2021, 49, 421–425. [Google Scholar] [CrossRef]
- Woodruff, J.M.; Busam, K.J. Histologically benign cutaneous dendritic cell tumor with pseudorosettes. Am. J. Surg. Pathol. 2003, 26, 1644–1645. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Wu, Y.-H.; Hsieh, Y.-J. Dendritic cell neurofibroma with pseudorosettes: One case report and literature review. Dermatol. Sin. 2012, 30, 75–77. [Google Scholar] [CrossRef]
- Saggini, A.; Held, L.; Kempter, W. Dendritic Cell Neurofibroma with Pseudorosettes: A Variant of Neurofibroma? Am. J. Dermatopathol. 2021, 43, 158–160. [Google Scholar] [CrossRef]
- Simpson, R.H.; Seymour, M.J. Dendritic cell neurofibroma with pseudorosettes: Two tumors in a patient with evidence of neurofibromatosis. Am. J. Surg. Pathol. 2001, 25, 1458–1459. [Google Scholar] [CrossRef]
- Shaktawat, S.S.; Golka, D. Floret-like multinucleated giant cells in neurofibroma. Diagn. Pathol. 2007, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Magro, G.; Scavo, S.; Ruggieri, M. Floretlike multinucleated giant cells in a neurofibroma from a patient with NF-1: An unusual finding for such a tumor. Virchows Arch. 2002, 441, 525–526. [Google Scholar] [CrossRef]
- Magro, G.; Amico, P.; Vecchio, G.M.; Caltabiano, R.; Castaing, M.; Kacerovska, D.; Kazakov, D.V.; Michal, M. Multinucleated floret-like giant cells in sporadic and NF1-associated neurofibromas: A clinicopathologic study of 94 cases. Virchows Arch. 2010, 456, 71–76. [Google Scholar] [CrossRef]
- Taungjaruwinai, W.M.; Goldberg, L.J. Multinucleate giant cells in neurofibromas: A clue to the diagnosis of neurofibromatosis. J. Cutan. Pathol. 2009, 36, 1164–1167. [Google Scholar] [CrossRef]
- Satter, E. MPH Floret-Like Multinucleated Giant Cells in a Neurofibroma Outside the Context of Neurofibromatosis Type 1. Am. J. Dermatopathol. 2009, 31, 724–725. [Google Scholar] [CrossRef]
- Rozza-de-Menezes, R.E.; Brum, C.d.A.I.; Gaglionone, N.C.; de Sousa Almeida, L.M.; Andrade-Losso, R.M.; Paiva, B.V.B.; Faveret, P.L.S.; da Silva, A.V.; Siqueira, O.H.K.; Riccard, V.M.; et al. Prevalence and clinicopathological characteristics of lipomatous neurofibromas in neurofibromatosis 1: An investigation of 229 cutaneous neurofibromas and a systematic review of the literature. J. Cutan. Pathol. 2018, 45, 743–753. [Google Scholar] [CrossRef]
- Sode, T.; Kunzler, E.; Uzoma, B.; McCollough, M.; Hosler, G.A. A meissnerian neurofibroma: Case report of a rare neurofibroma variant. J. Cutan. Pathol. 2020, 47, 967–969. [Google Scholar] [CrossRef]
- Weiser, G. An electron microscope study of “Pacinian neurofibroma”. Virchows Arch. A Pathol. Anat. Histol. 1975, 366, 331–340. [Google Scholar] [CrossRef]
- Smith, T.W.; Bhawan, J. Tactile-like structures in neurofibromas. An ultrastructural study. Acta Neuropathol. 1980, 50, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Watabe, K.; Kumanishi, T.; Ikuta, F.; Oyake, Y. Tactile-like corpuscles in neurofibromas: Immunohistochemical demonstration of S-100 protein. Acta Neuropathol. 1983, 61, 173–177. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, S.E.; Melín-Aldana, H.; Yakuboff, K.P. Pacinian neurofibroma of the hand: A case report and literature review. J Hand Surg. Am. 1999, 24, 413–416. [Google Scholar] [CrossRef]
- Nath, A.K.; Timshina, D.K.; Thappa, D.M.; Basu, D. Pacinian neurofibroma: A rare neurogenic tumor. Indian J. Dermatol. Venereol. Leprol. 2011, 77, 204–205. [Google Scholar] [CrossRef] [PubMed]
- Fassola, I.; Wenzke, L.; Ertel, W.; Krasnici, S. Pacinian neuromas and neurofibromas of the hands and fingers: A systematic review. J. Hand Surg. Eur. Vol. 2019, 44, 925–931. [Google Scholar] [CrossRef]
- Inaba, M.; Yamamoto, T.; Minami, R.; Ohbayashi, C.; Hanioka, K. Pigmented neurofibroma: Report of two cases and literature review. Pathol. Int. 2001, 51, 565–569. [Google Scholar] [CrossRef]
- Friedrich, R.E.; Hagel, C. Pigmented (melanotic) diffuse neurofibroma of the back in neurofibromatosis type 1. GMS Interdiscip. Plast. Reconstr. Surg. DGPW 2018, 7, Doc04. [Google Scholar] [CrossRef]
- Fetsch, J.F.; Michal, M.; Miettinen, M. Pigmented (melanotic) neurofibroma: A clinicopathologic and immunohistochemical analysis of 19 lesions from 17 patients. Am. J. Surg. Pathol. 2000, 24, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Novoa, R.A.; Kovarik, C.L.; Low, D.W.; Argenyi, Z. Cutaneous epithelioid melanocytic neurofibroma arising in a patient with neurofibromatosis 1. J. Cutan. Pathol. 2014, 41, 457–461. [Google Scholar] [CrossRef]
- Mesbah Ardakani, N.; Yap, F.; Wood, B.A. Cutaneous Atypical Neurofibroma: A Case Report and Review of Literature. Am. J. Dermatopathol. 2018, 40, 864–867. [Google Scholar] [CrossRef]
- Jokinen, C.H.; Argenyi, Z.B. Atypical neurofibroma of the skin and subcutaneous tissue: Clinicopathologic analysis of 11 cases. J. Cutan. Pathol. 2010, 37, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Mateo, M.d.C.; Compañ-Quilis, A.; Monteagudo, C. Microcystic pseudoglandular plexiform cutaneous neurofibroma. J. Cutan. Pathol. 2015, 42, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Pak, K.Y.; Pun, S.; Cassarino, D.S. Case of Symptomatic Dermal Neurofibroma With Microcystic Features. Am. J. Dermatopathol. 2021, 43, e290–e292. [Google Scholar] [CrossRef]
- Gmyrek, R.F.; Beer, R.; Silvers, D.N.; Reiffel, R.; Grossman, M.E. Periungual myxoid neurofibroma. Cutis 2002, 69, 54–56. [Google Scholar]
- Baran, R.; Haneke, E. Subungual myxoid neurofibroma on the thumb. Acta Derm. Venereol. 2001, 81, 210–211. [Google Scholar] [CrossRef]
- Trieu, D.; Drosou, A.; Schwartz, M.R.; Goldberg, L.H. Myxoid Neurofibroma Treated with Mohs Micrographic Surgery. Dermatol. Surg. 2015, 41, 166–168. [Google Scholar] [CrossRef]
- McHugh, K.E.; Sturgis, C.D.; Bergfeld, W.F. Hyalinized Neurofibromas: Not Just Rare Variants in Skin of the Female Breast. Am. J. Dermatopathol. 2019, 41, 718–721. [Google Scholar] [CrossRef]
- Nakashima, K.; Yamada, N.; Yoshida, Y.; Yamamoto, O. Solitary sclerotic neurofibroma of the skin. Am. J. Dermatopathol. 2008, 30, 278–280. [Google Scholar] [CrossRef]
- González-Vela, M.C.; Val-Bernal, J.F.; González-López, M.A.; Drake, M.; Fernández-Llaca, J.H. Pure sclerotic neurofibroma: A neurofibroma mimicking sclerotic fibroma. J. Cutan. Pathol. 2006, 33, 47–50. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, Y.C. Sclerosing segmental neurofibromatosis. J. Dermatol. 2005, 32, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Val-Bernal, J.F.; de la Dehesa, J.; Garijo, M.F.; Val, D. Cutaneous lipomatous neurofibroma. Am. J. Dermatopathol. 2002, 24, 246. [Google Scholar] [CrossRef] [PubMed]
- Val-Bernal, J.F.; González-Vela, M.C. Cutaneous lipomatous neurofibroma: Characterization and frequency. J. Cutan. Pathol. 2005, 32, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Gaurish, S.K.S.; Bhatt, N.C.; Joshi, A. A rare case of neurofibroma with lipomatous differentiation and floret like giant cells-cytologically disguising as pleomorphic lipoma. Indian J. Pathol. Microbiol. 2014, 57, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.K.; Ahn, H.J.; Kim, T.-H.; Hwang, S.M.; Choi, E.H.; Lee, S.H. Intratumoral fat in neurofibroma. Am. J. Dermatopathol. 2002, 24, 326. [Google Scholar] [CrossRef] [PubMed]
- Petronic-Rosic, V.; Shea, C.R. Lipomatous neurofibroma. A report of three cases. Am. J. Dermatopathol. 2004, 26, 137. [Google Scholar]
- Arbiser, J.L.; Flynn, E.; Barnhill, R.L. Analysis of vascularity of human neurofibromas. J. Am. Acad. Dermatol. 1998, 38, 950–954. [Google Scholar] [CrossRef]
- Friedrich, R.E.; Behrendt, C.A.; Glatzel, M.; Hagel, C. Vascular Innervation in Benign Neurofibromas of Patients with Neurofibromatosis Type 1. Anticancer Res. 2015, 35, 6509–6516. [Google Scholar]
- Saxer-Sekulic, N.; Kaya, G. Cutaneous Angioneurofibroma: A New Histopathological Variant of Neurofibroma. Dermatopathology 2014, 1, 7–10. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anatomic location | Cutaneous Cutaneous/subcutaneous Deep |
| Growth Pattern | Localized Diffuse/infiltrating |
| Relationship to nerve | Intraneural Extraneural |
| Pathogenesis | Endoneurial Perineurial Epineurial |
| Conventional/Classic cNF |
|---|
According to variations in cell morphology:
|
According to the variations in stroma:
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagrani, N.S.; Bhawan, J. Histopathological Variants of Cutaneous Neurofibroma: A Compendious Review. Dermatopathology 2023, 10, 1-19. https://doi.org/10.3390/dermatopathology10010001
Nagrani NS, Bhawan J. Histopathological Variants of Cutaneous Neurofibroma: A Compendious Review. Dermatopathology. 2023; 10(1):1-19. https://doi.org/10.3390/dermatopathology10010001
Chicago/Turabian StyleNagrani, Neha S., and Jag Bhawan. 2023. "Histopathological Variants of Cutaneous Neurofibroma: A Compendious Review" Dermatopathology 10, no. 1: 1-19. https://doi.org/10.3390/dermatopathology10010001