
Machine Learning in Chemical Product Engineering: The State of the Art and a Guide for Newcomers


Abstract
:1. Introduction
2. ML: Background
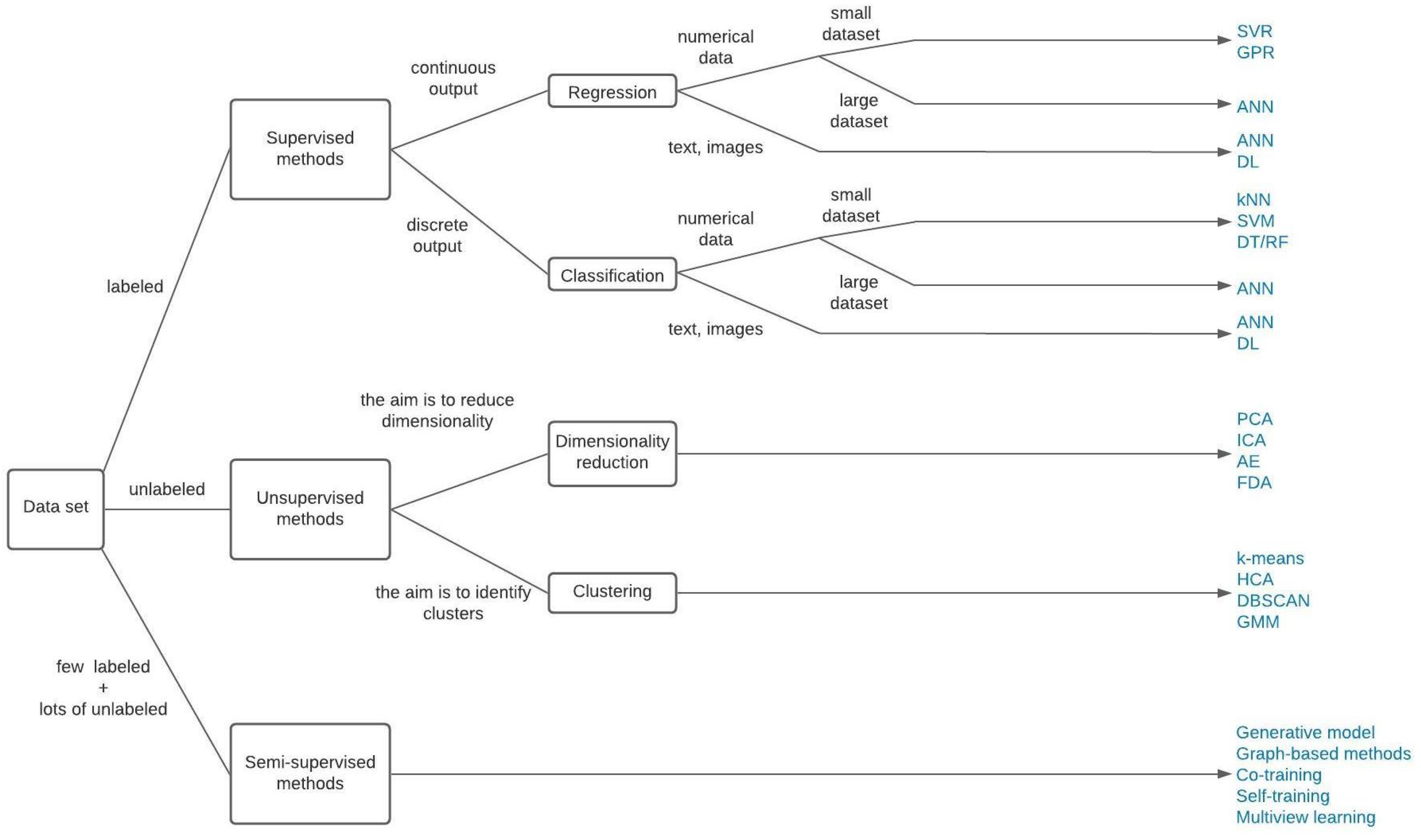
2.1. Categories of ML Algorithms
- Supervised learning
- Unsupervised learning
- Semi-supervised learning
- Reinforcement learning
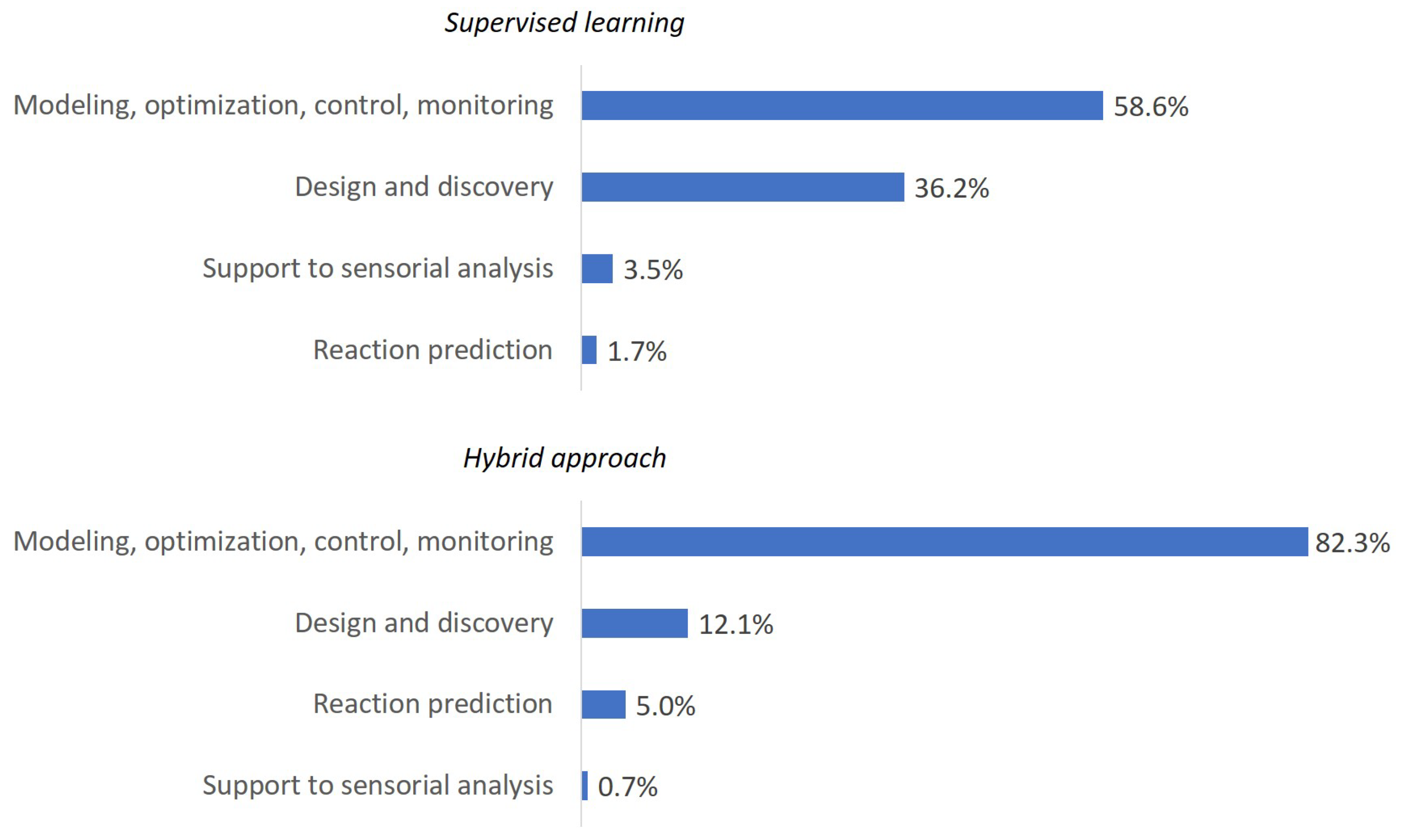
2.2. Hybrid and Combinatorial Approaches
3. ML in Chemical Product Engineering: State-of-the-Art
3.1. Current Challenges in Chemical Product Engineering and Role of AI/ML
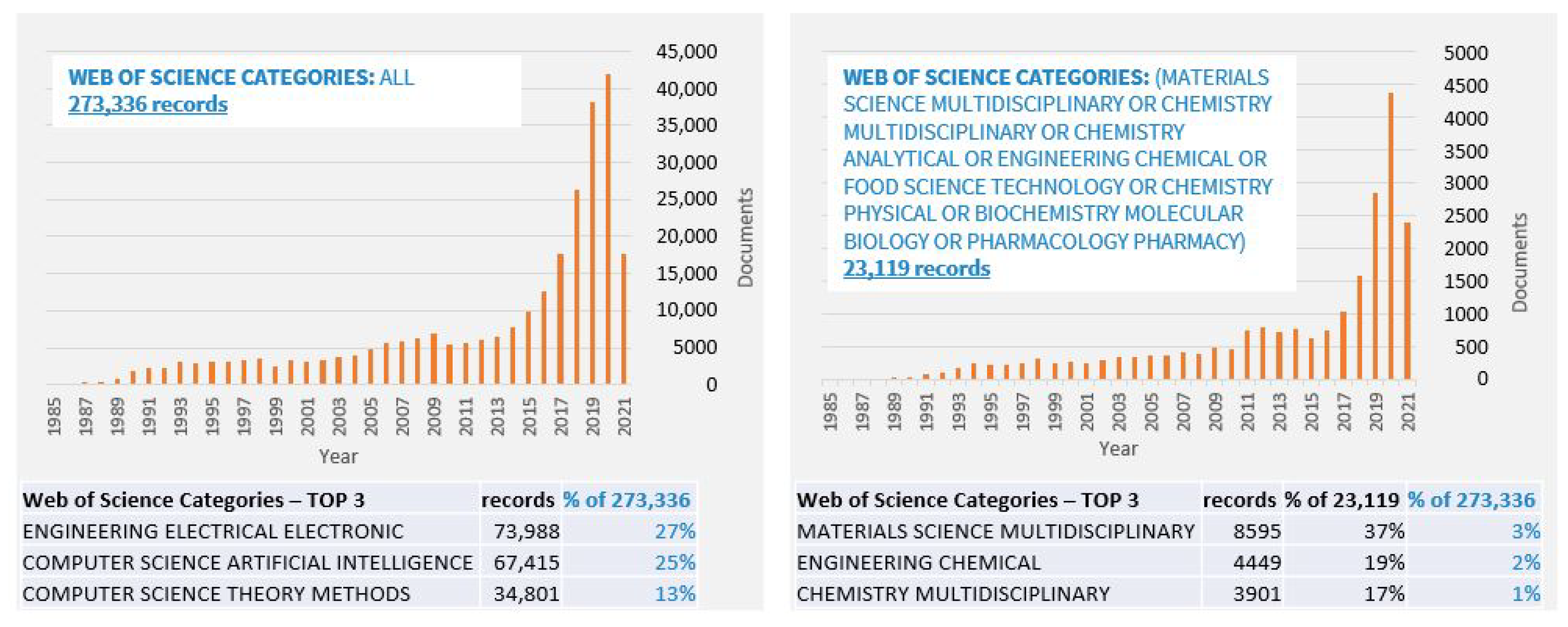
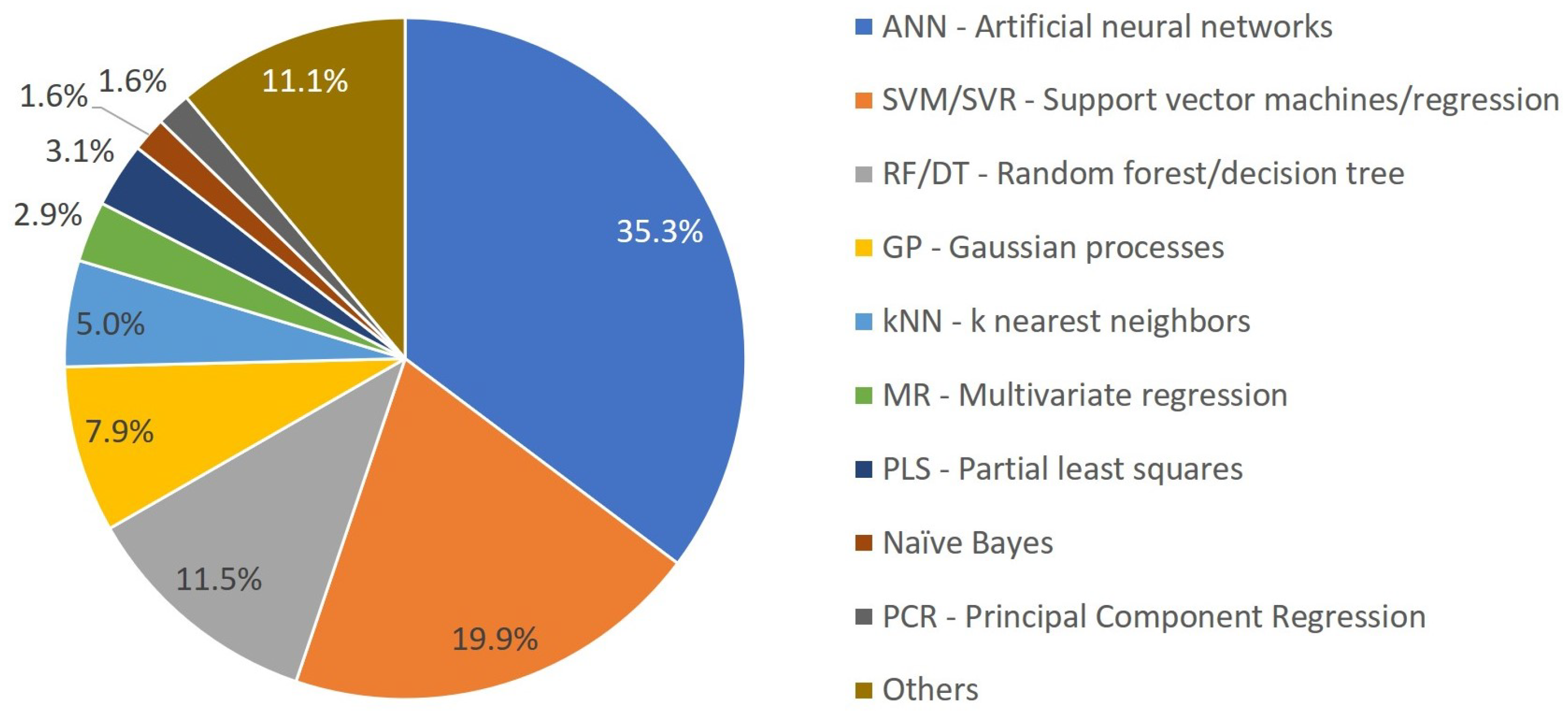
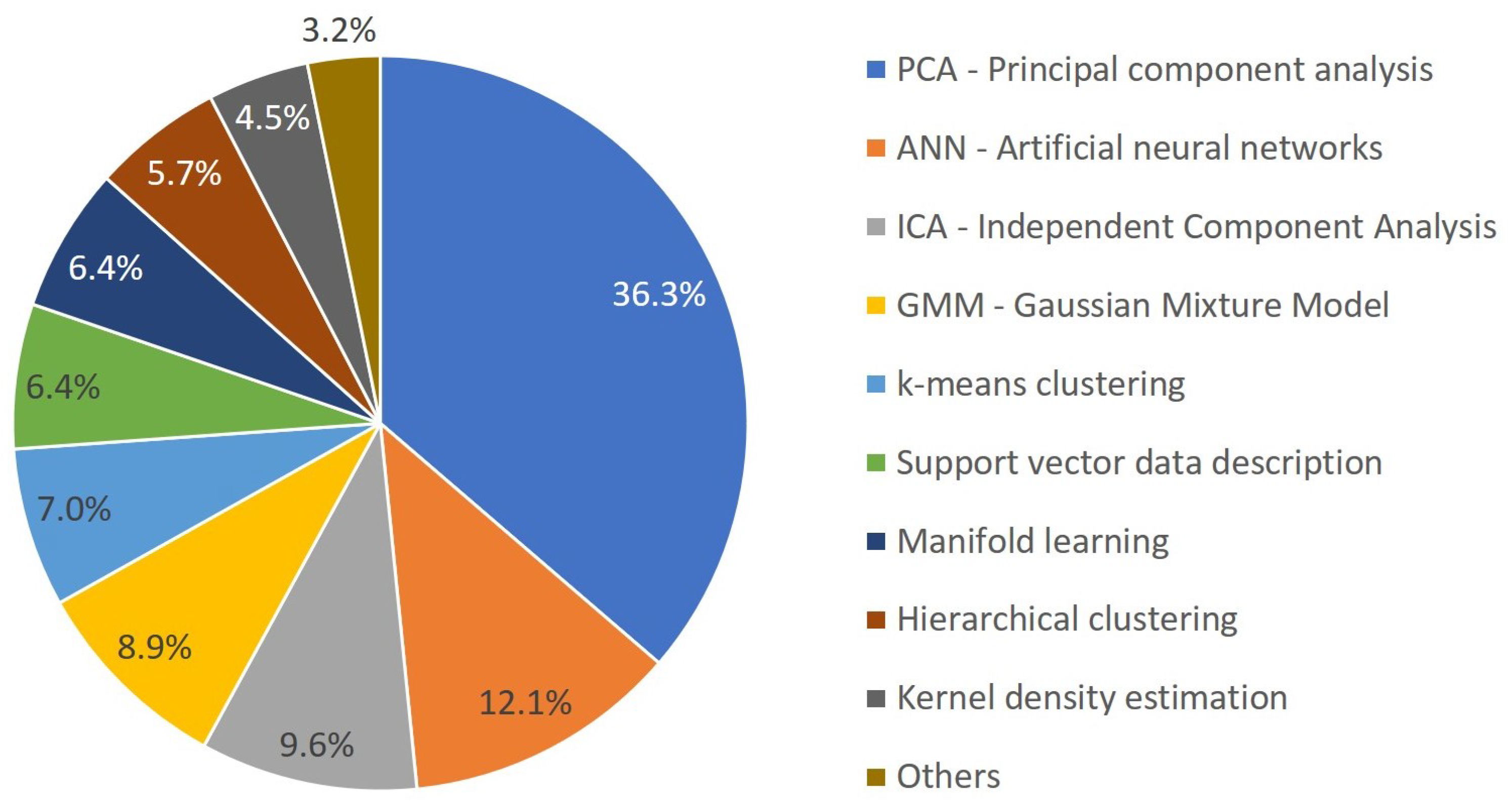
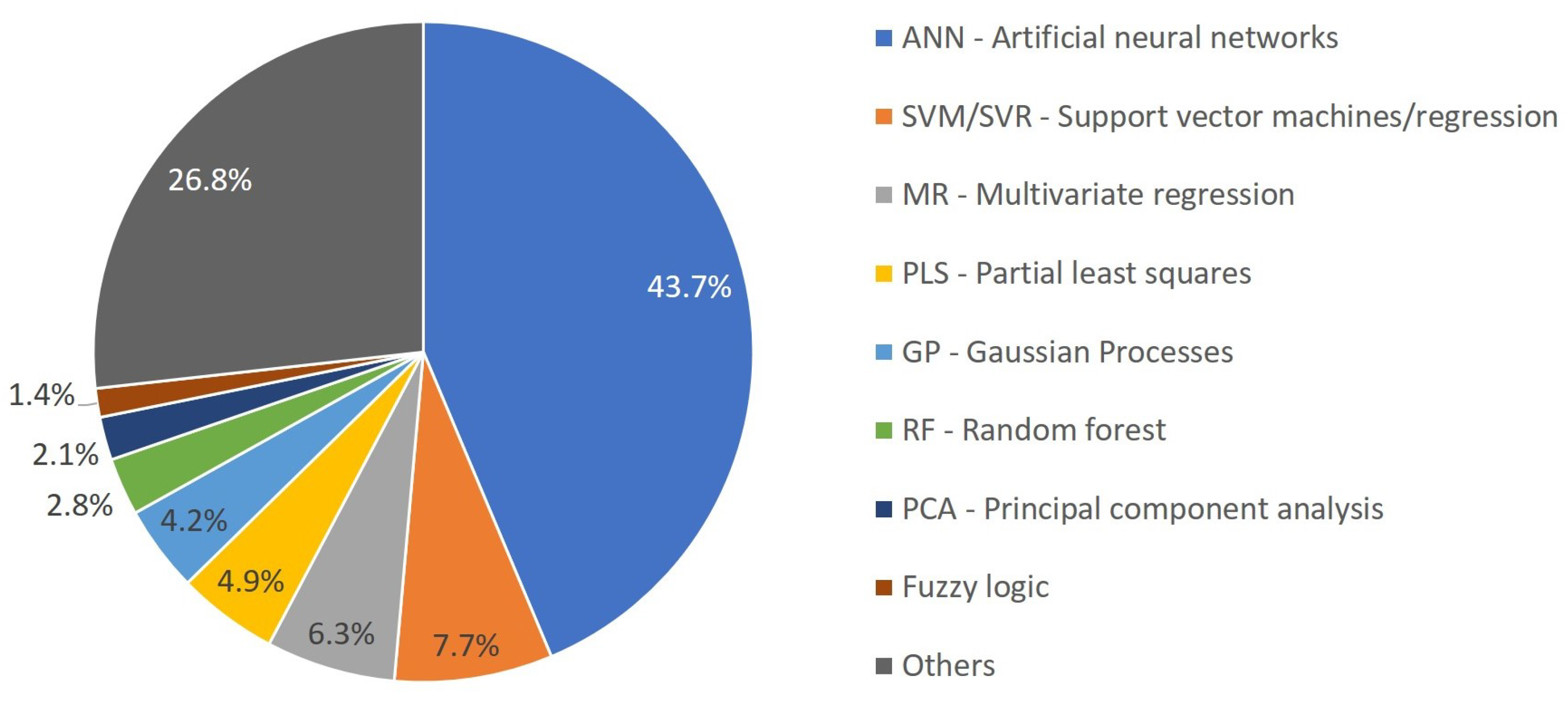
3.2. Overview of ML Methods in Chemical Product Engineering
3.3. Popular ML Applications in Chemical Product Engineering Problems
- Design and discovery of new molecules and materials
- Prediction of chemical reactions and retrosynthesis
- Modeling and optimization of process–properties relationship
- Support for sensorial analysis
4. Guidelines for Applying ML in Chemical Product Engineering Problems
4.1. General Principle of Some Popular ML Methods in Chemical Product Engineering
- ANN
- SVM
- GP
- PCA
- Other ML methods
4.2. Interest of Data-Driven Methods
4.3. Challenges and Solutions
- Data
- Lack of understanding
4.4. General Guidelines for the Selection of a ML Method
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAE | Adversarial AutoEncoders |
| AE | AutoEncoders |
| AENN | AutoEncoders Neural Network |
| AI | Artificial Intelligence |
| ANN | Artificial Neural Network |
| ANOVA | ANalysis Of VAriance |
| BFGS | Broyden–Fletcher–Goldfarb–Shanno |
| BL | Bayesian Learning |
| BN | Boron Nitride |
| BNN | Bayesian Neural Network |
| BO | Bayesian Optimization |
| BPNN | Back-Propagation Neural Network |
| C2V | Code2Vect |
| CAMD | Computer Aided Molecular Design |
| CFD | Computational Fluid Dynamics |
| CNN | Convolutional Neural Network |
| Co-ANN | Co-training Artificial Neural Network |
| COSMO-RS | COnductor like Screening MOdel for real solvents |
| CPE | Chemical Product Engineering |
| DBN | Deep Belief Network |
| DFT | Density Functional Theory |
| DNN | Deep Neural Network |
| DL | Deep Learning |
| DoE | Design of Experiments |
| DRL | Deep Reinforcement Learning |
| DT | Decision Tree |
| ELM | Extreme Learning Machine |
| FFNN | Feed-Forward Neural Network |
| FT-IR | Fourier Transform InfraRed |
| GAN | Generative Adversarial Network |
| GC | Group Contribution |
| GC-MS | Gas Chromatography–Mass Spectrometry |
| GCN | Graph Convolutional Network |
| GMM | Gaussian Mixture Model |
| GP | Gaussian Process |
| GCPR | G-Coupled Protein Receptor |
| HCA | Hierarchical Clustering Analysis |
| HNN | Hierarchical Neural Network |
| ICA | Independent Clustering Analysis |
| iDMD | inspired by Dynamic Model Decomposition |
| InChI | International Chemical Identifier |
| kNN | k-Nearest Neighbors |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| LDA | Linear Discriminant Analysis |
| LMNNR | Large Margin Nearest Neighbor for Regression |
| LSSVM | Least Squares Support Vector Machine |
| MD | Molecular Dynamics |
| MDPI | Multidisciplinary Digital Publishing Institute |
| ML | Machine Learning |
| MLP | Multi-Layer Perceptron |
| MR | Multivariate Regression |
| MSE | Mean Square Error |
| NIR | Near InfraRed |
| NIST | National Institute of Standards and Technology |
| NLP | Natural Language Processing |
| NN | Neural Network |
| PCA | Principal Component Analysis |
| PCR | Principal Component Regression |
| PLS | Partial Least Squares |
| QC | Quantum Chemistry |
| QM | Quantum Mechanics |
| QSAR | Quantitative Structure–Activity Relationship |
| QSPR | Quantitative Structure–Property Relationship |
| RBF | Radial Basis Function |
| RF | Random Forest |
| RI | Refractive Index |
| RL | Reinforcement learning |
| RNN | Recurrent Neural Network |
| sPGD | sparse Proper Generalized Decomposition |
| SMARTS | SMILES ARbitrary Target Specification |
| SMILES | Simplified Molecular Input Line Entry Specification |
| SVM | Support Vector Machine |
| SVR | Support Vector Regression |
| VAE | Variational AutoEncoders |
References
- Mitchell, T. Machine Learning; McGraw-Hill: New York, NY, USA, 1997. [Google Scholar]
- Butler, K.T.; Davies, D.W.; Cartwright, H.; Isayev, O.; Walsh, A. Machine learning for molecular and materials science. Nature 2018, 559, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Elton, D.C.; Boukouvalas, Z.; Fuge, M.D.; Chung, P.W. Deep learning for molecular design-A review of the state of the art. Mol. Syst. Des. Eng. 2019, 4, 828–849. [Google Scholar] [CrossRef] [Green Version]
- Pilania, G. Machine learning in materials science: From explainable predictions to autonomous design. Comput. Mater. Sci. 2021, 193, 110360. [Google Scholar] [CrossRef]
- Zhou, T.; Song, Z.; Sundmacher, K. Big Data Creates New Opportunities for Materials Research: A Review on Methods and Applications of Machine Learning for Materials Design. Engineering 2019, 5, 1017–1026. [Google Scholar] [CrossRef]
- Schmidt, J.; Marques, M.R.; Botti, S.; Marques, M.A. Recent advances and applications of machine learning in solid-state materials science. NPJ Comput. Mater. 2019, 5, 1–36. [Google Scholar] [CrossRef]
- Westermayr, J.; Gastegger, M.; Schütt, K.T.; Maurer, R.J. Deep integration of machine learning into computational chemistry and materials science. arXiv 2021, arXiv:2102.08435v1. [Google Scholar]
- Himanen, L.; Geurts, A.; Foster, A.S.; Rinke, P. Data-Driven Materials Science: Status, Challenges, and Perspectives. Adv. Sci. 2019, 6, 1900808. [Google Scholar] [CrossRef] [PubMed]
- Winkler, D.A. Chapter 9 Machine Learning at the (Nano)materials-biology Interface. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 206–226. [Google Scholar] [CrossRef]
- Bennett, S.; Tarzia, A.; Zwijnenburg, M.A.; Jelfs, K.E. Chapter 12 Artificial Intelligence Applied to the Prediction of Organic Materials. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 280–310. [Google Scholar] [CrossRef]
- Zhuo, Y.; Tehrani, A.M.; Brgoch, J. Chapter 13 A New Era of Inorganic Materials Discovery Powered by Data Science. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 311–339. [Google Scholar] [CrossRef]
- Ramprasad, R.; Batra, R.; Mannodi-Kanakkithodi, A.; Kim, C.; Pilania, G. Machine learning in materials informatics: Recent applications and prospects. NPJ Comput. Mater. 2017, 3, 54. [Google Scholar] [CrossRef]
- Chen, A.; Zhang, X.; Zhou, Z. Machine learning: Accelerating materials development for energy storage and conversion. InfoMat 2020, 2, 553–576. [Google Scholar] [CrossRef]
- Zhu, H. Big data and artificial intelligence modeling for drug discovery. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 573–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Wang, Y.; Byrne, R.; Schneider, G.; Yang, S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem. Rev. 2019, 119, 10520–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.C.; Ren, G.; Honda, H.; Davis, K.L. Artificial Intelligence-Based Drug Design and Discovery; Intech: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.; Ertl, P.; Lewis, R.; Luksch, T.; Reker, D.; Schneider, N. Artificial Intelligence in Chemistry and Drug Design; Springer Nature Swirzerland AG: Cham, Swirzerland, 2020. [Google Scholar] [CrossRef]
- Klambauer, G.; Hochreiter, S.; Rarey, M. Machine Learning in Drug Discovery. J. Chem. Inf. Model. 2019, 59, 945–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlexer Lamoureux, P.; Winther, K.T.; Garrido Torres, J.A.; Streibel, V.; Zhao, M.; Bajdich, M.; Abild-Pedersen, F.; Bligaard, T. Machine Learning for Computational Heterogeneous Catalysis. ChemCatChem 2019, 11, 3581–3601. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Kang, P.L.; Shang, C.; Liu, Z.P. Chapter 19 Machine Learning for Heterogeneous Catalysis: Global Neural Network Potential from Construction to Applications. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 488–511. [Google Scholar] [CrossRef]
- Yang, W.; Fidelis, T.T.; Sun, W.H. Machine Learning in Catalysis, From Proposal to Practicing. ACS Omega 2020, 5, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.H.; Schwaller, P.; Laino, T. Data-driven Chemical Reaction Prediction and Retrosynthesis. Chimia 2019, 73, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Coley, C.W.; Green, W.H.; Jensen, K.F. Machine Learning in Computer-Aided Synthesis Planning. Acc. Chem. Res. 2018, 51, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.L.; Redshaw, J.; Gaertner, T.; Taylor, A.; Mason, A.M.; Hirst, J.D. Chapter 7 Machine Learning for Chemical Synthesis. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 169–194. [Google Scholar] [CrossRef]
- Commenge, J.M. Big Data et Intelligence Artificielle pour le Génie des Procédés 2021. Available online: https://hal.univ-lorraine.fr/hal-03107557/document (accessed on 10 August 2021).
- Ge, Z.; Song, Z.; Ding, S.X.; Huang, B. Data Mining and Analytics in the Process Industry: The Role of Machine Learning. IEEE Access 2017, 5, 20590–20616. [Google Scholar] [CrossRef]
- Yan, Y.; Borhani, T.N.; Clough, P.T. Chapter 14 Machine Learning Applications in Chemical Engineering. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 340–371. [Google Scholar] [CrossRef]
- Wang, C.; Tan, X.P.; Tor, S.B.; Lim, C.S. Machine learning in additive manufacturing: State-of-the-art and perspectives. Addit. Manuf. 2020, 36, 101538. [Google Scholar] [CrossRef]
- DebRoy, T.; Mukherjee, T.; Wei, H.L.; Elmer, J.W.; Milewski, J.O. Metallurgy, mechanistic models and machine learning in metal printing. Nat. Rev. Mater. 2021, 6, 48–68. [Google Scholar] [CrossRef]
- Géron, A. Hands-on Machine Learning with Scikit-Learn, Keras, and TensorFlow: Concepts, Tools, and Techniques to Build Intelligent Systems; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2019. [Google Scholar]
- Burkov, A. The Hundred-Page Machine Learning Book; Andriy Burkov: Quebec City, QC, Canada, 2019. [Google Scholar]
- Nasery, S.; Hoseinpour, S.; Phung, L.T.K.; Bahadori, A. Prediction of the viscosity of water-in-oil emulsions. Pet. Sci. Technol. 2016, 34, 1972–1977. [Google Scholar] [CrossRef]
- Gola, J.; Webel, J.; Britz, D.; Guitar, A.; Staudt, T.; Winter, M.; Mücklich, F. Objective microstructure classification by support vector machine (SVM) using a combination of morphological parameters and textural features for low carbon steels. Comput. Mater. Sci. 2019, 160, 186–196. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, F.; Ye, Y.; Chen, L.; Liu, J.; Gui, A.; Zhang, J.; Dong, C. Application of machine learning algorithms in quality assurance of fermentation process of black tea— based on electrical properties. J. Food Eng. 2019, 263, 165–172. [Google Scholar] [CrossRef]
- Ge, Z.; Chen, T.; Song, Z. Quality prediction for polypropylene production process based on CLGPR model. Control. Eng. Pract. 2011, 19, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Gao, X.; Liu, Y.; Wang, L.; Yang, J.; Gao, Z. Industrial Mooney viscosity prediction using fast semi-supervised empirical model. Chemom. Intell. Lab. Syst. 2017, 171, 86–92. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, Z.; Liu, W. A co-training style semi-supervised artificial neural network modeling and its application in thermal conductivity prediction of polymeric composites filled with BN sheets. Energy AI 2021, 4, 100052. [Google Scholar] [CrossRef]
- Yan, W.; Guo, P.; Tian, Y.; Gao, J. A Framework and Modeling Method of Data-Driven Soft Sensors Based on Semisupervised Gaussian Regression. Ind. Eng. Chem. Res. 2016, 55, 7394–7401. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, C.; Gao, Z.; Yao, Y. Ensemble deep kernel learning with application to quality prediction in industrial polymerization processes. Chemom. Intell. Lab. Syst. 2018, 174, 15–21. [Google Scholar] [CrossRef]
- Yin, X.; Niu, Z.; He, Z.; Li, Z.; hee Lee, D. Ensemble deep learning based semi-supervised soft sensor modeling method and its application on quality prediction for coal preparation process. Adv. Eng. Inform. 2020, 46, 101136. [Google Scholar] [CrossRef]
- He, X.; Ji, J.; Liu, K.; Gao, Z.; Liu, Y. Soft sensing of silicon content via bagging local semi-supervised models. Sensors 2019, 19, 3814. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Cheng, F.; Xu, Y.; Wen, Q.; Liu, Y. Probabilistic Representation and Inverse Design of Metamaterials Based on a Deep Generative Model with Semi-Supervised Learning Strategy. Adv. Mater. 2019, 31, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okaro, I.A.; Jayasinghe, S.; Sutcliffe, C.; Black, K.; Paoletti, P.; Green, P.L. Automatic fault detection for laser powder-bed fusion using semi-supervised machine learning. Addit. Manuf. 2019, 27, 42–53. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Z. Exploring semi-supervised variational autoencoders for biomedical relation extraction. Methods 2019, 166, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, P.; Roy, I.; Wang, Z.; Mi, F.; Yu, L.; Balasubramani, P.; Khan, L.; Stoddart, J.F. MultiCon: A Semi-Supervised Approach for Predicting Drug Function from Chemical Structure Analysis. J. Chem. Inf. Model. 2020, 60, 5995–6006. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ji, J.; Li, P.; Shao, F.; Wu, S.; Sui, Y.; Li, S.; He, F.; Liu, J. Semi-Supervised Hybrid Local Kernel Regression for Soft Sensor Modelling of Rubber-Mixing Process. Adv. Polym. Technol. 2020, 2020, 6981302. [Google Scholar] [CrossRef]
- Zheng, J.; Du, J.; Liang, Y.; Liao, Q.; Li, Z.; Zhang, H.; Wu, Y. Deeppipe: A semi-supervised learning for operating condition recognition of multi-product pipelines. Process. Saf. Environ. Prot. 2021, 150, 510–521. [Google Scholar] [CrossRef]
- Ma, Y.; Zhu, W.; Benton, M.; Romagnoli, J. Continuous control of a polymerization system with deep reinforcement learning. J. Process. Control. 2019, 75, 40–47. [Google Scholar] [CrossRef]
- Singh, V.; Kodamana, H. Reinforcement learning based control of batch polymerisation processes. IFAC PapersOnLine 2020, 53, 667–672. [Google Scholar] [CrossRef]
- Nian, R.; Liu, J.; Huang, B. A review On reinforcement learning: Introduction and applications in industrial process control. Comput. Chem. Eng. 2020, 139, 106886. [Google Scholar] [CrossRef]
- Venkatasubramanian, V. The promise of artificial intelligence in chemical engineering: Is it here, finally? AIChE J. 2019, 65, 466–478. [Google Scholar] [CrossRef]
- Zendehboudi, S.; Rezaei, N.; Lohi, A. Applications of hybrid models in chemical, petroleum, and energy systems: A systematic review. Appl. Energy 2018, 228, 2539–2566. [Google Scholar] [CrossRef]
- Uhlemann, J.; Costa, R. Product Design and Engineering in Chemical Engineering: Past, Present State, and Future. Chem. Eng. Technol. 2019, 42, 2258–2274. [Google Scholar] [CrossRef]
- Meng, Y.; Yu, S.; Zhang, J.; Qin, J.; Dong, Z.; Lu, G. Hybrid modeling based on mechanistic and data-driven approaches for cane sugar crystallization. J. Food Eng. 2019, 257, 44–55. [Google Scholar] [CrossRef]
- Li, B.; Lin, Y.; Yu, W.; Wilson, D.I.; Young, B.R. Application of mechanistic modelling and machine learning for cream cheese fermentation pH prediction. J. Chem. Technol. Biotechnol. 2021, 96, 125–133. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, T.; Zhang, L.; Fung, K.Y.; Ng, K.M. Food Product Design: A Hybrid Machine Learning and Mechanistic Modeling Approach. Ind. Eng. Chem. Res. 2019, 58, 16743–16752. [Google Scholar] [CrossRef]
- von Stosch, M.; Davy, S.; Francois, K.; Galvanauskas, V.; Hamelink, J.M.; Luebbert, A.; Mayer, M.; Oliveira, R.; O’Kennedy, R.; Rice, P.; et al. Hybrid modeling for quality by design and PAT-benefits and challenges of applications in biopharmaceutical industry. Biotechnol. J. 2014, 9, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Drăgoi, E.N.; Curteanu, S.; Fissore, D.; Curteanu, S.; Fissore, D. On the Use of Artificial Neural Networks to Monitor a Pharmaceutical Freeze-Drying Process On the Use of Artificial Neural Networks to Monitor a Pharmaceutical Freeze-Drying Process. Dry. Technol. 2013, 31, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Gómez, J.M.; Ricardez-Sandoval, L.; Alvarez, O. Integrated design of emulsified cosmetic products: A review. Chem. Eng. Res. Des. 2020, 161, 279–303. [Google Scholar] [CrossRef]
- Sadowski, P.; Fooshee, D.; Subrahmanya, N.; Baldi, P. Synergies between quantum mechanics and machine learning in reaction prediction. J. Chem. Inf. Model. 2016, 56, 2125–2128. [Google Scholar] [CrossRef]
- Castéran, F.; Ibanez, R.; Argerich, C.; Delage, K.; Chinesta, F.; Cassagnau, P. Application of Machine Learning Tools for the Improvement of Reactive Extrusion Simulation. Macromol. Mater. Eng. 2020, 305, 2000375. [Google Scholar] [CrossRef]
- Ghiba, L.; Drăgoi, E.N.; Curteanu, S. Neural network-based hybrid models developed for free radical polymerization of styrene. Polym. Eng. Sci. 2021, 61, 716–730. [Google Scholar] [CrossRef]
- Curteanu, S.; Leon, F. Hybrid neural network models applied to a free radical polymerization process. Polym. Plast. Technol. Eng. 2006, 45, 1013–1023. [Google Scholar] [CrossRef]
- Ng, C.W.; Hussain, M.A. Hybrid neural network-prior knowledge model in temperature control of a semi-batch polymerization process. Chem. Eng. Process. Process. Intensif. 2004, 43, 559–570. [Google Scholar] [CrossRef]
- Qi, C.; Ly, H.B.; Chen, Q.; Le, T.T.; Le, V.M.; Pham, B.T. Flocculation-dewatering prediction of fine mineral tailings using a hybrid machine learning approach. Chemosphere 2020, 244, 125450. [Google Scholar] [CrossRef]
- Bi, K.; Qiu, T.; Huang, Y. A deep learning method for yogurt preferences prediction using sensory attributes. Processes 2020, 8, 518. [Google Scholar] [CrossRef]
- Chen, L.; Kim, C.; Batra, R.; Lightstone, J.P.; Wu, C.; Li, Z.; Deshmukh, A.A.; Wang, Y.; Tran, H.D.; Vashishta, P.; et al. Frequency-dependent dielectric constant prediction of polymers using machine learning. NPJ Comput. Mater. 2020, 6, 30–32. [Google Scholar] [CrossRef]
- Batra, R.; Dai, H.; Huan, T.D.; Chen, L.; Kim, C.; Gutekunst, W.R.; Song, L.; Ramprasad, R. Polymers for Extreme Conditions Designed Using Syntax-Directed Variational Autoencoders. Chem. Mater. 2020, 32, 10489–10500. [Google Scholar] [CrossRef]
- Zhou, T.; Gani, R.; Sundmacher, K. Hybrid data-driven and mechanistic modeling approaches for multiscale material and process design. Engineering 2021. [Google Scholar] [CrossRef]
- McBride, K.; Sanchez Medina, E.I.; Sundmacher, K. Hybrid Semi-parametric Modeling in Separation Processes: A Review. Chem. Ingenieur-Technik 2020, 92, 842–855. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, X.; Song, Z.; Zhou, T.; Sundmacher, K. Integrated ionic liquid and rate-based absorption process design for gas separation: Global optimization using hybrid models. AIChE J. 2021, e17340. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, H.; Liu, Q.; Gani, R. Chemical product design–recent advances and perspectives. Curr. Opin. Chem. Eng. 2020, 27, 22–34. [Google Scholar] [CrossRef]
- Costa, R.; Moggridge, G.D.; Saraiva, P.M. Chemical Product Engineering: An Emerging Paradigm within Chemical Engineering. Aiche J. 2006, 52, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Arrieta-Escobar, J.A.; Camargo, M.; Morel, L.; Orjuela, A. Current approaches on chemical product design: A study of opportunities identification for integrated methodologies. In Proceedings of the Towards the Digital World and Industry X.0-Proceedings of the 29th International Conference of the International Association for Management of Technology, IAMOT 2020, Cairo, Egypt, 13–17 September 2020; pp. 785–794. [Google Scholar]
- Ng, K.M.; Gani, R. Chemical product design: Advances in and proposed directions for research and teaching. Comput. Chem. Eng. 2019, 126, 147–156. [Google Scholar] [CrossRef]
- Cussler, E.L. Chemical Product Design; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2011. [Google Scholar]
- Hill, M. Chemical Product Engineering-The third paradigm. Comput. Chem. Eng. 2009, 33, 947–953. [Google Scholar] [CrossRef]
- Taifouris, M.; Martín, M.; Martínez, A.; Esquejo, N. Challenges in the design of formulated products: Multiscale process and product design. Curr. Opin. Chem. Eng. 2020, 27, 1–9. [Google Scholar] [CrossRef]
- Fischer, A. Artificial Intelligence Colloquium: Accelerating Chemistry with AI. 2019. Available online: https://theengineeringofconsciousexperience.com/artificial-intelligence-colloquium-accelerating-chemistry-with-ai/ (accessed on 10 August 2021).
- Goussard, V.; Duprat, F.; Ploix, J.L.; Dreyfus, G.; Nardello-Rataj, V.; Aubry, J.M. A New Machine-Learning Tool for Fast Estimation of Liquid Viscosity. Application to Cosmetic Oils. J. Chem. Inf. Model. 2020, 60, 2012–2023. [Google Scholar] [CrossRef]
- Dobbelaere, M.R.; Plehiers, P.P.; Van de Vijver, R.; Stevens, C.V.; Van Geem, K.M. Learning Molecular Representations for Thermochemistry Prediction of Cyclic Hydrocarbons and Oxygenates. J. Phys. Chem. A 2021, 125, 5166–5179. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Roitberg, A.E.; Isayev, O. Transforming Computational Drug Discovery with Machine Learning and AI. ACS Med. Chem. Lett. 2018, 9, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- David, L.; Thakkar, A.; Mercado, R.; Engkvist, O. Molecular representations in AI-driven drug discovery: A review and practical guide. J. Cheminform. 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lengeling, B.; Aspuru-Guzik, A. Inverse molecular design using machine learning: Generative models for matter engineering. Science 2018, 361, 360–365. [Google Scholar] [CrossRef] [Green Version]
- Staker, J.; Marques, G.; Dakka, J. Chapter 15 Representation Learning in Chemistry. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 372–397. [Google Scholar] [CrossRef]
- Alshehri, A.S.; Gani, R.; You, F. Deep learning and knowle dge-base d methods for computer-aided molecular design—Toward a unified approach: State-of-the-art and future directions. Comput. Chem. Eng. 2020, 141, 107005. [Google Scholar] [CrossRef]
- Audus, D.J.; De Pablo, J.J. Polymer Informatics: Opportunities and Challenges. ACS Macro Lett. 2017, 6, 1078–1082. [Google Scholar] [CrossRef]
- Wei, J.N. Exploring Machine Learning Applications to Enable Next-Generation Chemistry. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 2019. [Google Scholar]
- Luan, F. Classification of the fragrance properties of chemical compounds based on support vector machine and linear discriminant analysis. Flavour Fragr. J. 2007, 23, 311–316. [Google Scholar] [CrossRef]
- Kennedy, K.; Cal, R.; Casey, R.; Lopez, C.; Adelfio, A.; Molloy, B.; Wall, A.M.; Holton, T.A.; Khaldi, N. The anti-ageing effects of a natural peptide discovered by artificial intelligence. Int. J. Cosmet. Sci. 2020, 42, 388–398. [Google Scholar] [CrossRef]
- Lightstone, J.P.; Chen, L.; Kim, C.; Batra, R.; Ramprasad, R. Refractive index prediction models for polymers using machine learning. J. Appl. Phys. 2020, 127, 215105. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, X. Machine learning glass transition temperature of polymers. Heliyon 2020, 6, e05055. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.; Yin, J.; Yu, X. Rapid and Nondestructive On-Site Classification Method for Consumer-Grade Plastics Based on Portable NIR Spectrometer and Machine Learning. J. Spectrosc. 2020, 2020, 6631234. [Google Scholar] [CrossRef]
- Bieler, M.; Reutlinger, M.; Rodrigues, T.; Schneider, P.; Kriegl, J.M.; Schneider, G. Designing Multi-target Compound Libraries with Gaussian Process Models. Mol. Inform. 2016, 35, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Kim, C.; Chandrasekarn, A.; Everett, J.; Ramprasad, R.; Lively, R.P. Polymer genome-based prediction of gas permeabilities in polymers. J. Polym. Eng. 2020, 40, 451–457. [Google Scholar] [CrossRef]
- Song, Z.; Shi, H.; Zhang, X.; Zhou, T. Prediction of CO2 solubility in ionic liquids using machine learning methods. Chem. Eng. Sci. 2020, 223, 115752. [Google Scholar] [CrossRef]
- Doan Tran, H.; Kim, C.; Chen, L.; Chandrasekaran, A.; Batra, R.; Venkatram, S.; Kamal, D.; Lightstone, J.P.; Gurnani, R.; Shetty, P.; et al. Machine-learning predictions of polymer properties with Polymer Genome. J. Appl. Phys. 2020, 128, 171104. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Song, Z.; Zhou, T. Data-Driven Ionic Liquid Design for CO2 Capture: Molecular Structure Optimization and DFT Veri fi cation. Ind. Eng. Chem. Res. 2021, 60, 9992–10000. [Google Scholar] [CrossRef]
- Haghighatlari, M.; Hachmann, J. Advances of machine learning in molecular modeling and simulation. Curr. Opin. Chem. Eng. 2019, 23, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Pilania, G.; Wang, C.; Jiang, X.; Rajasekaran, S.; Ramprasad, R. Accelerating materials property predictions using machine learning. Sci. Rep. 2013, 3, 2810. [Google Scholar] [CrossRef]
- Yeo, C.S.H.; Xie, Q.; Wang, X.; Zhang, S. Understanding and optimization of thin film nanocomposite membranes for reverse osmosis with machine learning. J. Membr. Sci. 2020, 606, 118135. [Google Scholar] [CrossRef]
- Yan, Y.; Mattisson, T.; Moldenhauer, P.; Anthony, E.J.; Clough, P.T. Applying machine learning algorithms in estimating the performance of heterogeneous, multi-component materials as oxygen carriers for chemical-looping processes. Chem. Eng. J. 2020, 387, 124072. [Google Scholar] [CrossRef]
- Sun, Y.; Hewitt, M.; Wilkinson, S.C.; Davey, N.; Adams, R.G.; Gullick, D.R.; Moss, G.P. Development of a Gaussian Process–feature selection model to characterise (poly)dimethylsiloxane (Silastic®) membrane permeation. J. Pharm. Pharmacol. 2020, 72, 873–888. [Google Scholar] [CrossRef] [Green Version]
- Ju, S.; Shimizu, S.; Shiomi, J. Designing thermal functional materials by coupling thermal transport calculations and machine learning. J. Appl. Phys. 2020, 128, 161102. [Google Scholar] [CrossRef]
- Jiao, P.; Alavi, A.H. Artificial intelligence-enabled smart mechanical metamaterials: Advent and future trends. Int. Mater. Rev. 2021, 66, 365–393. [Google Scholar] [CrossRef]
- Chen, C.; Zuo, Y.; Ye, W.; Li, X.; Deng, Z.; Ong, S.P. A Critical Review of Machine Learning of Energy Materials. Adv. Energy Mater. 2020, 10, 1903242. [Google Scholar] [CrossRef]
- Christensen, T.; Loh, C.; Picek, S.; Jakobović, D.; Jing, L.; Fisher, S.; Ceperic, V.; Joannopoulos, J.D.; Soljačić, M. Predictive and generative machine learning models for photonic crystals. Nanophotonics 2020, 9, 4183–4192. [Google Scholar] [CrossRef]
- Xie, Y.; Zhang, C.; Hu, X.; Zhang, C.; Kelley, S.P.; Atwood, J.L.; Lin, J. Machine Learning Assisted Synthesis of Metal-Organic Nanocapsules. J. Am. Chem. Soc. 2020, 142, 1475–1481. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Han, J.; Cao, T.; Lam, W.; Fan, B.; Tang, W.; Chen, S.; Fok, K.L.; Li, L. Design of self-assembly dipeptide hydrogels and machine learning via their chemical features. Proc. Natl. Acad. Sci. USA 2019, 166, 11259–11264. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.H.; Noh, J.; Kim, I.; Jung, Y. Machine learning for renewable energy materials. J. Mater. Chem. A 2019, 7, 17096–17117. [Google Scholar] [CrossRef]
- Conduit, B.D.; Illston, T.; Baker, S.; Duggappa, D.V.; Harding, S.; Stone, H.J.; Conduit, G.J. Probabilistic neural network identification of an alloy for direct laser deposition. Mater. Des. 2019, 168, 107644. [Google Scholar] [CrossRef]
- Balachandran, P.V. Machine learning guided design of functional materials with targeted properties. Comput. Mater. Sci. 2019, 164, 82–90. [Google Scholar] [CrossRef]
- Noack, M.M.; Doerk, G.S.; Li, R.; Streit, J.K.; Vaia, R.A.; Yager, K.G.; Fukuto, M. Autonomous materials discovery driven by Gaussian process regression with inhomogeneous measurement noise and anisotropic kernels. Sci. Rep. 2020, 10, 17663. [Google Scholar] [CrossRef]
- Mansouri Tehrani, A.; Oliynyk, A.O.; Parry, M.; Rizvi, Z.; Couper, S.; Lin, F.; Miyagi, L.; Sparks, T.D.; Brgoch, J. Machine Learning Directed Search for Ultraincompressible, Superhard Materials. J. Am. Chem. Soc. 2018, 140, 9844–9853. [Google Scholar] [CrossRef]
- Tabor, D.P.; Roch, L.M.; Saikin, S.K.; Kreisbeck, C.; Sheberla, D.; Montoya, J.H.; Dwaraknath, S.; Aykol, M.; Ortiz, C.; Tribukait, H.; et al. Accelerating the discovery of materials for clean energy in the era of smart automation. Nat. Rev. Mater. 2018, 3, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Saeki, A. Evaluation-oriented exploration of photo energy conversion systems: From fundamental optoelectronics and material screening to the combination with data science. Polym. J. 2020, 52, 1307–1321. [Google Scholar] [CrossRef]
- Kayala, M.A.; Baldi, P. ReactionPredictor: Prediction of complex chemical reactions at the mechanistic level using machine learning. J. Chem. Inf. Model. 2012, 52, 2526–2540. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, P.; Gaudin, T.; Lányi, D.; Bekas, C.; Laino, T. “Found in Translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. [Google Scholar] [CrossRef] [Green Version]
- Kayala, M.A.; Azencott, C.A.; Chen, J.H.; Baldi, P. Learning to predict chemical reactions. J. Chem. Inf. Model. 2011, 51, 2209–2222. [Google Scholar] [CrossRef] [Green Version]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segler, M.H.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem. Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef]
- Segler, M.H.; Waller, M.P. Modelling Chemical Reasoning to Predict and Invent Reactions. Chem. Eur. J. 2017, 23, 6118–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segler, M.H.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Cent. Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, P.; Petraglia, R.; Zullo, V.; Nair, V.H.; Haeuselmann, R.A.; Pisoni, R.; Bekas, C.; Iuliano, A.; Laino, T. Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy. Chem. Sci. 2020, 11, 3316–3325. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, P.; Hoover, B.; Reymond, J.L.; Strobelt, H.; Laino, T. Extraction of organic chemistry grammar from unsupervised learning of chemical reactions. Sci. Adv. 2021, 7, eabe4166. [Google Scholar] [CrossRef]
- Gao, H.; Struble, T.J.; Coley, C.W.; Wang, Y.; Green, W.H.; Jensen, K.F. Using Machine Learning to Predict Suitable Conditions for Organic Reactions. ACS Cent. Sci. 2018, 4, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.N.; Duvenaud, D.; Aspuru-Guzik, A. Neural networks for the prediction of organic chemistry reactions. ACS Cent. Sci. 2016, 2, 725–732. [Google Scholar] [CrossRef]
- Ishida, S.; Terayama, K.; Kojima, R.; Takasu, K.; Okuno, Y. Prediction and Interpretable Visualization of Retrosynthetic Reactions Using Graph Convolutional Networks. J. Chem. Inf. Model. 2019, 59, 5026–5033. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Kim, J. Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions. arXiv 2016, arXiv:1612.09529. [Google Scholar]
- McCoy, J.T.; Auret, L. Machine learning applications in minerals processing: A review. Miner. Eng. 2019, 132, 95–109. [Google Scholar] [CrossRef]
- Curteanu, S.; Leon, F.; Mircea-Vicoveanu, A.M.; Logofătu, D. Regression methods based on nearest neighbors with adaptive distance metrics applied to a polymerization process. Mathematics 2021, 9, 547. [Google Scholar] [CrossRef]
- Curteanu, S. Chapter 10 Machine Learning Techniques Applied to a Complex Polymerization Process. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 227–250. [Google Scholar] [CrossRef]
- Meimaroglou, D.; Florez, D.; Hu, G.H. A kinetic modeling framework for the peroxide-initiated radical polymerization of styrene in the presence of rubber particles from recycled tires. Chem. Eng. Sci. 2020. under review. [Google Scholar]
- Khayyam, H.; Jazar, R.N.; Nunna, S.; Golkarnarenji, G.; Badii, K.; Fakhrhoseini, S.M.; Kumar, S.; Naebe, M. PAN precursor fabrication, applications and thermal stabilization process in carbon fiber production: Experimental and mathematical modelling. Prog. Mater. Sci. 2020, 107, 100575. [Google Scholar] [CrossRef]
- Kramer, A.; Morgado-Dias, F. Artificial intelligence in process control applications and energy saving: A review and outlook. Greenh. Gases Sci. Technol. 2020, 10, 1133–1150. [Google Scholar] [CrossRef]
- Dong, E.L.; Song, J.H.; Song, S.O.; En, S.Y. Weighted support vector machine for quality estimation in the polymerization process. Ind. Eng. Chem. Res. 2005, 44, 2101–2105. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, J.; Dong, Z.; Huang, J.; Zhou, H.; Fu, J.; Turng, L.S. Intelligent Injection Molding on Sensing, Optimization, and Control. Adv. Polym. Technol. 2020, 2020, 7023616. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Shamsi, M.H.; Choi, T.S. Correlating dynamical mechanical properties with temperature and clay composition of polymer-clay nanocomposites. Comput. Mater. Sci. 2009, 45, 257–265. [Google Scholar] [CrossRef]
- Li, C.; Rubín De Celis Leal, D.; Rana, S.; Gupta, S.; Sutti, A.; Greenhill, S.; Slezak, T.; Height, M.; Venkatesh, S. Rapid Bayesian optimisation for synthesis of short polymer fiber materials. Sci. Rep. 2017, 7, 5683. [Google Scholar] [CrossRef]
- Ibañez, R.; Casteran, F.; Argerich, C.; Ghnatios, C.; Hascoet, N.; Ammar, A.; Cassagnau, P.; Chinesta, F. On the data-driven modeling of reactive extrusion. Fluids 2020, 5, 94. [Google Scholar] [CrossRef]
- Curteanu, S.; Leon, F.; Galea, D. Neural network models for free radical polymerization of methyl methacrylate Neural Network Models for Free Radical Polymerization of Methyl Methacrylate. Eurasian Chemtech. J. 2003, 5, 225–231. [Google Scholar]
- Curteanu, S. Direct and inverse neural network modeling in free radical polymerization. Cent. Eur. J. Chem. 2004, 2, 113–140. [Google Scholar] [CrossRef]
- Rodríguez-Dorado, R.; Landín, M.; Altai, A.; Russo, P.; Aquino, R.P.; Del Gaudio, P. A novel method for the production of core-shell microparticles by inverse gelation optimized with artificial intelligent tools. Int. J. Pharm. 2018, 538, 97–104. [Google Scholar] [CrossRef]
- Rouco, H.; Diaz-Rodriguez, P.; Rama-Molinos, S.; Remuñán-López, C.; Landin, M. Delimiting the knowledge space and the design space of nanostructured lipid carriers through Artificial Intelligence tools. Int. J. Pharm. 2018, 553, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Han, L.; Mustakis, J.; Li, B.; Magano, J.; Damon, D.B.; Dion, A.; Maloney, M.T.; Post, R.; Li, R. Kinetic and Data-Driven Reaction Analysis for Pharmaceutical Process Development. Ind. Eng. Chem. Res. 2020, 59, 2409–2421. [Google Scholar] [CrossRef]
- Sarmadi, M.; Behrens, A.M.; McHugh, K.J.; Contreras, H.T.; Tochka, Z.L.; Lu, X.; Langer, R.; Jaklenec, A. Modeling, design, and machine learning-based framework for optimal injectability of microparticle-based drug formulations. Sci. Adv. 2020, 6, abb6594. [Google Scholar] [CrossRef]
- Jhamb, S.; Enekvist, M.; Liang, X.; Zhang, X.; Dam-Johansen, K.; Kontogeorgis, G.M. A review of computer-aided design of paints and coatings. Curr. Opin. Chem. Eng. 2020, 27, 107–120. [Google Scholar] [CrossRef]
- Jasso-Salcedo, A.B.; Hoppe, S.; Pla, F.; Escobar-Barrios, V.A.; Camargo, M.; Meimaroglou, D. Modeling and optimization of a photocatalytic process: Degradation of endocrine disruptor compounds by Ag/ZnO. Chem. Eng. Res. Des. 2017, 128, 174–191. [Google Scholar] [CrossRef]
- Jeguirim, S.E.G.; Dhouib, A.B.; Sahnoun, M.; Cheikhrouhou, M.; Schacher, L.; Adolphe, D. The use of fuzzy logic and neural networks models for sensory properties prediction from process and structure parameters of knitted fabrics. J. Intell. Manuf. 2011, 22, 873–884. [Google Scholar] [CrossRef]
- Golkarnarenji, G.; Naebe, M.; Badii, K.; Milani, A.S.; Jazar, R.N.; Khayyam, H. A machine learning case study with limited data for prediction of carbon fiber mechanical properties. Comput. Ind. 2019, 105, 123–132. [Google Scholar] [CrossRef]
- Wang, Q.; Lonergan, S.M.; Yu, C. Rapid determination of pork sensory quality using Raman spectroscopy. Meat Sci. 2012, 91, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, D. Intelligent Sensory Evaluation: Methodologies and Applications; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Zeng, X.; Ruan, D.; Koehl, L. Intelligent sensory evaluation: Concepts, implementations, and applications. Math. Comput. Simul. 2008, 77, 443–452. [Google Scholar] [CrossRef]
- Ouyang, Q.; Chen, Q.; Zhao, J. Intelligent sensing sensory quality of Chinese rice wine using near infrared spectroscopy and nonlinear tools. Spectrochim. Acta Part Mol. Biomol. Spectrosc. 2016, 154, 42–46. [Google Scholar] [CrossRef]
- Gunaratne, T.M.; Viejo, C.G.; Gunaratne, N.M.; Torrico, D.D.; Dunshea, F.R.; Fuentes, S. Chocolate quality assessment based on chemical fingerprinting using near infra-red and machine learning modeling. Foods 2019, 8, 426. [Google Scholar] [CrossRef] [Green Version]
- Sanahuja, S.; Fédou, M.; Briesen, H. Classification of puffed snacks freshness based on crispiness-related mechanical and acoustical properties. J. Food Eng. 2018, 226, 53–64. [Google Scholar] [CrossRef]
- Bahamonde, A.; Díez, J.; Quevedo, J.R.; Luaces, O.; del Coz, J.J. How to learn consumer preferences from the analysis of sensory data by means of support vector machines (SVM). Trends Food Sci. Technol. 2007, 18, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Zhi, R.; Zhao, L.; Shi, J. Improving the sensory quality of flavored liquid milk by engaging sensory analysis and consumer preference. J. Dairy Sci. 2016, 99, 5305–5317. [Google Scholar] [CrossRef]
- Krishnamurthy, R.; Srivastava, A.K.; Paton, J.E.; Bell, G.A.; Levy, D.C. Prediction of consumer liking from trained sensory panel information: Evaluation of neural networks. Food Qual. Prefer. 2007, 18, 275–285. [Google Scholar] [CrossRef]
- Rocha, R.S.; Calvalcanti, R.N.; Silva, R.; Guimarães, J.T.; Balthazar, C.F.; Pimentel, T.C.; Esmerino, E.A.; Freitas, M.Q.; Granato, D.; Costa, R.G.; et al. Consumer acceptance and sensory drivers of liking of Minas Frescal Minas cheese manufactured using milk subjected to ohmic heating: Performance of machine learning methods. LWT 2020, 126, 109342. [Google Scholar] [CrossRef]
- Fuentes, S.; Torrico, D.D.; Tongson, E.; Viejo, C.G. Machine learning modeling of wine sensory profiles and color of vertical vintages of pinot noir based on chemical fingerprinting, weather and management data. Sensors 2020, 20, 3618. [Google Scholar] [CrossRef]
- Liu, P.; Zhu, X.; Hu, X.; Xiong, A.; Wen, J.; Li, H.; Ai, S.; Wu, R. Local tangent space alignment and relevance vector machine as nonlinear methods for estimating sensory quality of tea using NIR spectroscopy. Vib. Spectrosc. 2019, 103, 102923. [Google Scholar] [CrossRef]
- Vigneau, E.; Courcoux, P.; Symoneaux, R.; Guérin, L.; Villière, A. Random forests: A machine learning methodology to highlight the volatile organic compounds involved in olfactory perception. Food Qual. Prefer. 2018, 68, 135–145. [Google Scholar] [CrossRef]
- Viejo, C.G.; Fuentes, S. A Digital Approach to Model Quality and Sensory Traits of Beers Fermented under Sonication Based on Chemical Fingerprinting. Fermentation 2020, 6, 73. [Google Scholar] [CrossRef]
- Nozaki, Y.; Nakamoto, T. Correction: Predictive modeling for odor character of a chemical using machine learning combined with natural language processing (PLoS ONE (2018) 13, 6 (e0198475) DOI: 10.1371/journal.pone.0198475). PLoS ONE 2018, 13, e0208962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhou, T.; Ng, K.M. Optimization-based cosmetic formulation: Integration of mechanistic model, surrogate model, and heuristics. AIChE J. 2021, 67, 1–16. [Google Scholar] [CrossRef]
- Gonzalez Viejo, C.; Fuentes, S.; Torrico, D.; Lee, M.; Hu, Y.; Chakraborty, S.; Dunshea, F. The Effect of Soundwaves on Foamability Properties and Sensory of Beers with a Machine Learning Modeling Approach. Beverages 2018, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Lerma-García, M.J.; Cerretani, L.; Cevoli, C.; Simó-Alfonso, E.F.; Bendini, A.; Toschi, T.G. Use of electronic nose to determine defect percentage in oils. Comparison with sensory panel results. Sens. Actuators B Chem. 2010, 147, 283–289. [Google Scholar] [CrossRef]
- Goodfellow, I. Deep Learning; The MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Martynenko, A.; Misra, N.N. Machine learning in drying. Dry. Technol. 2020, 38, 596–609. [Google Scholar] [CrossRef]
- Lu, N.V.; Tansuchat, R.; Yuizono, T.; Huynh, V.N. Incorporating active learning into machine learning techniques for sensory evaluation of food. Int. J. Comput. Intell. Syst. 2020, 13, 655–662. [Google Scholar] [CrossRef]
- Al-Jamimi, H.A.; Al-Azani, S.; Saleh, T.A. Supervised machine learning techniques in the desulfurization of oil products for environmental protection: A review. Process. Saf. Environ. Prot. 2018, 120, 57–71. [Google Scholar] [CrossRef]
- Azencott, C.A. Introduction au Machine Learning; Dunod: Malakoff, Malaysia, 2018. [Google Scholar]
- Asante-Okyere, S.; Shen, C.; Ziggah, Y.Y.; Rulegeya, M.M.; Zhu, X. Investigating the predictive performance of Gaussian process regression in evaluating reservoir porosity and permeability. Energies 2018, 11, 3261. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Yabansu, Y.C.; Collins, P.C.; Kalidindi, S.R. Evaluation of Ti – Mn Alloys for Additive Assays and Gaussian Process Regression. Materials 2020, 13, 4641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Wang, S.; Ying, J.; Fu, J. Non-destructive measurement of cavity pressure during injection molding process based on ultrasonic technology and Gaussian process. Polym. Test. 2013, 32, 1436–1444. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, Z. Real-time property prediction for an industrial rubber-mixing process with probabilistic ensemble Gaussian process regression models. J. Appl. Polym. Sci. 2015, 132, 1–9. [Google Scholar] [CrossRef]
- Liu, H.; Ong, Y.S.; Shen, X.; Cai, J. When Gaussian Process Meets Big Data: A Review of Scalable GPs. IEEE Trans. Neural Netw. Learn. Syst. 2020, 31, 4405–4423. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, C. Gaussian Processes for Machine Learning; MIT Press: Cambridge, MA, USA, 2006. [Google Scholar]
- Burkov, A. Machine Learning Engineering; True Positive, Inc.: Québec, QC, Canada, 2020. [Google Scholar]
- Cartwright, H.M. Chapter 5 Machine Learning in Science – A Role for Mechanical Sympathy? In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 109–135. [Google Scholar] [CrossRef]
- Irwin, B.W.; Levell, J.R.; Whitehead, T.M.; Segall, M.D.; Conduit, G.J. Practical Applications of Deep Learning to Impute Heterogeneous Drug Discovery Data. J. Chem. Inf. Model. 2020, 60, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, T.M.; Irwin, B.W.; Hunt, P.; Segall, M.D.; Conduit, G.J. Imputation of Assay Bioactivity Data Using Deep Learning. J. Chem. Inf. Model. 2019, 59, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Stukenbroeker, T.; Clausen, J. Chapter 6 A Prediction of Future States: AI-powered Chemical Innovation for Defense Applications. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 136–168. [Google Scholar] [CrossRef]
- Sha, W.; Li, Y.; Tang, S.; Tian, J.; Zhao, Y.; Guo, Y.; Zhang, W.; Zhang, X.; Lu, S.; Cao, Y.; et al. Machine learning in polymer informatics. InfoMat 2021, 3, 353–361. [Google Scholar] [CrossRef]
- Cai, J.; Chu, X.; Xu, K.; Li, H.; Wei, J. Machine learning-driven new material discovery. Nanoscale Adv. 2020, 2, 3115–3130. [Google Scholar] [CrossRef]
- Luo, L.; Yao, Y.; Gao, F.; Zhao, C. Mixed-effects Gaussian process modeling approach with application in injection molding processes. J. Process. Control. 2017, 62, 37–43. [Google Scholar] [CrossRef]
- Haghighatlari, M.; Li, J.; Heidar-Zadeh, F.; Liu, Y.; Guan, X.; Head-Gordon, T. Learning to Make Chemical Predictions: The Interplay of Feature Representation, Data, and Machine Learning Methods. Chem 2020, 6, 1527–1542. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Liu, C.; Wu, S.; Koyama, Y.; Ju, S.; Shiomi, J.; Morikawa, J.; Yoshida, R. Predicting Materials Properties with Little Data Using Shotgun Transfer Learning. ACS Cent. Sci. 2019, 5, 1717–1730. [Google Scholar] [CrossRef] [Green Version]
- Geiger, A.C.; Cao, Z.; Song, Z.; Ulcickas, J.R.W.; Simpson, G.J. Chapter 18 Autonomous Science: Big Data Tools for Small Data Problems in Chemistry. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 450–487. [Google Scholar] [CrossRef]
- Stein, H.S.; Gregoire, J.M. Progress and prospects for accelerating materials science with automated and autonomous workflows. Chem. Sci. 2019, 10, 9640–9649. [Google Scholar] [CrossRef] [Green Version]
- Mitchell B.O., J.B. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Roscher, R.; Bohn, B.; Duarte, M.F.; Garcke, J. Explainable Machine Learning for Scientific Insights and Discoveries. IEEE Access 2020, 8, 42200–42216. [Google Scholar] [CrossRef]
- Voosen, P. The AI detectives. Science 2017, 357, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.G.; Lawrence, R. Chapter 3 MedChemInformatics: An Introduction to Machine Learning for Drug Discovery. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 37–75. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Domain | References |
|---|---|
| Molecular and material science | [2,3,4,5,6,7,8,9,10,11,12,13] |
| Drug design and discovery | [14,15,16,17,18] |
| Catalysis | [19,20,21] |
| Chemical synthesis | [22,23,24] |
| Chemical and process engineering | [25,26,27] |
| Additive manufacturing | [28,29] |
| Learning Category | Training Data Set Configuration | Objective | Examples in Chemical Product Engineering | Examples of Algorithms |
|---|---|---|---|---|
| Supervised | Labeled data | The algorithm describes the relationship between inputs x and outputs y |
| ANN SVM/SVR GP DT RF kNN MR Logistic regression |
| Unsupervised | Unlabeled data | The algorithm explores and extracts hidden patterns within the input features x |
| PCA k-means clustering ANN HCA AE ICA GMM |
| Semi-supervised | Few labeled data with a large amount of unlabeled data | The algorithm explores the information hidden in unlabeled data in order to improve the prediction performance of the supervised learning model constructed with the labeled data | ANN Generative models Graph-based methods Co-training Self-training Multiview learning | |
| Reinforcement | Input data are the states and the feedback signals of environment; output is action | The algorithm learns an optimal policy that selects which is the best action to execute given the state of the environment | Control of polymerization processes [48,49] | Dynamic programming Monte Carlo methods Temporal difference |
| References | ML Method | Inputs | Outputs | Data Set |
|---|---|---|---|---|
| • Forward design (property/activity prediction from chemical structure) | ||||
| [90] Fragrance | LDA, SVM | Molecules structural descriptors | Fragrance class (apple, pineapple, rose) | 91 organic compounds with their fragrance class from database |
| [91] Cosmetics | ANN | Peptides | Anti-age properties | Data set from papers and patents (unstructured data) and from public databases (structured data), processed respectively by NLP and graph-based techniques |
| [92] Polymers | PCA/LASSO for data visualisation and feature reduction + GP for regression | Polymer relevant features | Refractive index (RI) | 500 polymers from publicly available sources with their experimentally measured RI |
| [93] Polymers | GP + Lower confidence bound Bayesian optimizations | Molecular traceless quadrupole moment, molecule average hexadecapole moment | Glass transition temperature | 60 polymers with their transition temperature from database |
| [94] Homogeneous catalysis | Hybrid: MR, Kernel ridge regression, RF, ANN and QM/DFT calculations | Energy, atomic, molecular, vibrational, structural descriptors (DFT) | Catalytic activity, reaction yield | 4600–18,062 catalysts/reactions from libraries |
| [94] Heterogeneous catalysis | Hybrid: ANN, MR, RF, SVM, GP and QM/DFT calculations | Fingerprint features, structural and charge descriptors (DFT) | Adsorption, formation, binding, activation, reaction barrier energies, catalytic activity (DFT) | 315–788 catalysts/reactions from libraries |
| [87] Molecules | Generative model for latent space creation (RNN, VAE, AAE, GAN, RL, BL and BNN) + predictive model for mapping latent variables and properties (RNN, RL, DNN, SVM, GP, BO and BNN) | Molecular representations (numerical, text-based or graph-based) | Physical, chemical or biological properties | 5k–1800k molecules from databases |
| [67] Polymers | PCA/LASSO for data visualization and feature reduction + GP for regression | 53–57 Relevant features from three hierarchical levels (atomic, block and chain) | Frequency-dependent dielectric constant, glass transition temperature | 738 polymers and their 1210 experimentally measured properties at various frequencies |
| [95] Pharmaceutical compounds | GP and ant colony optimization algorithm (activity prediction followed by automated top scoring compounds picking from virtual combinatorial library) | Structure | Activity of ligand binding to 11 pharmaceutical relevant GCPRs (G-coupled protein receptor) drug target | 3519 compounds with affinity annotations for 11 diverse GCPR targets (from libraries) |
| [83] Pharmaceutical compounds | ANN | Small molecules conformations | Energy of much larger systems | 22 M small molecules conformations |
| [96] Polymers | GP (Polymer Genome) | Structure | Gas permeability | 315 polymers and their associated 1501 permeability data |
| [97] Ionic liquids | ANN, SVM | Groups present in ionic liquid molecule | CO solubility | 10,116 CO solubility data in various ionic liquids |
| [80] Cosmetics | Graph machine; Hybrid: ANN and COSMO-RS | SMILES, moments (COSMO-RS) | Viscosity | 300 liquid compounds with known viscosities |
| [98] Polymers | GP, ANN, Kriging (Polymer Genome) | Polymer name, SMILES | Polymer properties | 80–6721 polymers and associated properties obtained from first principles and experimental measurements |
| • Inverse design (generation of candidates molecules/materials given target properties) | ||||
| [88] Polymers | ANN | Lightweight, strong, chemical resistant | Candidates structures/patterns | Large database from experiments and simulations |
| [99] Ionic liquids | ANN | Ionic liquid maximized solubility | Top ionic liquids candidates for CO capture | 10,116 CO solubility data in various ionic liquids |
| [100] Molecules | ANN SVM and Kernel ridge regression | Specifications, properties, reagents | Candidates (structures and products) | Not identified |
| References |
|---|
| [101] Polymers |
| [102] Thin film nanocomposite membranes |
| [103] Heterogeneous, multicomponent materials |
| [104] Memristors materials |
| [105] Thermal functional materials |
| [106] Mechanical metamaterials |
| [107] Energy materials |
| [108] Photonic crystals |
| [109] Metal-organic nanocapsules |
| [110] Hydrogels |
| [111] Renewable energy materials |
| [112] Alloys |
| [113] Functional materials |
| [114] Polymers |
| [115] Ultraincompressible, superhard materials |
| [116] Materials for clean energy |
| [117] Photo energy conversion systems |
| Method | Advantages | Limitations |
|---|---|---|
| Physical-based |
|
|
| Rule-based expert systems |
|
|
| Machine learning |
|
|
| Application Category | References | ML Method | Inputs | Outputs |
|---|---|---|---|---|
| Reaction conditions prediction | [128] | HNN (classification and regression) | Reaction (difference between reactants and products fingerprints) | Reaction conditions (catalyst, solvent, reagent and temperature) |
| Ranking templates | [122,129,130] | ANN/DL/GCN (classification) | Reactants, reagents or product fingerprints | Most probable reaction template |
| Generating products | [119,131] | ANN/DL (encoder/decoder translation model) | Reactant SMILES | Product SMILES |
| Classifying reaction feasibility | [124] | ANN/DL | Product | Likely reactions |
| Predicting mechanistic pathway | [118,120] | ANN | Reactants, conditions, products | Reaction, mechanistic pathway |
| Ranking products | [121] | ANN | Possible reactions given reactants | Major product |
| References | ML Method | Inputs | Outputs | Data Set |
|---|---|---|---|---|
| Polymer science | ||||
| [136] | ANN | Dwell time, oven temperature, tension applied on filaments | Yield, final properties of carbon fibers | Not identified |
| [39] | Semi- supervised: DBN and kernel learning | Reactor pressure/temperature, liquid level and catalysts flowrate | Melt index | 1900 unlabeled + 310 labeled |
| [137] | ANN | Process parameters | Monomer conversion, average molecular weight and viscosity, reaction time, dispersion and thermal stability | Not identified |
| [138] | SVM | Temperature, feed rates, reaction time and catalyst quantities | Viscosity | 120 labeled |
| [139] | ANN, SVR, GP | Injection speed/pressure, packing duration/pressure, mold temperature, cooling time, shot size, screw rotation speed, cylinder pressure, barrel temperature, coolant temperature and sensor measurements | Product quality (deformation, defects), melt state, process parameters, fiber orientation distribution, physical/mechanical properties, skin layer and surface roughness | Not identified |
| [35] | PCA + GP | Hydrogen concentration, feed rate and reaction temperature | Process conditions and product quality | 300 labeled |
| [140] | ANN | Temperature and clay composition | Dynamic mechanical properties (storage modulus and loss tangent) | More than 1500 labeled |
| [141] | GP | Process parameters (position, constriction angle, channel width, polymer and solvent flows) | Product parameters (median length, median diameter and quality of fibers) | Not identified |
| [142] | ANN, C2V, sPGD, SVM, DT and iDMD | Material and process parameters (rotation speed, exit flowrate, temperature and compositions) | Properties and performance (Young modulus, yield stress, stress at break, strain at break and impact strength) | 59 labeled |
| [61] | Hybrid: knowledge-based and C2V and sGPD | Flowrate and rotation speed | Torque, pressure, engine power and exit temperature | 47 labeled |
| [133] | LMNNR, Nearest Neighbor Regression with adaptive metrics | Reation conditions (initiator concentration, temperature and time) | Monomer conversion and average molecular weight | 337–414 labeled |
| [62] | Hybrid: knowledge-based and ANN | Reation conditions (initiator concentration, temperature and time) | Monomer conversion and average molecular weight | 3363 labeled |
| [143,144] | ANN | Reation conditions (initiator concentration and temperature) | Monomer conversion, average molecular weight and mass reaction viscosity | Not identified |
| Food industry | ||||
| [54] | Hybrid: knowledge-based and SVR, SVM or ANN | Easy measurements (massecuite temperature/volume/level, vacuum degree, steam pressure/temperature and feeding rate) | Difficult measurements (mother liquor purity/supersaturation) | 210 labeled |
| [56] | Hybrid: knowledge-based and RF | Food ingredients (selection and composition), processing conditions (baking time and temperature) | Sensory properties (color, crispiness and flavors) | 446–462 labeled |
| [66] | Hierarchical clustering | Intrinsic characteristics of yogurt product | Brand and storage conditions | 36 unlabeled |
| Pharmaceutical industry | ||||
| [145] | ANN/Fuzzy logic | Flowrates, frequency of vibration and concentrations | Microparticles properties (shape, oil content and distribution) | 41 labeled |
| [146] | ANN/Fuzzy logic | Compositions, stirring speed | Properties of nanoparticles (size, size distribution, zeta potential, encapsulation efficiency and drug loading) | 15 labeled |
| [147] | MR | Base equivalents, water equivalents and solvent loading | Dynamic profile of starting materials, product and key impurity | 25 labeled |
| [148] | CFD and DoE and ANN | Dimensionless parameters based on material properties, concentration of the particles, viscosity of the injection solution and ratio needle diameter over the greatest dimension of the particles | Drug injectability | 319 labeled |
| Paints | ||||
| [149] | ANN, MR | Formulation parameters | Thermodynamic and functional properties (elasticity, hardness and barrier properties) | Not identified |
| Catalysis | ||||
| [150] | ANN | Nominal silver concentration, pH, reaction time, actual amount of Ag attached on ZnO surface, initial contaminant concentration and light wavelength | Actual amount of Ag attached on ZnO surface and photodegradation performance | 27–63 labeled |
| Minerals | ||||
| [132] | PCR | Fast and easy measurements (flowrate, pressure, temperature and spectra) | Slow and difficult measurements (composition, size distribution, mill load and equipment failure) | Not identified |
| Textile | ||||
| [151] | ANN | Process and structure parameters (bleaching or dyeing, bio-polishing, softening, emerizing, calendering, material and count of yarn) | Sensory properties (bipolar, surface and handle attributes) | 23 labeled |
| Materials science | ||||
| [152] | SVR, ANN | Structure and process parameters (temperature, stretching ration and space velocity) | Mechanical property (Young’s modulus and tensile strength) | 30 labeled |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trinh, C.; Meimaroglou, D.; Hoppe, S. Machine Learning in Chemical Product Engineering: The State of the Art and a Guide for Newcomers. Processes 2021, 9, 1456. https://doi.org/10.3390/pr9081456
Trinh C, Meimaroglou D, Hoppe S. Machine Learning in Chemical Product Engineering: The State of the Art and a Guide for Newcomers. Processes. 2021; 9(8):1456. https://doi.org/10.3390/pr9081456
Chicago/Turabian StyleTrinh, Cindy, Dimitrios Meimaroglou, and Sandrine Hoppe. 2021. "Machine Learning in Chemical Product Engineering: The State of the Art and a Guide for Newcomers" Processes 9, no. 8: 1456. https://doi.org/10.3390/pr9081456