CO2 Hydrogenation to Methanol by a Liquid-Phase Process with Alcoholic Solvents: A Techno-Economic Analysis

Abstract
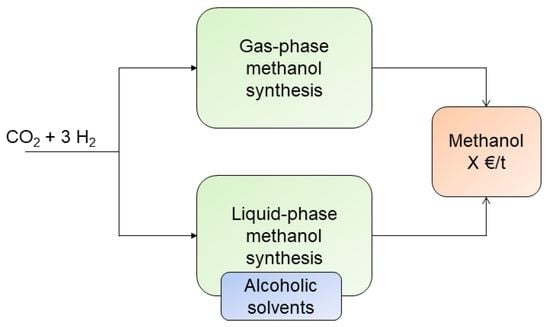
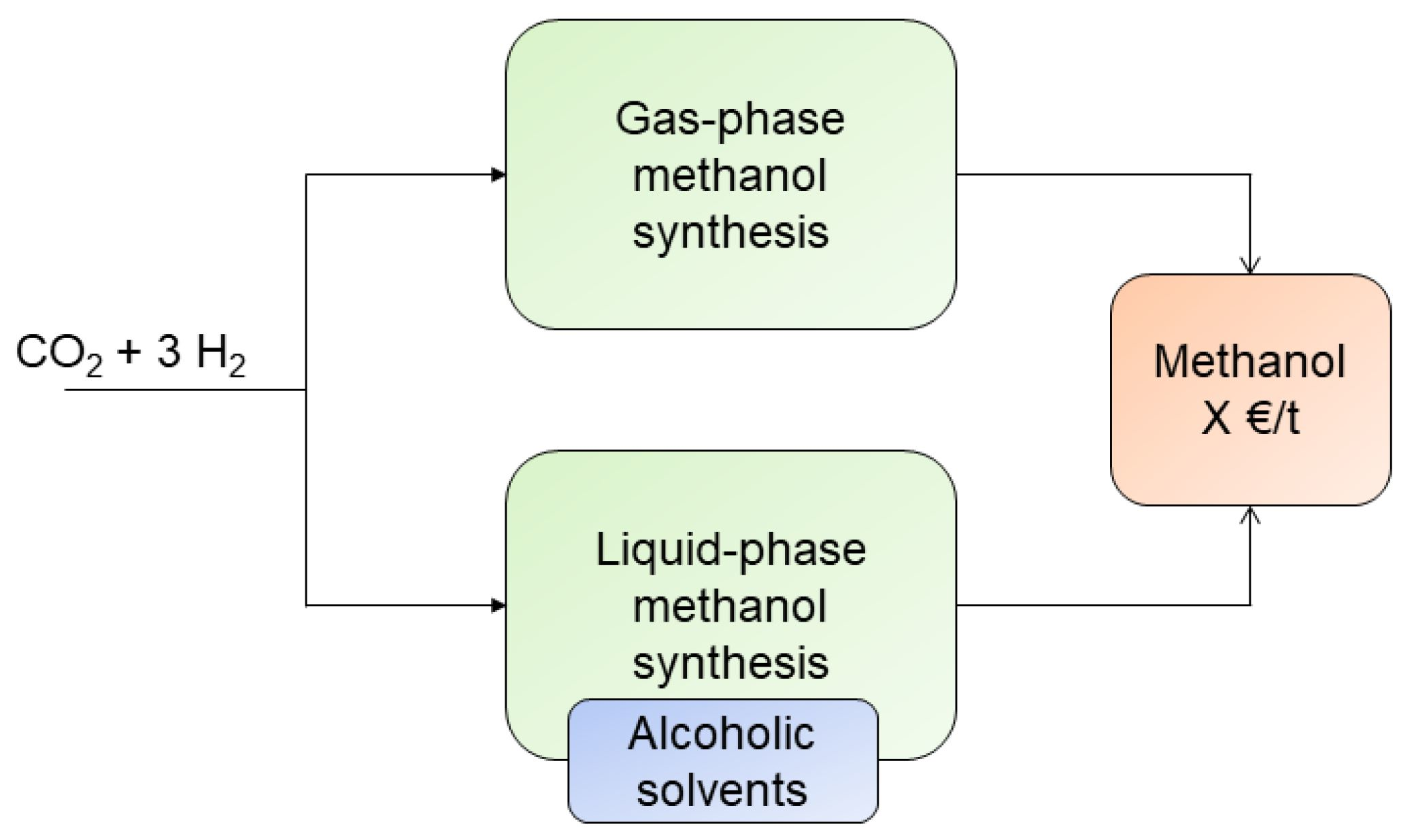
:
1. Introduction
2. Materials and Methods
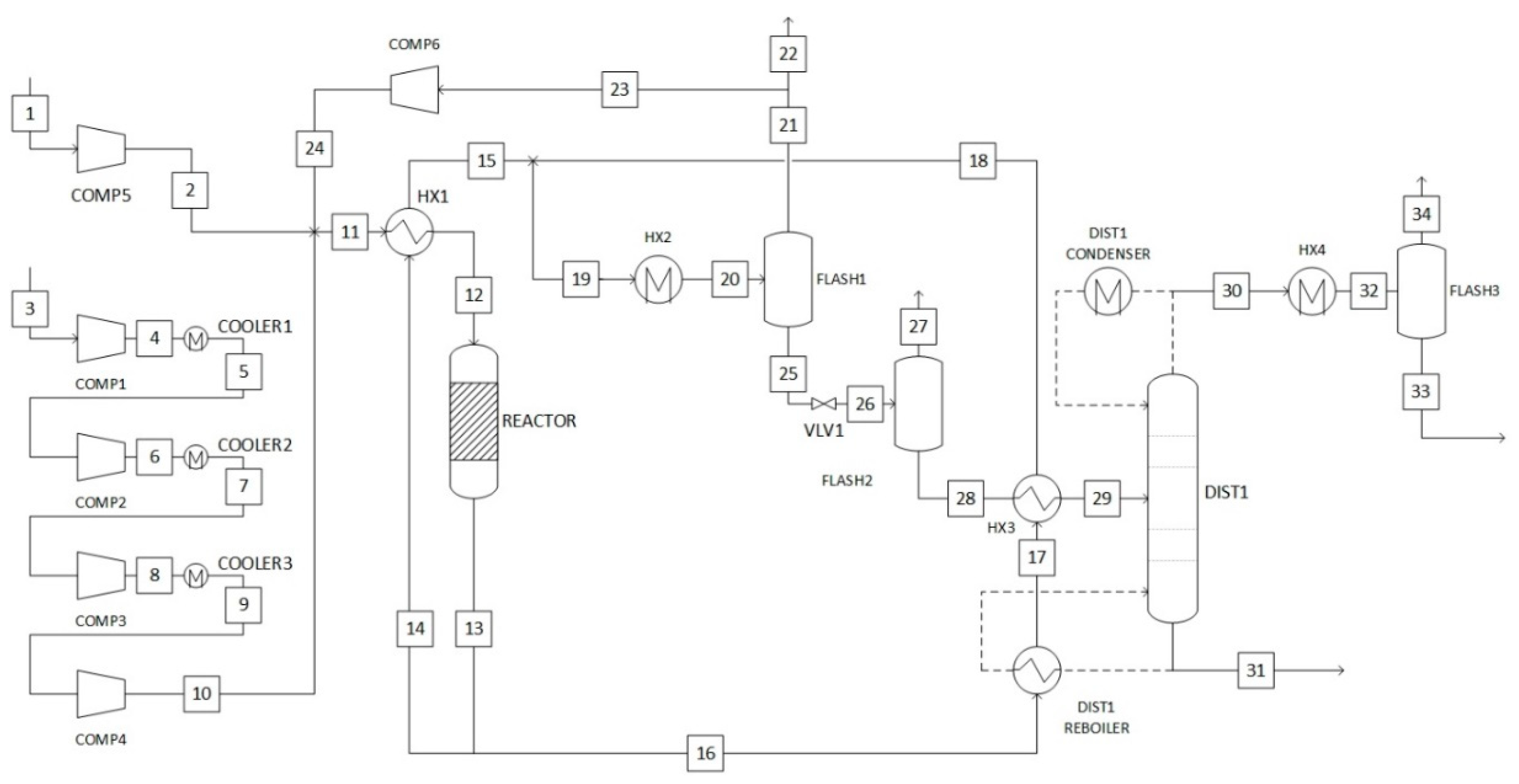
2.1. Modelling Details
2.2. Environmental Impact Analysis
2.3. Cost Estimation
2.3.1. Capital Costs
2.3.2. Variable and Fixed Operating Costs
2.3.3. Revenues
2.4. Economic Analysis
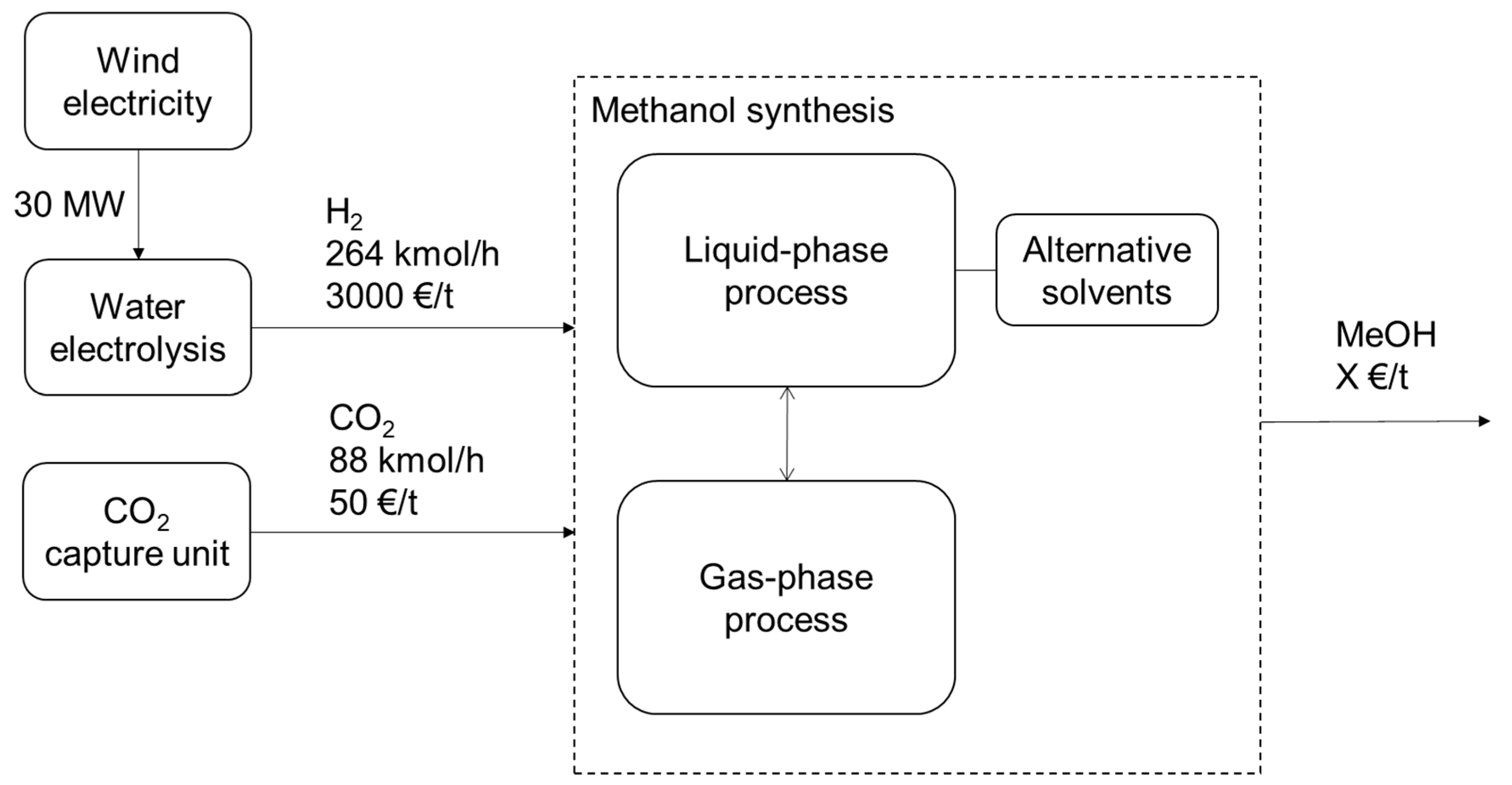
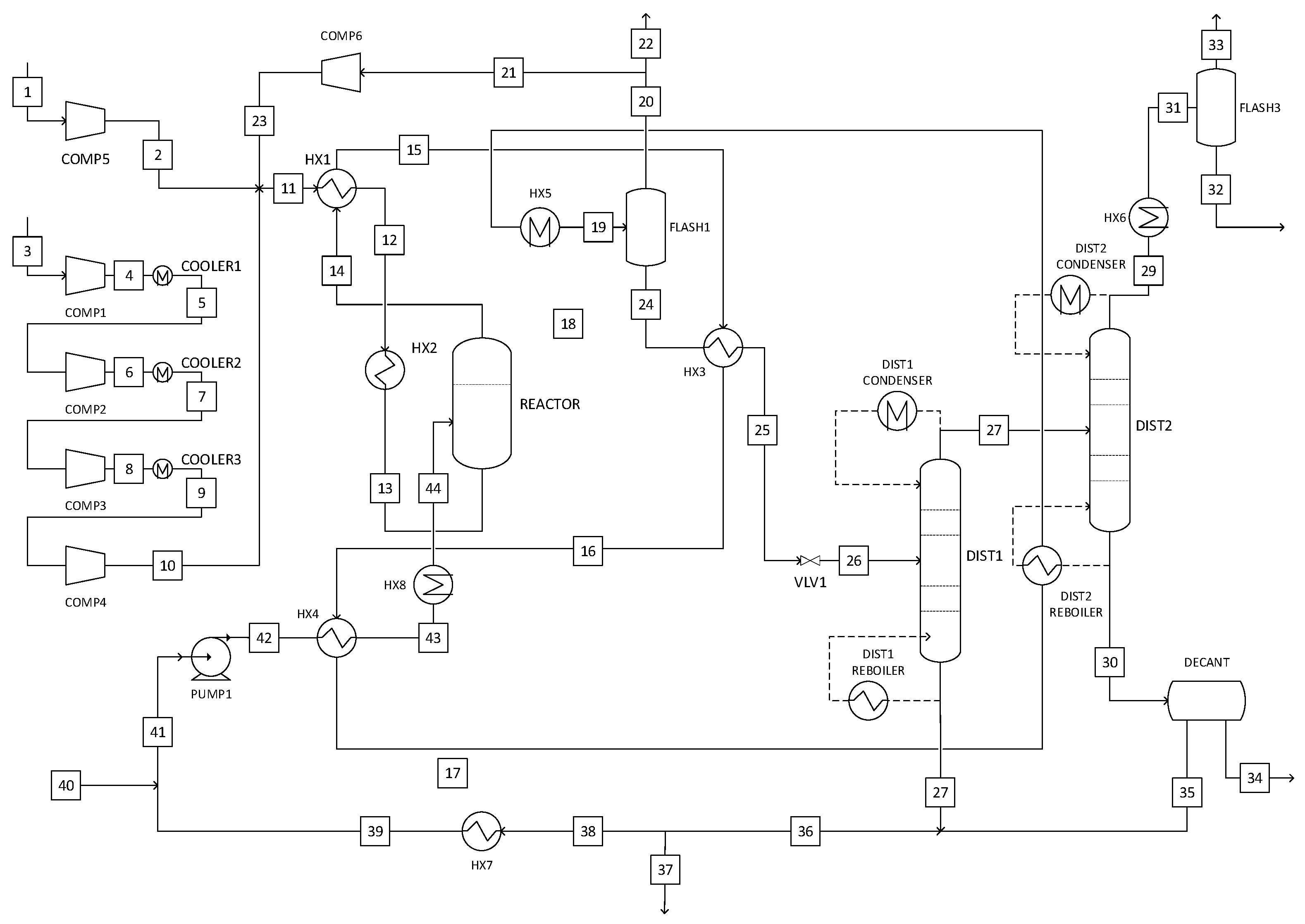
2.5. Process Descriptions
2.5.1. Electrolysis and Wind Electricity
2.5.2. Gas-Phase Methanol Synthesis
2.5.3. Liquid-Phase Methanol Synthesis
3. Results and Discussion
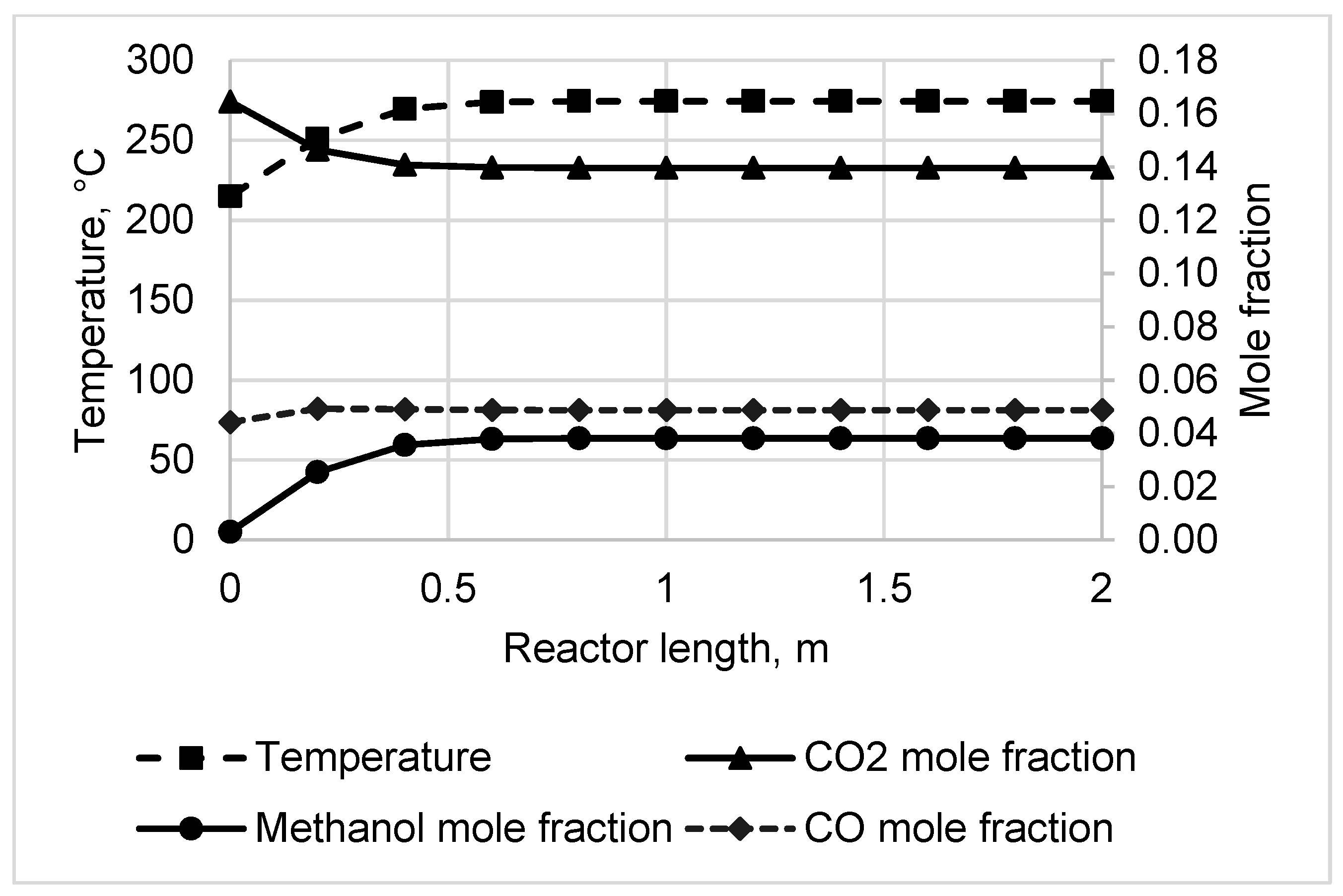
3.1. Reactor Sizing and Performance
3.2. Mass and Energy Balances
3.3. Environmental Analysis
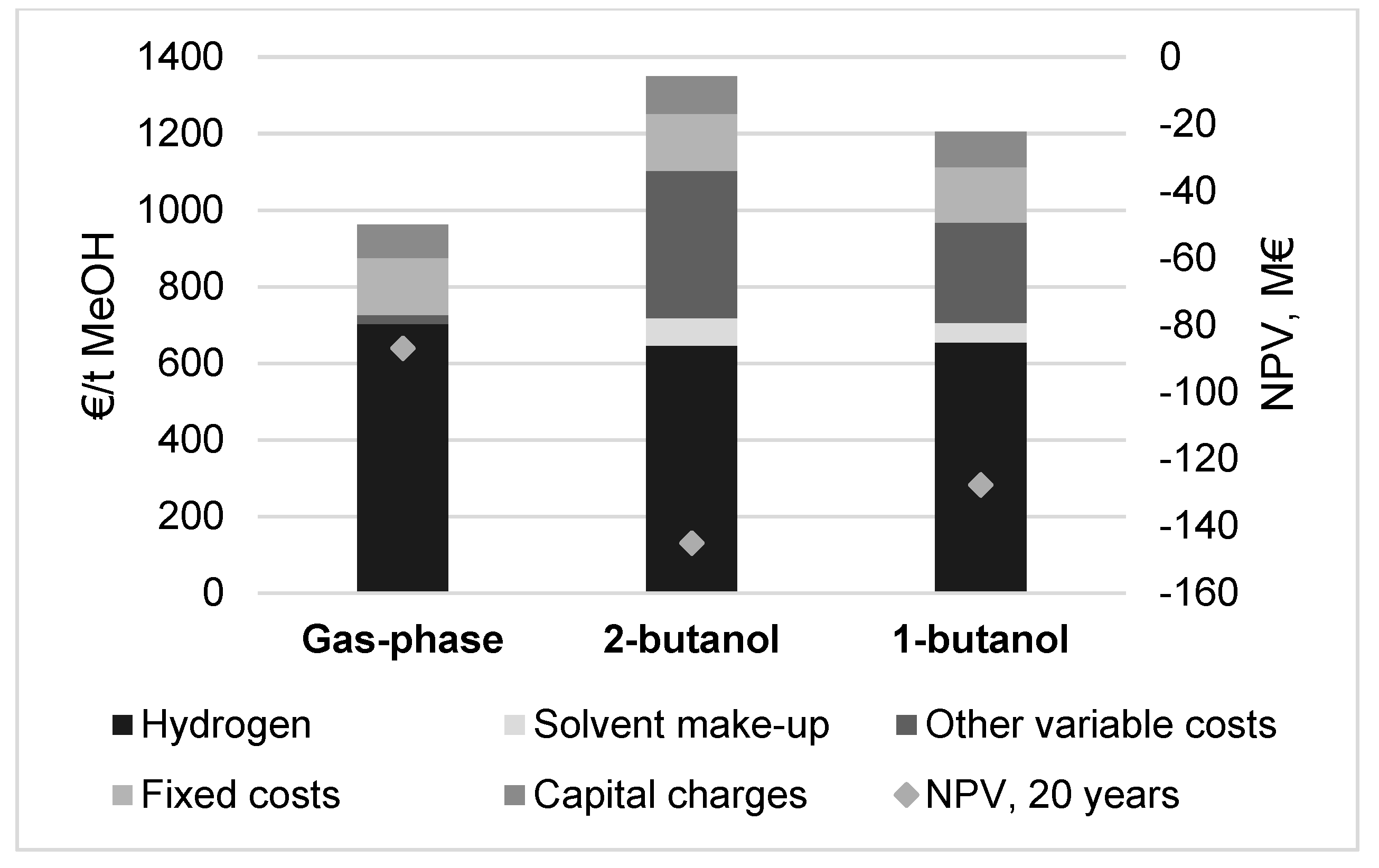
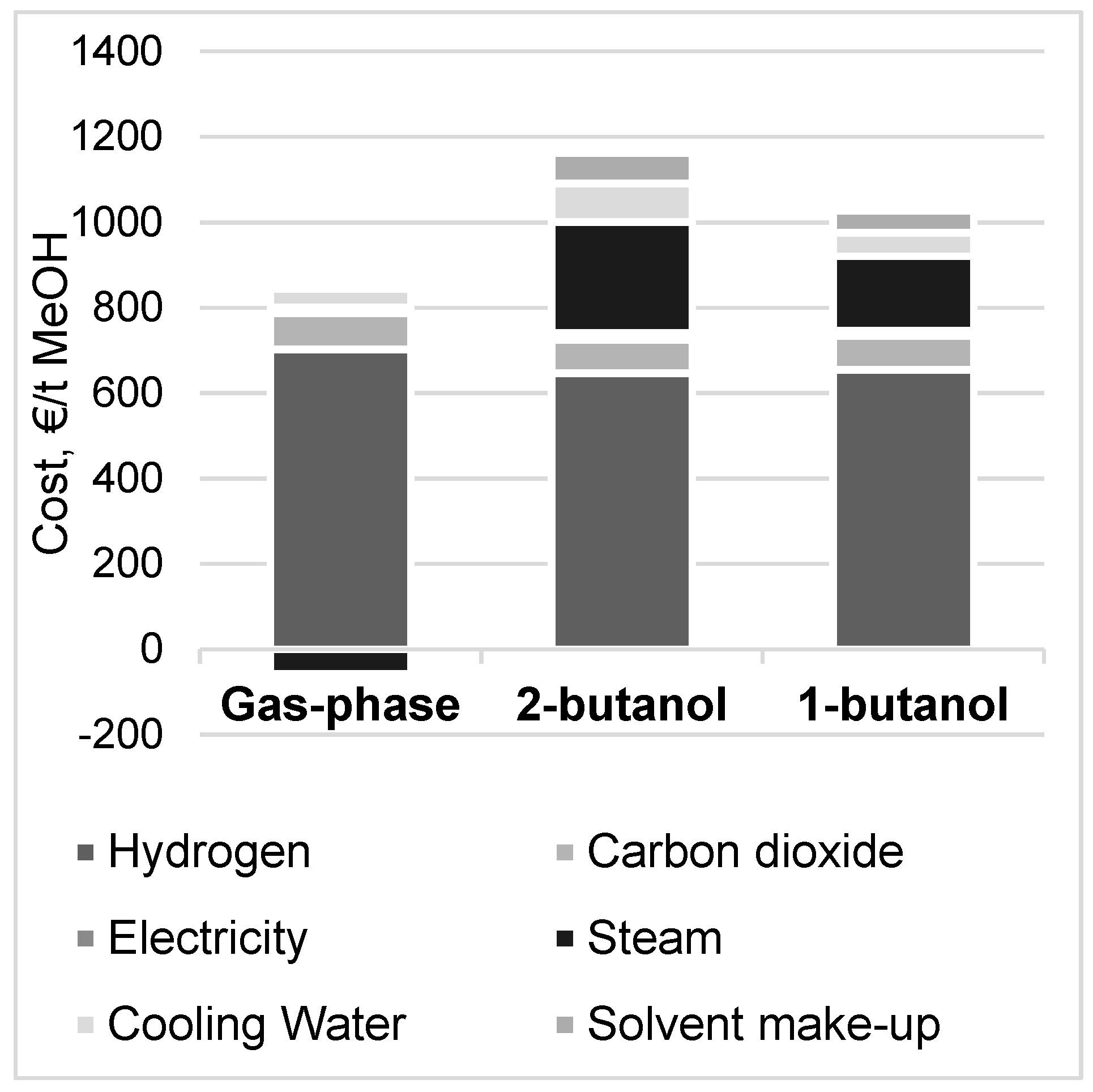
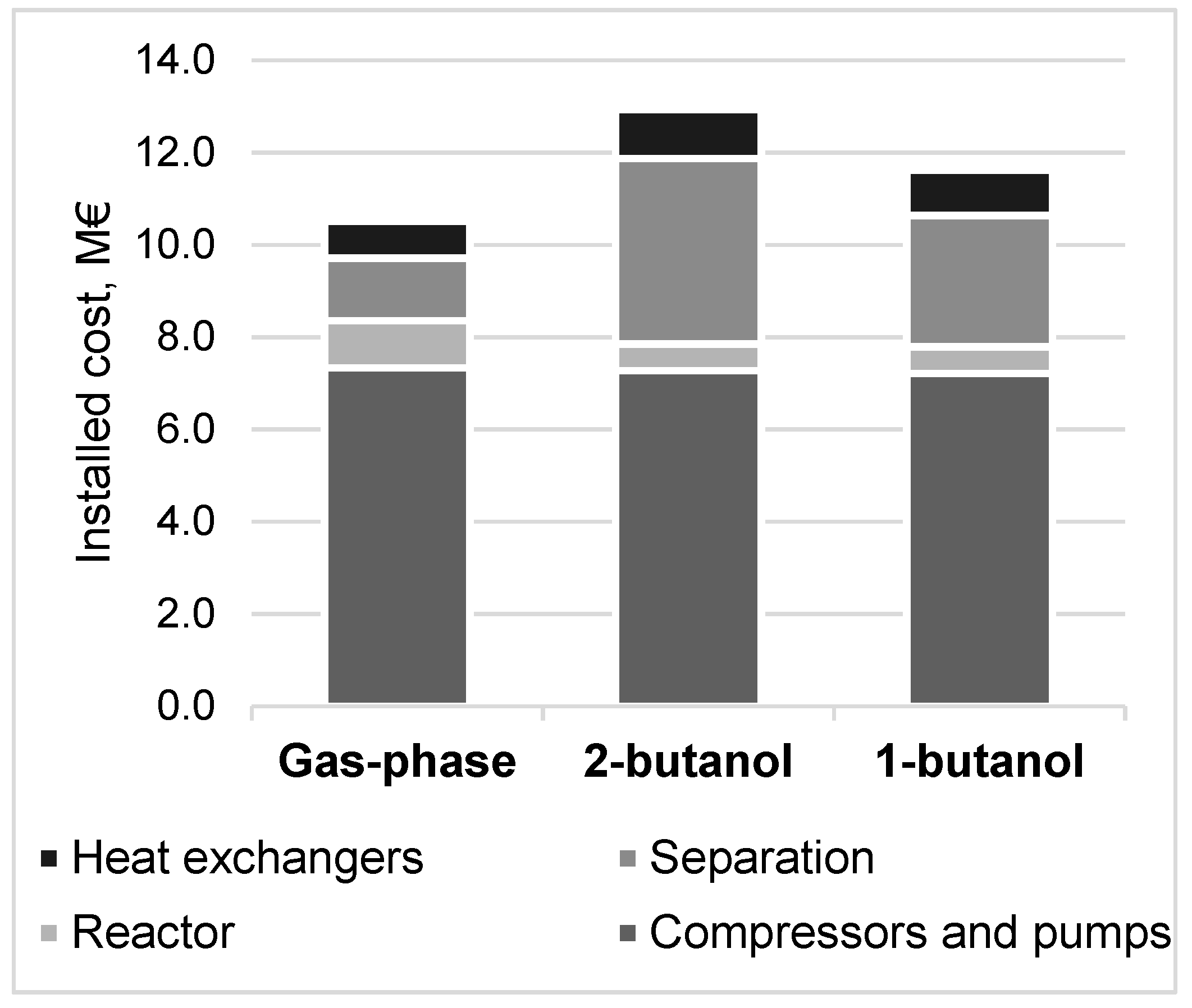
3.4. Methanol Production Cost and Net Present Value
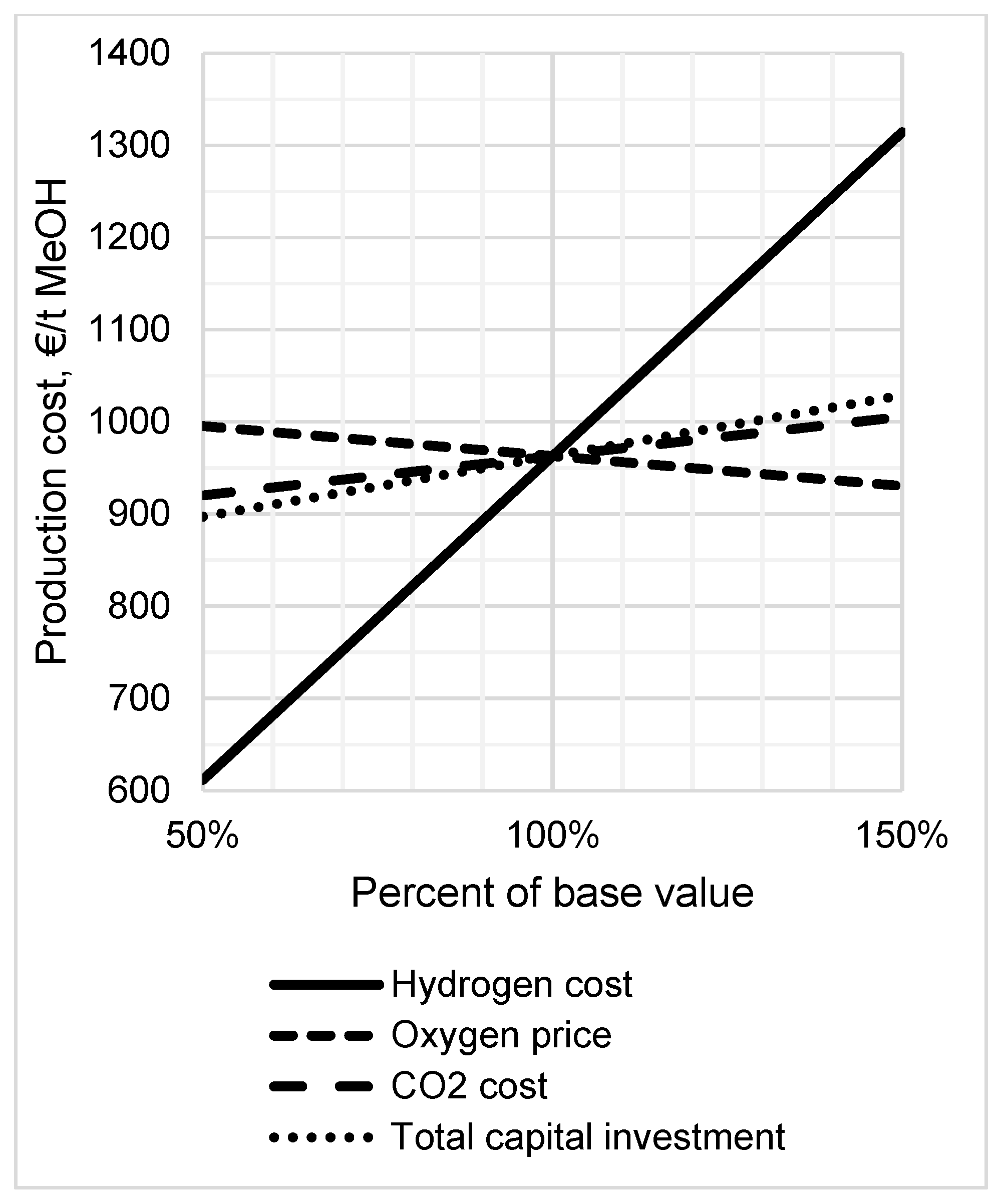
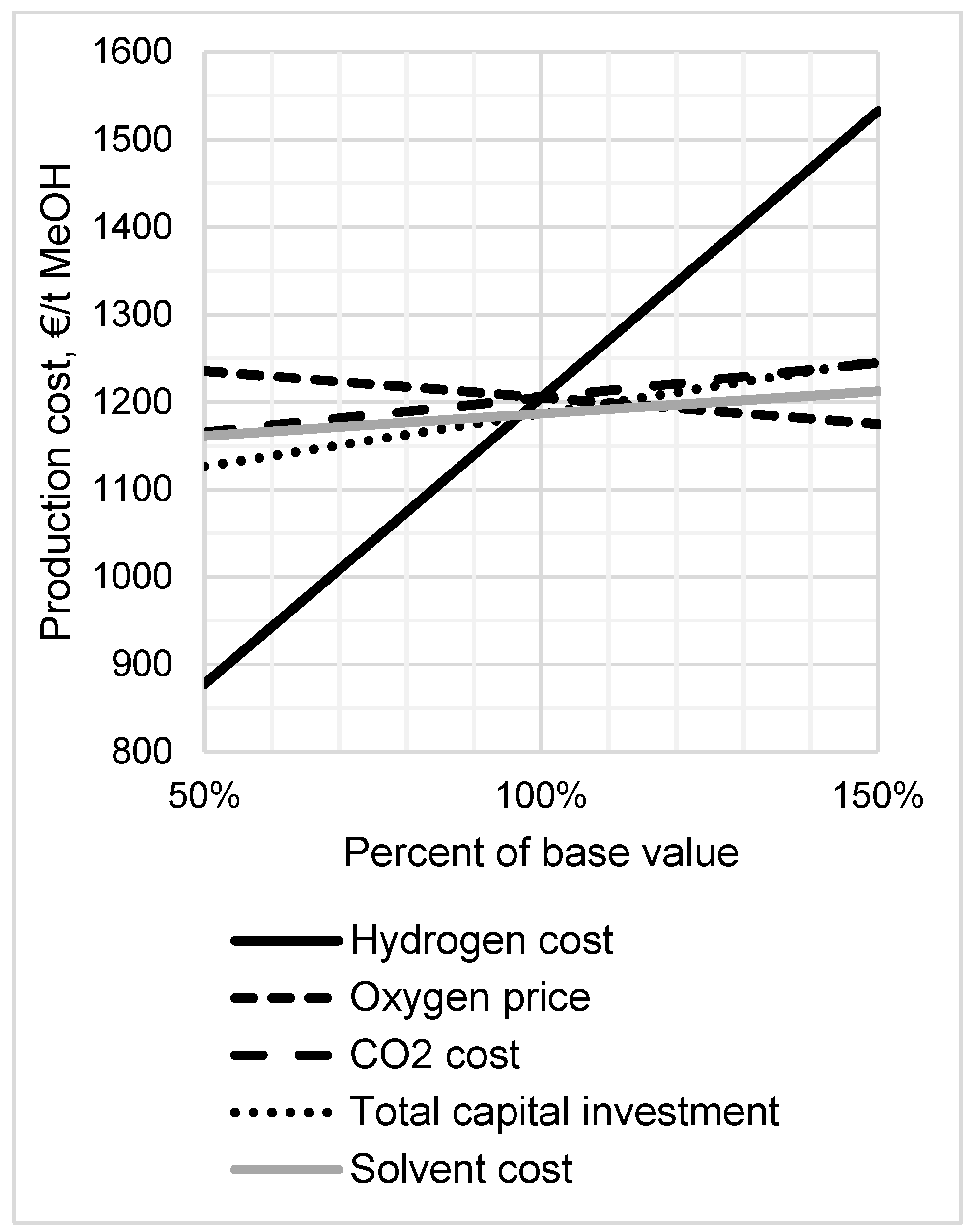
3.5. Sensitivity Analysis
3.6. Summary
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Schlögl, R. The Solar Refinery. In Chemical Energy Storage; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2013; pp. 1–34. [Google Scholar]
- Olah, G.; Goeppert, A.; Prakash, G. Beyond Oil and Gas: The Methanol Economy, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Goeppert, A.; Czaun, M.; Jones, J.P.; Prakash, G.K.S.; Olah, G.A. Recycling of carbon dioxide to methanol and derived products—Closing the loop. Chem. Soc. Rev. 2014, 43, 7995–8048. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, H.; Plass, L.; Bertau, M. From Raw Materials to Methanol, Chemicals and Fuels. In Methanol: The Basic Chemical and Energy Feedstock of the Future; Springer: Heidelberg/Berlin, Germany, 2014; pp. 1–7. [Google Scholar]
- Reichelt, L.; Schmidt, F. Methanol-to-Gasoline Process. In Methanol: The Basic Chemical and Energy Feedstock of the Future; Springer: Heidelberg/Berlin, Germany, 2015; pp. 440–453. [Google Scholar]
- Schmidt, F.; Pätzold, C. 6.4.2 Methanol-to-Olefins Processes. In Methanol: The Basic Chemical and Energy Feedstock of the Future; Springer: Heidelberg/Berlin, Germany, 2015; pp. 454–472. [Google Scholar]
- Ott, J.; Gronemann, V.; Pontzen, F.; Fiedler, E.; Grossmann, G.; Kersebohm, D.B.; Weiss, G.; Witte, C. Methanol. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 1–27. [Google Scholar]
- Pontzen, F.; Liebner, W.; Gronemann, V.; Rothaemel, M. BAhlers, CO2-based methanol and DME—Efficient technologies for industrial scale production. Catal. Today 2011, 171, 242–250. [Google Scholar] [CrossRef]
- Kunkes, E.; Behrens, M. Methanol Chemistry. In Chemical Energy Storage; Walter de Gruyter GmbH: Berlin, Germany, 2013; pp. 413–435. [Google Scholar]
- Sahibzada, M.; Metcalfe, I.S.; Chadwick, D. Methanol Synthesis from CO/CO2/H2 over Cu/ZnO/Al2O3 at Differential and Finite Conversions. J. Catal. 1998, 174, 111–118. [Google Scholar] [CrossRef]
- Martin, O.; Pérez-Ramírez, J. New and revisited insights into the promotion of methanol synthesis catalysts by CO2. Catal. Sci. Technol. 2013, 3, 3343–3352. [Google Scholar] [CrossRef]
- Mignard, D.; Sahibzada, M.; Duthie, J.M.; Whittington, H.W. Methanol synthesis from flue-gas CO2 and renewable electricity: A feasibility study. Int. J. Hydrog. Energy 2003, 28, 455–464. [Google Scholar] [CrossRef]
- Van-Dal, È.; Bouallou, C. Design and simulation of a methanol production plant from CO2 hydrogenation. J. Clean. Prod. 2013, 57, 38–45. [Google Scholar] [CrossRef]
- Anicic, B.; Trop, P.; Goricanec, D. Comparison between two methods of methanol production from carbon dioxide. Energy 2014, 77, 279–289. [Google Scholar] [CrossRef]
- Pérez-Fortes, M.; Schöneberger, J.C.; Boulamanti, A.; Tzimas, E. Methanol synthesis using captured CO2 as raw material: Techno-economic and environmental assessment. Appl. Energy 2016, 161, 718–732. [Google Scholar] [CrossRef]
- Rivera-Tinoco, R.; Farran, M.; Bouallou, C.; Auprêrte, F.; Valentin, S.; Millet, P.; Ngameni, J.R. Investigation of power-to-methanol processes coupling electrolytic hydrogen production and catalytic CO2 reduction. Int. J. Hydrogen Energy 2016, 41, 4546–4559. [Google Scholar] [CrossRef]
- Atsonios, K.; Panopoulos, K.D.; Kakaras, E. Investigation of technical and economic aspects for methanol production through CO2 hydrogenation. Int. J. Hydrogen. Energy 2016, 41, 2202–2214. [Google Scholar] [CrossRef]
- Rivarolo, M.; Bellotti, D.; Magistri, L.; Massardo, A.F. Feasibility study of methanol production from different renewable sources and thermo-economic analysis. Int. J. Hydrogen Energy 2016, 41, 2105–2116. [Google Scholar] [CrossRef]
- Varone, A.; Ferrari, M. Power to liquid and power to gas: An option for the German Energiewende. Renew. Sustain. Energy Rev. 2015, 45, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Matzen, M.; Alhajji, M.; Demirel, Y. Chemical storage of wind energy by renewable methanol production: Feasibility analysis using a multi-criteria decision matrix. Energy 2015, 93, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Matzen, M.; Demirel, Y. Methanol and dimethyl ether from renewable hydrogen and carbon dioxide: Alternative fuels production and life-cycle assessment. J. Clean. Prod. 2016, 139, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Tremel, A.; Wasserscheid, P.; Baldauf, M.; Hammer, T. Techno-economic analysis for the synthesis of liquid and gaseous fuels based on hydrogen production via electrolysis. Int. J. Hydrogen Energy 2015, 40, 11457–11464. [Google Scholar] [CrossRef]
- Lee, S.; Sardesai, A. Liquid phase methanol and dimethyl ether synthesis from syngas. Top. Catal. 2005, 32, 197–207. [Google Scholar] [CrossRef]
- Fan, L.; Sakaiya, Y.; Fujimoto, K. Low-temperature methanol synthesis from carbon dioxide and hydrogen via formic ester. Appl. Catal. A 1999, 180, L11–L13. [Google Scholar] [CrossRef]
- Xu, B.; Yang, R.; Meng, F.; Reubroycharoen, P.; Vitidsant, T.; Zhang, Y.; Yoneyama, Y.; Tsubaki, N. A New Method of Low Temperature Methanol Synthesis. Catal. Surv. Asia 2009, 13, 147–163. [Google Scholar] [CrossRef]
- Tsubaki, N.; Zeng, J.; Yoneyama, Y.; Fujimoto, K. Continuous synthesis process of methanol at low temperature from syngas using alcohol promoters. Catal. Commun. 2001, 2, 213–217. [Google Scholar] [CrossRef]
- Reubroycharoen, P.; Yamagami, T.; Vitidsant, T.; Yoneyama, Y.; Ito, M.; Tsubaki, N. Continuous Low-Temperature Methanol Synthesis from Syngas Using Alcohol Promoters. Energy Fuels 2003, 17, 817–823. [Google Scholar] [CrossRef]
- Finnish Wind Power Association. Wind Power Projects in Finland, March 2017. Available online: http://www.tuulivoimayhdistys.fi/filebank/969–968-STY_hankelista_2017_web_final.xls (accessed on 28 February 2018).
- Liu, G.; Larson, E.D.; Williams, R.H.; Kreutz, T.G.; Guo, X. Online Supporting Material, Making Fischer-Tropsch Fuels and Electricity from Coal and Biomass: Performance and Cost Analysis. Energy Fuels 2011, 25, 415–437. [Google Scholar] [CrossRef]
- Bussche, K.M.V.; Froment, G.F. A Steady-State Kinetic Model for Methanol Synthesis and the Water Gas Shift Reaction on a Commercial Cu/ZnO/Al2O3 Catalyst. J. Catal. 1996, 161, 1–10. [Google Scholar] [CrossRef]
- Mignard, D.; Pritchard, C. On the use of electrolytic hydrogen from variable renewable energies for the enhanced conversion of biomass to fuels. Chem. Eng. Res. Des. 2008, 86, 473–487. [Google Scholar] [CrossRef]
- Brown, W.R.; Drown, D.P.; Frenduto, F.S. Commercial-Scale Demonstration of the Liquid Phase Methanol (LPMeOH) Process—Public Design Report; Air Products, Chemicals, Inc., Eastman Chemical Company: Kingsport, TN, USA, 2000. [Google Scholar]
- Tsubaki, N.; Ito, M.; Fujimoto, K. A New Method of Low-Temperature Methanol Synthesis. J. Catal. 2001, 197, 224–227. [Google Scholar] [CrossRef]
- Tomorrow Electricity Map. Available online: https://www.electricitymap.org/?page=country&solar=false&remote=true&wind=false&countryCode=FI (accessed on 6 July 2018).
- Towler, G.; Sinnott, R. Capital Cost Estimating. In Chemical Engineering Design; Elsevier: Oxford, UK, 2013; pp. 307–354. [Google Scholar]
- Levene, J.; Kroposki, B.; Sverdrup, G. Wind Energy and Production of Hydrogen and Electricity—Opportunities for Renewable Hydrogen; Preprint; National Renewable Energy Laboratory (NREL): Golden, CO, USA, 2006.
- Vakkilainen, E.; Kivistö, A. Comparison of Electricity Generation Costs; Lappeenranta University of Technology: Lappeenranta, Finland, 2017. [Google Scholar]
- Smolinka, T.; Günther, M.; Garche, J. Stand und Entwicklungspotenzial der Wasserelektrolyse zur Herstellung von Wasserst off aus Regenerativen Energien; NOW-Studie: Freiburg im Breisgau, Germany, 2011. [Google Scholar]
- Eurostat. Electricity Prices for Non-Household Consumers-bi-Annual Data 23 November 2017. Available online: http://appsso.eurostat.ec.europa.eu/nui/show.do?dataset=nrg_pc_205&lang=en (accessed on 28 February 2018).
- Finkenrath, M. Cost and Performance of Carbon Dioxide Capture from Power Generation; IEA: Paris, France, 2011. [Google Scholar]
- Trippe, F.; Fröhling, M.; Schultmann, F.; Stahl, R.; Henrich, E.; Dalai, A. Comprehensive techno-economic assessment of dimethyl ether (DME) synthesis. Fuel Process. Technol. 2013, 106, 577–586. [Google Scholar] [CrossRef]
- Statistics Finland, Energy Prices Grew in the Third Quarter, 7 December 2017. Available online: https://tilastokeskus.fi/til/ehi/2017/03/ehi_2017_03_2017–12-07_tie_001_en.html (accessed on 28 February 2018).
- Hall, S. Energy Conservation. In Rules of Thumb for Chemical Engineers; Elsevier: Oxford, UK, 2012; pp. 375–385. [Google Scholar]
- The Engineering ToolBox, Fuels—Higher and Lower Calorific Values, 2003. Available online: https://www.engineeringtoolbox.com/fuels-higher-calorific-values-d_169.html (accessed on 10 April 2019).
- Towler, G.; Sinnott, R. Estimating Revenues and Production Costs. In Chemical Engineering Design; Elsevier: Oxford, UK, 2013; pp. 355–387. [Google Scholar]
- Methanex Corporation, Methanex Monthly Average Regional Posted Contract Price History, February 2018. Available online: https://www.methanex.com/sites/default/files/methanol-price/MxAvgPrice_Feb%2028%2C%202018.pdf (accessed on 12 March 2018).
- VLE—Calc, Azeotrope Database for Organic Solvent Mixtures. Available online: http://vle-calc.com/azeotrope.html (accessed on 12 March 2018).
- Kaneko, T.; Derbyshire, F.; Makino, E.; Gray, D.; Tamura, M.; Li, K. Coal Liquefaction. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; p. 32. [Google Scholar]
- IREN. Renewable Power Generation Costs in 2017; International Renewable Energy Agency: Abu Dhabi, UAE, 2018. [Google Scholar]
- Schmidt, O.; Gambhir, A.; Staffell, I.; Hawkes, A.; Nelson, J.; Few, S. Future cost and performance of water electrolysis:An expert elicitation study. Int. J. Hydrogen Energy 2017, 42, 30470–30492. [Google Scholar] [CrossRef]
- Chen, Y.; Choi, S.; Thompson, L.T. Low-Temperature CO2 Hydrogenation to Liquid Products via a Heterogeneous Cascade Catalytic System. ACS Catal. 2015, 5, 1717–1725. [Google Scholar] [CrossRef]
- Bayat, M.; Dehghani, Z.; Hamidi, M.; Rahimpour, M. Methanol synthesis via sorption-enhanced reaction process: Modeling, multi-objective optimization. J. Taiwan Inst. Chem. Eng. 2014, 45, 481–494. [Google Scholar] [CrossRef]
- Wu, W.; Xie, K.; Sun, D.; Li, X.; Fang, F. CuO/ZnO/Al2O3 Catalyst Prepared by Mechanical-Force-Driven Solid-State Ion Exchange and Its Excellent Catalytic Activity under Internal Cooling Condition. Ind. Eng. Chem. Res. 2017, 56, 8216–8223. [Google Scholar] [CrossRef]
- Kourkoumpas, D.S.; Papadimou, E.; Atsonios, K.; Karellas, S.; Grammelis, P.; Kakaras, E. Implementation of the Power to Methanol concept by using CO2 from lignite power plants: Techno-economic investigation. Int. J. Hydrogen Energy 2016, 41, 16674–16687. [Google Scholar] [CrossRef]
- Bellotti, D.; Rivarolo, M.; Magistri, L.; Massardo, A.F. Feasibility study of methanol production plant from hydrogen and captured carbon dioxide. J. CO2 Util. 2017, 21, 132–138. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Basis |
|---|---|
| ISBL capital cost | Installed equipment cost from the Aspen Plus Exchange rate of 0.89 €/USD Material factor 1.3 (304 stainless steel) Location factor 1.043 (Western Europe) |
| OSBL capital cost | 25% of ISBL |
| Engineering cost | 20% of ISBL and OSBL |
| Contingency | 30% of ISBL and OSBL |
| Item | Cost and Details |
|---|---|
| Hydrogen | 3000 €/t, based on alkaline electrolysis powered by 30 MW of wind electricity (cost includes electricity production and hydrogen storage) [37,39] |
| Grid electricity | 60 €/MWh [40] |
| CO2 | 50 €/t [41] |
| Steam | 35 €/t for MP (20 bar) and LP (6 bar) steam, based on natural gas cost of 30 €/MWh [43] |
| Cooling water | 0.26 €/m3 [42] |
| Waste water | 0.32 €/m3 [42] |
| Catalyst | 95.24 €/kg [16], assumed lifetime 4 years |
| Solvent make-up | 500 €/t for all alcohols |
| Gas-Phase | 2-Butanol | 1-Butanol | |
|---|---|---|---|
| Distillation feed flow rate, kg/h | 3660 | 26,641.1 | 14,650 |
| Column #1 | |||
| Number of ideal stages | 30 | 15 | 10 |
| Reflux ratio (molar) | 1.1 | 1.0 | 1.0 |
| Reboiler duty, kW | 940 | 7282 | 5445 |
| Column #2 | |||
| Number of stages | - | 50 | 30 |
| Reflux ratio | - | 6.5 | 4.0 |
| Reboiler duty, kW | - | 4210 | 2265 |
| Methanol purity * (wt%) | 99.3% | 99.2% | 99.2% |
| Reactor Inlet | Reactor Outlet | ||||
|---|---|---|---|---|---|
| Gas/Vapor Phase | Liquid Phase | Gas/Vapor Phase | |||
| Component | Flow, kmol/h | Component | Flow, kmol/h | Component | Flow, kmol/h |
| CO2 | 118.6 | CO2 | 0.0 | CO2 | 36.2 |
| CO | 0.8 | CO | 0.0 | CO | 0.8 |
| Methanol | 1.4 | Methanol | 0.4 | Methanol | 84.2 |
| Hydrogen | 934.9 | Hydrogen | 1.4 | Hydrogen | 689.0 |
| Water | 0.7 | Water | 23.1 | Water | 106.2 |
| 1-butanol | 0.2 | 1-butanol | 132.7 | 1-butanol | 132.8 |
| Total | 1056.5 | Total | 157.6 | Total | 1049.3 |
| Flow, kg/h | Gas-Phase | 2-Butanol | 1-Butanol |
|---|---|---|---|
| Hydrogen in | 533 | 533 | 533 |
| CO2 in | 3882 | 3882 | 3882 |
| Methanol out | 2275 | 2311 | 2474 |
| Methanol losses | 44 | 248 | 167 |
| CO2 losses | 560 | 369 | 255 |
| Hydrogen losses | 89 | 51 | 35 |
| CO2 conversion per pass | 20% | 79% | 81% |
| Methanol yield | 81% | 82% | 88% |
| Solvent loss, kg/h | - | 352 | 249 |
| Fractional solvent loss | - | 2% | 3% |
| Energy, kWh/t MeOH | Gas-Phase | 2-Butanol | 1-Butanol |
|---|---|---|---|
| Hot utility | 0 | 6668 | 3912 |
| Cold utility | 2960 | 15,313 | 9604 |
| Heat integrated within process | 5104 | 5376 | 4047 |
| Waste heat generated | 2697 | 4048 | 2366 |
| Electricity | 624 | 683 | 625 |
| Gas-Phase | 2-Butanol | 1-Butanol | |
|---|---|---|---|
| CO2 balance, kg/h | |||
| Inlet streams | −3882 | −3882 | −3882 |
| Outlet streams | 560 | 369 | 255 |
| Hot utility (natural gas) | 0.0 | 2777 | 1837 |
| Waste heat combustion | 170 | 836 | 448 |
| Electricity (grid) | 106 | 116 | 106 |
| Net emissions | −3046 | 216 | −1239 |
| Water balance | |||
| Cooling water input, t/h | 379 | 516 | 516 |
| Solvent/water waste, kg/h | 1371 | 1574 | 1644 |
| Alcohol in waste, wt% * | 1% | 9% | 9% |
| Benefits | Challenges | Comments and Outlook |
|---|---|---|
| Lower reaction temperature leads to higher equilibrium conversion and lower reactant recycle and losses | - | Reaction temperature could be further lowered with catalyst development (e.g., Chen et al. [52]) |
| - | Complicated and energy-intensive separation leads to higher overall production cost and less favorable energy and CO2 balances | The amount of solvent recycle should be minimized by utilizing high-boiling alcohols and improved reactor design (e.g., reactive distillation?); energy consumption could be minimized by improved heat integration |
| - | Formation of azeotropic alcohol-water mixtures further complicates solvent separation and recovery | Solvent recovery improved by phase separation of water and higher alcohols |
| Liquid-phase reaction potentially allows improved reactor temperature control and catalyst stability | - | Previously demonstrated in liquid-phase methanol synthesis using inert solvents [24] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieminen, H.; Laari, A.; Koiranen, T. CO2 Hydrogenation to Methanol by a Liquid-Phase Process with Alcoholic Solvents: A Techno-Economic Analysis. Processes 2019, 7, 405. https://doi.org/10.3390/pr7070405
Nieminen H, Laari A, Koiranen T. CO2 Hydrogenation to Methanol by a Liquid-Phase Process with Alcoholic Solvents: A Techno-Economic Analysis. Processes. 2019; 7(7):405. https://doi.org/10.3390/pr7070405
Chicago/Turabian StyleNieminen, Harri, Arto Laari, and Tuomas Koiranen. 2019. "CO2 Hydrogenation to Methanol by a Liquid-Phase Process with Alcoholic Solvents: A Techno-Economic Analysis" Processes 7, no. 7: 405. https://doi.org/10.3390/pr7070405