
Diffusion Coefficients in Systems Related to Reservoir Fluids: Available Data and Evaluation of Correlations


Abstract
:1. Introduction
1.1. Importance for Petroleum Production
1.2. Diffusion Theories and Correlations
1.3. Motivation, Scope, and Structure
2. Diffusion Coefficients for Binary Mixtures Related to Reservoir Fluids
2.1. Overview
2.2. Experimental Methods Involved
- Diaphragm cell (DC)
- Constant volume diffusion (CVD)
- Constant pressure (CP)
- Taylor dispersion (TD)
- Capillary method (CM)
- Interferometery (IF)
- Other methods
- Summary
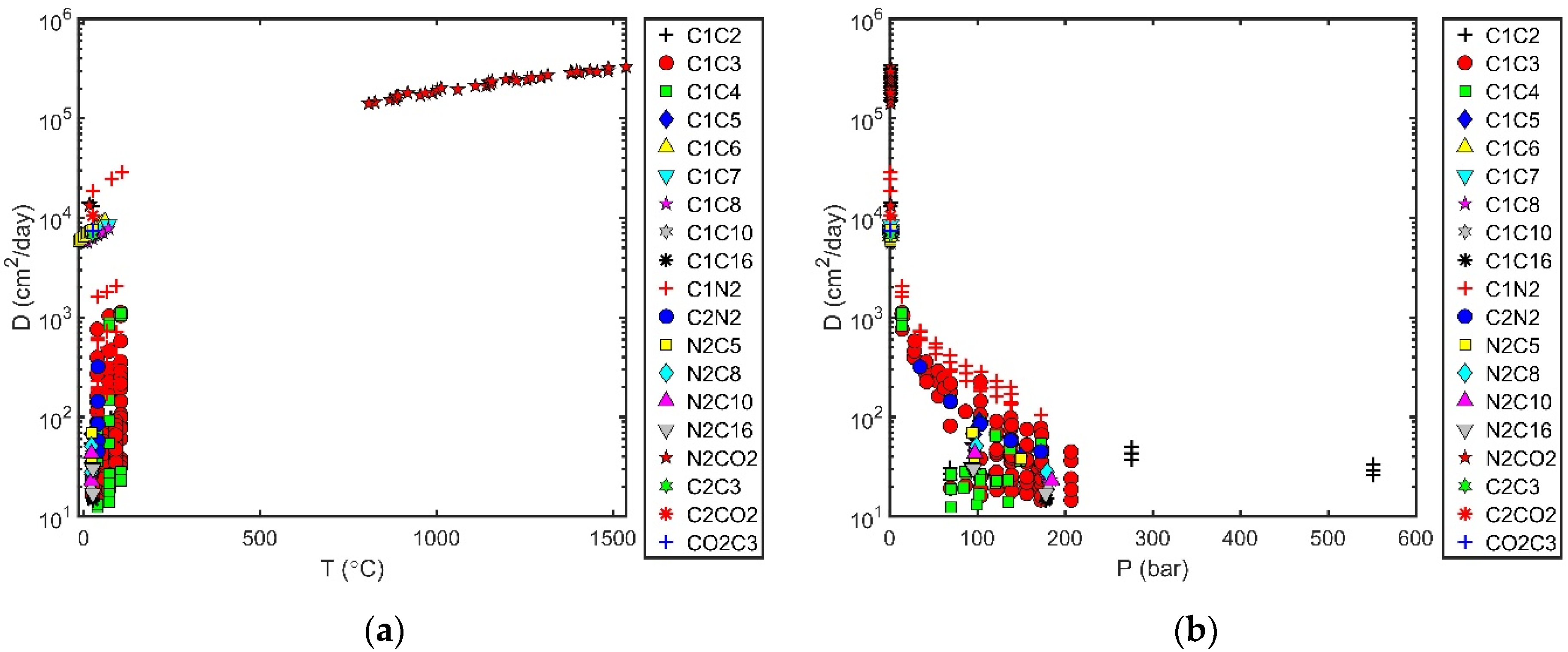
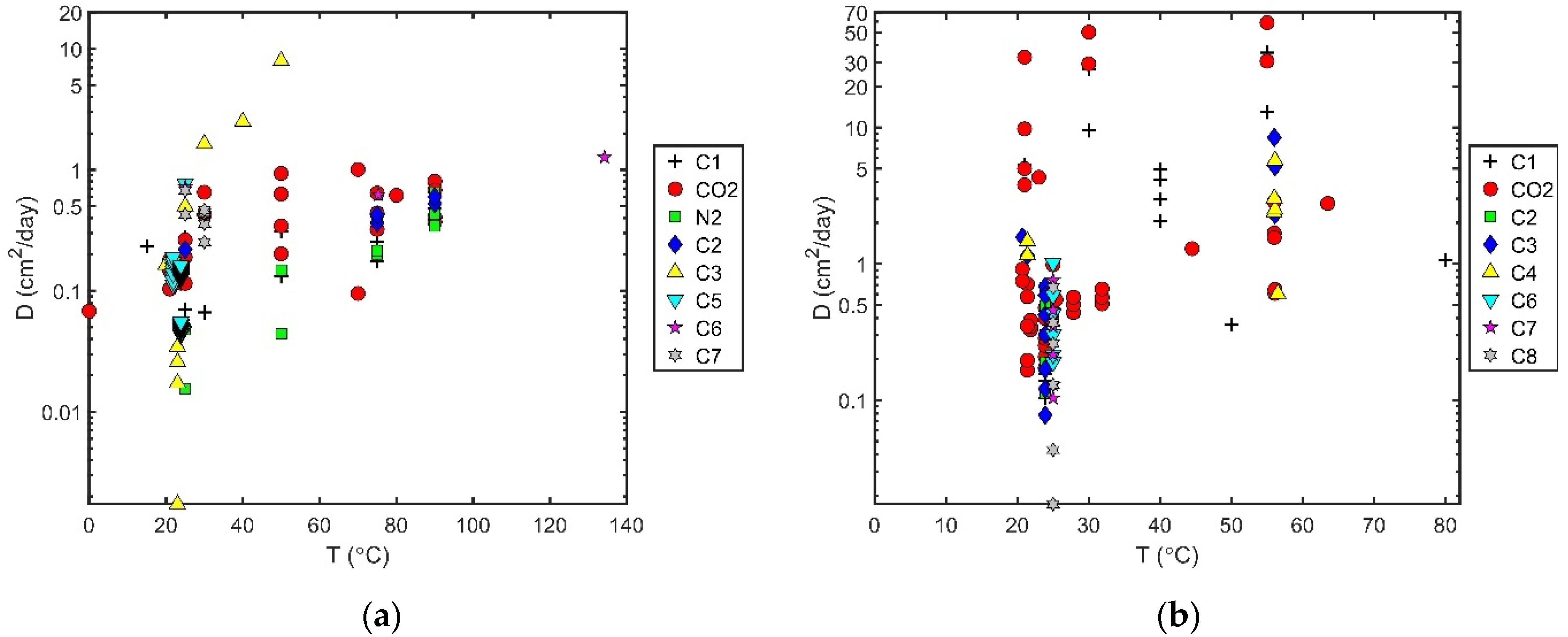
2.3. Trends in Diffusion Coefficients
- Trends with temperature or pressure for different classes
3. Diffusion Coefficients in Oil Systems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Visc. (cP) | Method | Np | P (bar) | T (K) | Source |
|---|---|---|---|---|---|---|
| C1 + Bitumen (Athabasca) | - | CVD | 3 | 40–80 | 298–363 | [146] |
| C1 + Bitumen (Athabasca) | 3165 | CVD | 2 | 40–80 | 348 | [146] |
| C1 + Bitumen (Athabasca) | 70,475 | CVD | 1 | 80 | 323 | [146] |
| C1 + bitumen (Athabasca) | 100,000 | CP | 1 | 38 | 323 | [82] |
| C1 + bitumen (Canadian) | - | CVD | 2 | 34.6–35.9 | 288–298 | [81] |
| C1 + bitumen (MacKay) | 82,160 | CVD | 1 | 35.5 | 303 | [170] |
| C1 + heavy oil | - | CVD | 2 | 63.5–63.9 | 323–353 | [83] |
| C1 + heavy oil (Lloydminster) | 20,267 | CVD | 1 | 41.8 | 297 | [171] |
| C1 + heavy oil (Lloydminster) | 23,000 | PDM | 5 | 59.6–141.2 | 297 | [172] |
| C1 + heavy oil (Venezuela) | 5000 | CVD | 1 | 34.71 | 294 | [173] |
| C1 + heavy oil sample1 | 21,285 | CVD | 2 | 14 | 303–328 | [165] |
| C1 + heavy oil sample2 | 8154 | CVD | 2 | 14 | 303–328 | [165] |
| C1 + heavy oil (Henan) | 5824 | CP | 4 | 40–100 | 313 | [174] |
| C2 + Bitumen (Athabasca) | - | CVD | 3 | 40–80 | 298–363 | [146] |
| C2 + Bitumen (Athabasca) | 3165 | CVD | 2 | 40–80 | 348 | [146] |
| C2 + heavy oil (Lloydminster) | 23,000 | PDM | 5 | 15.2–35 | 297 | [172] |
| C3 + heavy oil (Lloydminster) | - | CP | 1 | 55.4 | 329 | [175] |
| C3 + heavy oil (Lloydminster) | - | CP | 1 | 55.4 | 329 | [176] |
| C3 + heavy oil (Lloydminster) | 13,924 | CP | 1 | 39.5 | 294 | [177] |
| C3 + heavy oil (Lloydminster) | - | CP | 1 | 55 | 329 | [176] |
| C3 + heavy oil (Lloydminster) | 12,854 | CVD | 1 | 38 | 295 | [178] |
| C3 + bitumen (Athabasca) | 473,000 | Microchip | 2 | 6.6–7.5 | 293–298 | [164] |
| C3 + bitumen (Athabasca) | - | Microchip | 3 | 8.4–13.3 | 303–323 | [164] |
| C3 + bitumen E2 | 79,433 | X-ray CT | 1 | 6.3 | 296 | [161] |
| C3 + bitumen L4 | 501,187 | X-ray CT | 1 | 6.3 | 296 | [161] |
| C3 + bitumen M3 | 125,893 | X-ray CT | 1 | 6.3 | 296 | [161] |
| C3 + bitumen N1 | 3162 | X-ray CT | 1 | 6.3 | 296 | [161] |
| C3 + heavy oil (Lloydminster) | 20,267 | CVD | 1 | 7.6 | 297 | [171] |
| C3 + heavy oil (Lloydminster) | 23,000 | PDM | 6 | 4–9 | 297 | [172] |
| C4 + heavy oil (Lloydminster) | - | CP | 1 | 55.4 | 329 | [175] |
| C4 + heavy oil (Lloydminster) | - | CP | 1 | 55.4 | 329 | [176] |
| C4 + heavy oil (Lloydminster) | - | CP | 1 | 55.2 | 329 | [175] |
| C4 + heavy oil (Lloydminster) | 12,854 | CP | 1 | 37.4 | 295 | [176] |
| C4 + heavy oil (Lloydminster) | - | CP | 1 | 55.2 | 329 | [176] |
| C4 + heavy oil (Lloydminster) | 12,854 | CVD | 1 | 11.3 | 295 | [178] |
| C4 + heavy oil (Lloydminster) | - | CP | 1 | 5.3 | 330 | [175] |
| C5 + bitumen (Athabasca) | 18,000 | X-ray CT | 1 | 1 | 295 | [158] |
| C5 + bitumen (Athabasca) | 18,000 | X-ray CT | 17 | 1 | 295 | [159] |
| C5 + Bitumen (Cold Lake) | 130,000 | NMR | 1 | 1 | 298 | [179] |
| C5 + bitumen 1 (Athabasca) | 18,000 | X-ray CT | 17 | 1 | 297 | [160] |
| C5 + bitumen 2 (Athabasca) | 2,600,000 | X-ray CT | 12 | 1 | 297 | [160] |
| C6 + bitumen (Athabasca) | - | X-ray CT | 2 | 34.4–35.1 | 348–407 | [162] |
| C6 + Bitumen (Cold Lake) | 130,000 | NMR | 1 | 1 | 298 | [179] |
| C6 + heavy oil | - | X-ray CT | 6 | 1 | 298 | [180] |
| C7 + bitumen (Atlee Buffalo) | 6000 | NMR | 1 | 1 | 298 | [179] |
| C7 + bitumen (Cold Lake) | 130,000 | NMR | 6 | 1 | 303 | [179] |
| C7 + bitumen (Peace River) | 670,000 | NMR | 1 | 1 | 298 | [179] |
| C7 + heavy oil | - | X-ray CT | 6 | 1 | 298 | [180] |
| C8 + heavy oil | - | X-ray CT | 6 | 1 | 298 | [180] |
| CO2 + heavy oil (Lloydminster) | - | CP | 1 | 55.4 | 329 | [175] |
| CO2 + heavy oil (Lloydminster) | - | CP | 1 | 55.4 | 329 | [176] |
| CO2 + heavy oil (Lloydminster) | 13,924 | CP | 1 | 39.5 | 294 | [177] |
| CO2 + heavy oil (Lloydminster) | - | CP | 1 | 55 | 329 | [176] |
| CO2 + heavy oil (Lloydminster) | - | CP | 1 | 55.2 | 329 | [175] |
| CO2 + heavy oil (Lloydminster) | 12,854 | CP | 1 | 37.4 | 295 | [176] |
| CO2 + heavy oil (Lloydminster) | - | CP | 1 | 55.2 | 329 | [176] |
| CO2 + heavy oil (Lloydminster) | 12,854 | CVD | 1 | 38 | 295 | [178] |
| CO2 + heavy oil (Lloydminster) | 12,854 | CVD | 1 | 11.3 | 295 | [178] |
| CO2 + bitumen | - | CVD | 1 | 48.3 | 353 | [181] |
| CO2 + bitumen | - | CVD | 6 | 22.4–50.1 | 303–343 | [182] |
| CO2 + bitumen (Athabasca) | 250 | CVD | 1 | 40 | 363 | [183] |
| CO2 + bitumen (Athabasca) | - | CVD | 1 | 40 | 298 | [183] |
| CO2 + Bitumen (Athabasca) | 3165 | CVD | 2 | 40–80 | 348 | [146] |
| CO2 + Bitumen (Athabasca) | 70,475 | CVD | 2 | 40–80 | 323 | [146] |
| CO2 + Bitumen (Athabasca) | - | CVD | 3 | 40–80 | 298–363 | [146] |
| CO2 + bitumen (Athabasca) | 10,000 | CP | 1 | 32.4 | 348 | [82] |
| CO2 + bitumen (Athabasca) | 2,000,000 | other | 3 | 31–56 | 294 | [163] |
| CO2 + bitumen (Canadian) | - | CVD | 2 | 34.5–34.6 | 273–298 | [81] |
| CO2 + bitumen (MacKay) | 127,868 | CVD | 1 | 35.3 | 297 | [170] |
| CO2 + heavy oil (Aberfeldy) | 1058 | CP | 1 | 10 | 296 | [184] |
| CO2 + heavy oil (Lloydminster) | 12,854 | CVD | 1 | 37.4 | 295 | [178] |
| CO2 + heavy oil (Lloydminster) | - | CP | 1 | 54 | 318 | [175] |
| CO2 + heavy oil (Lloydminster) | 13,924 | CP | 1 | 39.5 | 294 | [177] |
| CO2 + heavy oil (Lloydminster) | - | CP | 3 | 54 | 299–337 | [185] |
| CO2 + heavy oil (Lloydminster) | 12,854 | CP | 1 | 37.4 | 295 | [176] |
| CO2 + heavy oil (Lloydminster) | 20,267 | CVD | 1 | 50.3 | 297 | [171] |
| CO2 + heavy oil (Lloydminster) | 23,000 | PDM | 5 | 20–60 | 297 | [151] |
| CO2 + heavy oil (Manatokan) | 179 | CVD | 1 | 50 | 294 | [186] |
| CO2 + heavy oil (Ontario) | - | PDM | 1 | 29 | 298 | [187] |
| CO2 + heavy oil (Venezuela) | 5000 | CVD | 1 | 35.1 | 294 | [173] |
| CO2 + heavy oil A (Saskatchewan) | 5000 | CVD | 1 | 17.3 | 298 | [188] |
| CO2 + heavy oil A (Saskatchewan) | - | CVD | 11 | 17.2–44.9 | 295–305 | [188] |
| CO2 + heavy oil 1 | 21,285 | CVD | 2 | 28.5 | 303–328 | [165] |
| CO2 + heavy oil 2 | 8154 | CVD | 2 | 26 | 303–328 | [165] |
| N2 + Bitumen (Athabasca) | - | CVD | 4 | 40–80 | 298–363 | [146] |
| N2 + Bitumen (Athabasca) | 3165 | CVD | 2 | 40–80 | 348 | [146] |
| N2 + Bitumen (Athabasca) | 70,475 | CVD | 2 | 40–80 | 323 | [146] |
| System | Visc. (cP) | Method | Np | P (bar) | T (K) | Source |
|---|---|---|---|---|---|---|
| C1 + crude oil | - | CVD | 3 | 200 | 333 | [166] |
| C1 + crude oil (Bakken) | - | CVD | 1 | 137.9 | 294 | [189] |
| C1 + crude oil (Sante Fe Springs) | - | CP | 19 | 23.4–286.7 | 278–411 | [190] |
| C1 + crude oil (typical Iranian) | - | CP | 16 | 35–275 | 298–323 | [80] |
| C1 + white oil | 3.8 | CP | 5 | 37.8–330.5 | 378 | [191] |
| C1 + white oil | - | CP | 15 | 35.4–347.2 | 278–444 | [191] |
| C1 + condensate oil | - | CVD | 1 | 200 | 333 | [167] |
| C2 + crude oil | 4.1 | CVD | 1 | 35.5 | 353 | [192] |
| C2 + white oil | 3.8 | CP | 4 | 9.8–76.8 | 378 | [193] |
| C2 + white oil | - | CP | 14 | 5.5–41.4 | 278–478 | [193] |
| C3 + kerosene | 0.9 | CP | 5 | 2.1–13.9 | 333 | [194] |
| C3 + kerosene | 1.2 | CP | 3 | 1.8–10.3 | 318 | [194] |
| C3 + kerosene | 1.4 | CP | 11 | 2.1–7.2 | 303 | [194] |
| C3 + spray oil | 5.1 | CP | 4 | 2.2–14 | 333 | [194] |
| C3 + spray oil | 7.9 | CP | 4 | 1.8–9.1 | 318 | [194] |
| C3 + spray oil | 13.5 | CP | 4 | 2.2–7.2 | 303 | [194] |
| CO2 + light oil (Bakken) | - | CP | 1 | 21.7 | 336 | [195] |
| CO2 + crude oil | - | CVD | 3 | 200 | 333 | [166] |
| CO2 + crude oil (Maljamar) | - | HCT | 1 | 52.1 | 298 | [114] |
| CO2 + crude oil (Shengli) | 151.0 | CVD | 3 | 39.3–142.8 | 323 | [169] |
| CO2 + crude oil (Weyburn) | - | PDM | 5 | 3.2–43.9 | 300 | [196] |
| CO2 + crude oil (Daqing) | - | CVD | 8 | 32–82.8 | 318 | [168] |
| CO2 + gas oil BP 200–300 | 3.9 | other | 1 | 1 | 298 | [119] |
| CO2 + gas oil BP 300–400 | 26.5 | other | 1 | 1 | 298 | [119] |
| CO2 + kerosine | 1.8 | other | 1 | 1 | 298 | [119] |
| CO2 + white oil no. 15 | 135.0 | other | 1 | 1 | 298 | [197] |
| CO2 + white oil no. 7 | 56.0 | other | 1 | 1 | 298 | [197] |
| CO2 + crude oil (Shengli)) | 151.0 | CVD | 3 | 49.8–145.9 | 323 | [169] |
| CO2 + condensate oil | - | CVD | 1 | 200 | 333 | [167] |
| CO2 + crude oil (Bakken) | 2.2 | CVD | 4 | 165.6–210.8 | 293 | [198] |
| CO2 + crude oil (Bakken) | - | CVD | 3 | 185.2–187.9 | 313 | [198] |
| N2 + light oil (Bakken) | - | CP | 1 | 52.8 | 336 | [195] |
| N2 + crude oil | - | CVD | 2 | 200 | 333 | [166] |
| N2 + RP-3 jet fuel | - | IF | 14 | 1 | 278–343 | [199] |
| N2 + RP-5 jet fuel | - | IF | 14 | 1 | 278–343 | [199] |
| N2 + condensate oil | - | CVD | 1 | 200 | 333 | [167] |
4. Diffusion Coefficient Correlations
4.1. Wilke–Chang (WC) Correlation (1955)
4.2. Hayduk–Minhas (HM) Correlation (1982)
4.3. Extended Sigmund (ES) Correlation (1976, 1989)
4.4. Riazi-Whitson (RW) Correlation (1993)
4.5. Leahy-Dios and Firoozabadi (LDF) Correlation (2007)
5. Results for Binary Mixtures
5.1. Property Models Used in the Viscosity Correlations
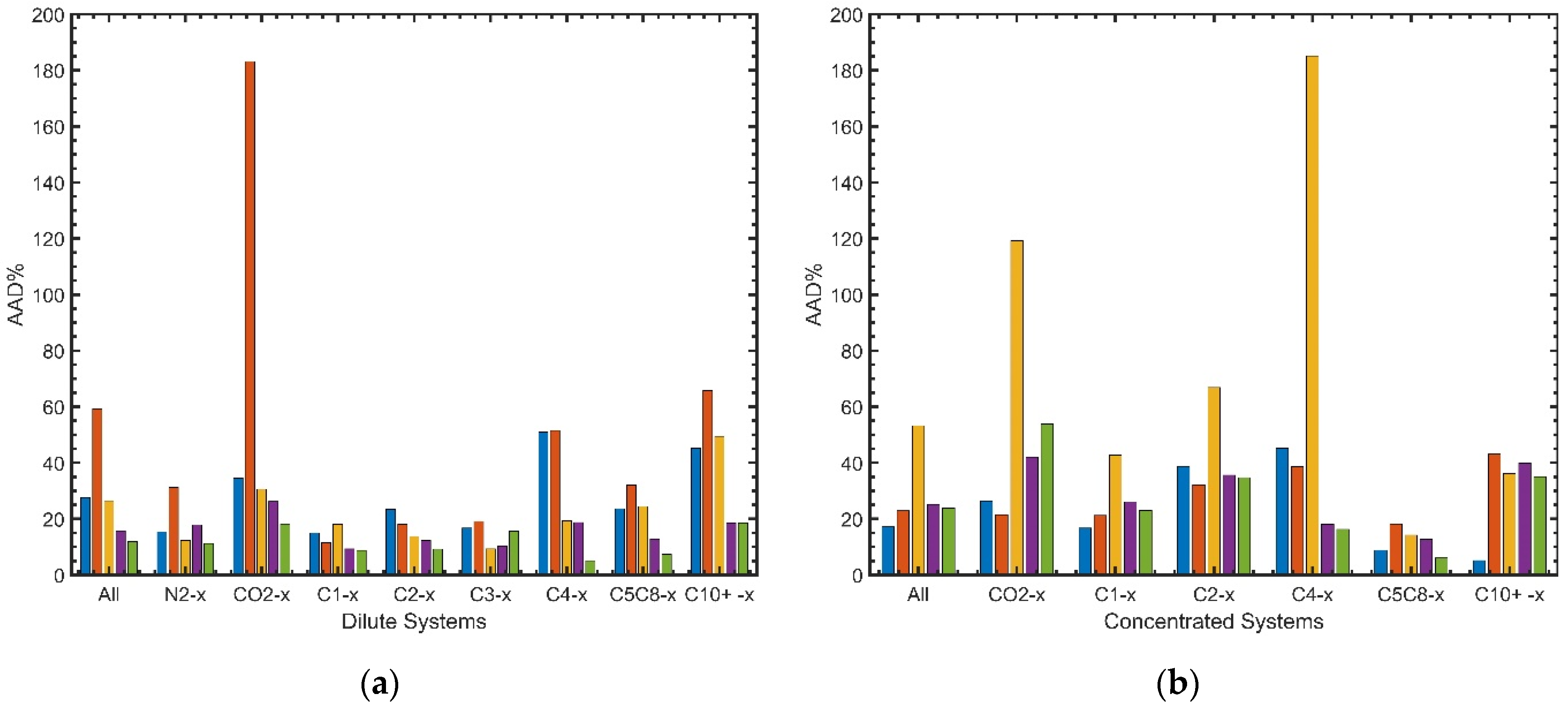
5.2. Overall Results from the Base Case Calculation
- Whole concentration range
- Different concentration ranges
5.3. Sensitivity to the Property Models
- Influence of the density models
- Influence of the viscosity models
- Options for LDF
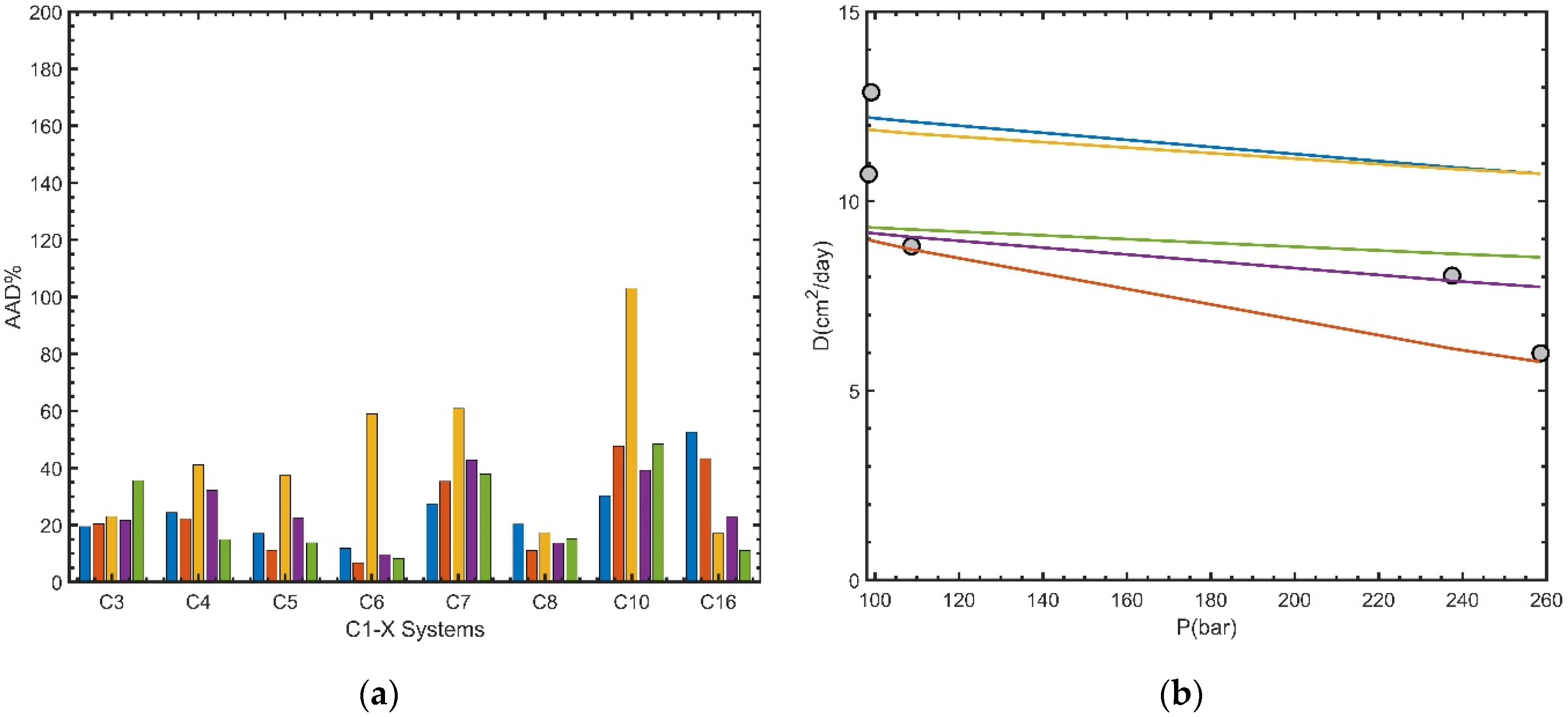
5.4. Results for Selected Groups
6. Results for Oil Mixtures
7. Conclusions
- The binary diffusion coefficients database has over 1600 data points, with around 1300 liquid-phase diffusion data points. These liquid-phase diffusion coefficients are influenced by factors including the component type, composition, temperature, and pressure. In the database, the liquid-phase diffusion coefficient for methane can reach 50 cm2/day, and the coefficient for CO2 can reach 200 cm2/day, while the minimum values are around 0.5–1 cm2/day for C1 and CO2. In general, the diffusion coefficient increases with increasing temperature, decreasing pressure, and increasing concentration of the light solute. There were over 400 data points in the oil diffusion database, with over half of them for heavy oils. The diffusion coefficients for C1/CO2/N2-heavy oil were mostly in the range of 0.01–50 cm2/day. Those for C1/CO2/N2-non-heavy oils were mostly in the range of 0.01–150 cm2/day.
- There was a lack of data in both the binary mixtures and oil systems. The number of available binary diffusion data collected here is just a small fraction (>20× smaller) of the available density data for the similar systems. Anyway, the quality of the diffusion coefficients is an issue, in terms of accuracy and completeness—many diffusion data did not have the corresponding composition. The oil diffusion coefficients were even more scarce, and their quality was more questionable. The reported oil diffusion coefficients may differ in orders of magnitude. Only a limited number of studies report oil compositions, and the studies on live oils were rare.
- The scarcity of diffusion data is in a noteworthy contrast to the significance of diffusion in various reservoir processes. It underscores the need for more experimental measurement. The data for binary mixtures, especially for gas-liquid mixtures at high pressures, are crucial for developing the fundamental diffusion models. For oil mixtures, it is worthwhile to include diffusion measurement as part of the PVT study, especially for gas injection. Publishing more oil diffusion data in the open literature should also be encouraged.
- Among the five correlations studied here, HM and WC have the same theoretical basis (the Stokes–Einstein equation); both ES and RW use an empirical correction to calculate the real fluid diffusion coefficient from the dilute gas one, and LDF is the only one using the MS framework. In comparison with large databases, no single correlation showed a consistent and dominant superiority over the others. Nevertheless, it is possible to find some general trends or identify some more suitable models for specific regions or systems. These details, presented in Section 5 and Section 6, are useful when selecting a correlation for specific applications.
- For binary mixtures, HM provides the lowest deviations, and it is particularly good for diluted solutions and heavier systems. WC provides a similar, but somewhat inferior, performance. However, it should be noted that both HM and WC had a consistency issue because both were originally developed for the diffusion of solute in solvent, and the definitions of solute and solvent are ambiguous at high concentrations. In concentrated composition range, ES seems to be the best choice, and RW is also a good choice. It should be noted that RW has a problem for systems containing a heavy component with a large acentric factor at high-reduced pressures, thus causing large deviations and abnormal pressure dependence for systems such as CO2-C16. RW will become more attractive if this problem is fixed. LDF, despite its strong theoretical basis, did not really show any obvious advantage over the other correlations. It provides good results in the dilute range. Its results at any non-zero concentrations were essentially from the Vignes mixing rule. The correlation tended to provide large deviations for gas-liquid systems such as C1-C10. Although the comparison of the five correlations was based on an extensive binary database, it should be noted that the conclusion regarding the correlation performance depended much on the data selected in the comparison. The recommendation for the best model depends on the system and range of temperature, pressure, and composition. The comparison here was more to reveal the strengths and limitations of these correlations.
- The results for diffusion coefficients also depend on the models selected for density, viscosity, and other properties. A sensitivity study using the binary database has shown that our selection in the base case is reasonable. We can always select the most accurate models for these properties, even though the models used in developing the original correlations may be different. Furthermore, if the modeled density and viscosity are beyond a certain accuracy, the influence of the models become unimportant. For example, GERG, PR-VT, SRK-VT, and SAFT-VT generally provided comparable results, and FT-PR, FT-SRK, and LBC-PRVT provided comparable results. The sensitivity analysis also reveals different degrees of sensitivity of these correlations to the property models. HM and WC were sensitive to viscosity, and ES was sensitive to density. RW and LDF had weak dependence on viscosity.
- For oil systems, HM was the easiest to use, since it requires the smallest set of input parameters. Many of the collected oils could only be tested with HM. HM seemed to provide reasonable results for ordinary oils and light oil products but had a tendency to under-estimate the gas diffusion in heavy oils. Only a limited number (10) of oil systems have sufficient composition information that allow for the use of an EoS model. Among the four correlations (ES, RW, HM, and WC) tested, ES tended to under-predict the diffusion coefficients for light oils but over-predict the results for heavy oils, whereas RW, HM, and WC seemed to have the opposite tendency. For heavy oils, RW, HM, and WC tended to under-estimate the diffusion coefficients by one to two orders of magnitude. The findings based on the small set of data should be used with caution.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poling, B.E.; Prausnitz, J.M.; O’connell, J.P. The Properties of Gases and Liquids; McGraw-Hill Education: New York, NY, USA, 2001. [Google Scholar]
- Medvedev, O. Diffusion Coefficients in Multicomponent Mixtures. Ph.D. Thesis, Technical University of Denmark, Kgs. Lyngby, Denmark, 2005. [Google Scholar]
- Oelkers, E.H.; Helgeson, H.C. Calculation of the Thermodynamic and Transport Properties of Aqueous Species at High Pressures and Temperatures: Aqueous Tracer Diffusion Coefficients of Ions to 1000 °C and 5 Kb. Geochim. Cosmochim. Acta 1988, 52, 63–85. [Google Scholar] [CrossRef]
- Desai, T.A.; Hansford, D.J.; Kulinsky, L.; Nashat, A.H.; Rasi, G.; Tu, J.; Wang, Y.; Zhang, M.; Ferrari, M. Nanopore Technology for Biomedical Applications. Biomed. Microdevices 1999, 2, 11–40. [Google Scholar] [CrossRef]
- Chordia, M. Diffusion in Naturally Fractured Reservoirs—A Review. In Proceedings of the SPE Asia Pacific Oil & Gas Conference and Exhibition, Brisbane, QLD, Australia, 18 October 2010. [Google Scholar]
- Jia, B.; Tsau, J.-S.; Barati, R. A Review of the Current Progress of CO2 Injection EOR and Carbon Storage in Shale Oil Reservoirs. Fuel 2019, 236, 404–427. [Google Scholar] [CrossRef]
- Hunt, E.B., Jr.; Berry, V.J., Jr. Evolution of Gas from Liquids Flowing through Porous Media. AIChE J. 1956, 2, 560–567. [Google Scholar] [CrossRef]
- Li, X.; Yortsos, Y. Theory of Multiple Bubble Growth in Porous Media by Solute Diffusion. Chem. Eng. Sci. 1995, 50, 1247–1271. [Google Scholar] [CrossRef]
- Campbell, B.T.; Orr, F.M. Flow Visualization for CO2/Crude-Oil Displacements. SPE J. 1985, 25, 665–678. [Google Scholar] [CrossRef]
- Grogan, A.; Pinczewski, W. The Role of Molecular Diffusion Processes in Tertiary CO2 Flooding. J. Pet. Technol. 1987, 39, 591–602. [Google Scholar] [CrossRef]
- Da Silva, F.V.; Belery, P. Molecular Diffusion in Naturally Fractured Reservoirs: A Decisive Recovery Mechanism. In Proceedings of the 6th Annual Technical Conference and Exhibition of the Society of Petroleum Engineers, San Antonio, TX, USA, 8 October 1989. [Google Scholar]
- Ghasemi, M.; Suicmez, V. Upscaling of CO2 Injection in a Fractured Oil Reservoir. J. Nat. Gas Sci. Eng. 2019, 63, 70–84. [Google Scholar] [CrossRef]
- Trivedi, J.; Babadagli, T. Experimental and Numerical Modeling of the Mass Transfer between Rock Matrix and Fracture. Chem. Eng. J. 2009, 146, 194–204. [Google Scholar] [CrossRef]
- Alavian, S.A.; Whitson, C.H. Modeling CO2 Injection Including Diffusion in a Fractured-Chalk Experiment. In Proceedings of the SPE Annual Technical Conference and Exhibition, Florence, Italy, 19 September 2010. [Google Scholar]
- Darvish, G.R.; Lindeberg, E.G.; Holt, T.; Utne, S.A.; Kleppe, J. Reservoir Conditions Laboratory Experiments of CO2 Injection into Fractured Cores. In Proceedings of the SPE Europec/EAGE Annual Conference and Exhibition, Vienna, Austria, 12 June 2006. [Google Scholar]
- Hoteit, H.; Firoozabadi, A. Numerical Modeling of Diffusion in Fractured Media for Gas-Injection and-Recycling Schemes. SPE J. 2009, 14, 323–337. [Google Scholar] [CrossRef]
- Alavian, S.A.; Whitson, C.H. Scale Dependence of Diffusion in Naturally Fractured Reservoirs for CO2 Injection. In Proceedings of the SPE Improved Oil Recovery Symposium, Tulsa, OK, USA, 24 April 2010. [Google Scholar]
- Yanze, Y.; Clemens, T. The Role of Diffusion for Nonequilibrium Gas Injection into a Fractured Reservoir. SPE Res. Eval. Eng. 2012, 15, 60–71. [Google Scholar] [CrossRef]
- Burrows, L.C.; Haeri, F.; Cvetic, P.; Sanguinito, S.; Shi, F.; Tapriyal, D.; Goodman, A.; Enick, R.M. A Literature Review of CO2, Natural Gas, and Water-Based Fluids for Enhanced Oil Recovery in Unconventional Reservoirs. Energy Fuels 2020, 34, 5331–5380. [Google Scholar] [CrossRef]
- Hawthorne, S.B.; Gorecki, C.D.; Sorensen, J.A.; Steadman, E.N.; Harju, J.A.; Melzer, S. Hydrocarbon Mobilization Mechanisms from Upper, Middle, and Lower Bakken Reservoir Rocks Exposed to CO2. In Proceedings of the SPE Unconventional Resources Conference-Canada, Calgary, AB, Canada, 5 November 2013. [Google Scholar]
- Yu, W.; Lashgari, H.R.; Wu, K.; Sepehrnoori, K. CO2 Injection for Enhanced Oil Recovery in Bakken Tight Oil Reservoirs. Fuel 2015, 159, 354–363. [Google Scholar] [CrossRef]
- Wan, T.; Sheng, J. Compositional Modelling of the Diffusion Effect on EOR Process in Fractured Shale-Oil Reservoirs by Gasflooding. J. Can. Pet. Technol. 2015, 54, 107–115. [Google Scholar] [CrossRef]
- Jin, L.; Hawthorne, S.; Sorensen, J.; Kurz, B.; Pekot, L.; Smith, S.; Bosshart, N.; Azenkeng, A.; Gorecki, C.; Harju, J. A Systematic Investigation of Gas-Based Improved Oil Recovery Technologies for the Bakken Tight Oil Formation. In Proceedings of the Unconventional Resources Technology Conference, San Antonio, TX, USA, 1 August 2016. [Google Scholar]
- Alharthy, N.; Teklu, T.; Kazemi, H.; Graves, R.; Hawthorne, S.; Braunberger, J.; Kurtoglu, B. Enhanced Oil Recovery in Liquid–Rich Shale Reservoirs: Laboratory to Field. SPE Res. Eval. Eng. 2018, 21, 137–159. [Google Scholar] [CrossRef]
- Das, S.K. Vapex: An Efficient Process for the Recovery of Heavy Oil and Bitumen. SPE J. 1998, 3, 232–237. [Google Scholar] [CrossRef]
- Jiang, Q.; Butler, R.M. Selection of Well Configurations in Vapex Process. In Proceedings of the SPE International Conference on Horizontal Well Technology, Calgary, AB, Canada, 18 November 1996. [Google Scholar]
- Butler, R.; Jiang, Q. Improved Recovery of Heavy Oil by VAPEX with Widely Spaced Horizontal Injectors and Producers. J. Can. Pet. Technol. 2000, 39, PETSOC-00-01-04. [Google Scholar] [CrossRef]
- Cussler, E.L. Diffusion: Mass Transfer in Fluid Systems; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Leahy-Dios, A.; Firoozabadi, A. Unified Model for Nonideal Multicomponent Molecular Diffusion Coefficients. AIChE J. 2007, 53, 2932–2939. [Google Scholar] [CrossRef]
- Fick, A.E. Ueber Diffusion. Ann. Phys. 1855, 170, 59–86. [Google Scholar] [CrossRef]
- Maxwell, J.C., IV. On the Dynamical Theory of Gases. Philos. Trans. R. Soc. 1867, 157, 49–88. [Google Scholar] [CrossRef]
- Onsager, L. Theories and Problems of Liquid Diffusion. Ann. N. Y. Acad. Sci. 1945, 46, 241–265. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.; Krishna, R. Multicomponent Mass Transfer; John Wiley & Sons: New York, NY, USA, 1993. [Google Scholar]
- Marrero, T.R.; Mason, E.A. Gaseous Diffusion Coefficients. J. Phys. Chem. Ref. Data 1972, 1, 3–118. [Google Scholar] [CrossRef]
- Chapman, S.; Cowling, T.G. The Mathematical Theory of Non-Uniform Gases; Cambridge University Press: Cambridge, UK, 1991. [Google Scholar]
- Einstein, A. Über Die von Der Molekularkinetischen Theorie Der Wärme Geforderte Bewegung von in Ruhenden Flüssigkeiten Suspendierten Teilchen. Ann. Phys. 1905, 322, 549–560. [Google Scholar] [CrossRef]
- Neufeld, P.D.; Janzen, A.; Aziz, R. Empirical Equations to Calculate 16 of the Transport Collision Integrals Omega(l, s) for the Lennard-Jones (12–6) Potential. J. Chem. Phys. 1972, 57, 1100–1102. [Google Scholar] [CrossRef]
- Dawson, R.; Khoury, F.; Kobayashi, R. Self-Diffusion Measurements in Methane by Pulsed Nuclear Magnetic Resonance. AIChE J. 1970, 16, 725–729. [Google Scholar] [CrossRef]
- Sigmund, P.M. Prediction of Molecular Diffusion at Reservoir Conditions. Part 1-Measurement and Prediction of Binary Dense Gas Diffusion Coefficients. J. Can. Pet. Technol. 1976, 15, PETSOC-76-02-05. [Google Scholar] [CrossRef]
- Arnold, J.H. Studies in Diffusion. II. A Kinetic Theory of Diffusion in Liquid Systems. J. Am. Chem. Soc. 1930, 52, 3937–3955. [Google Scholar] [CrossRef]
- Aljeshi, Y.A.; Taib, M.B.M.; Trusler, J.M. Modelling the Diffusion Coefficients of Dilute Gaseous Solutes in Hydrocarbon Liquids. Int. J. Thermophys. 2021, 42, 140. [Google Scholar] [CrossRef]
- Wilke, C.; Chang, P. Correlation of Diffusion Coefficients in Dilute Solutions. AIChE J. 1955, 1, 264–270. [Google Scholar] [CrossRef]
- Hayduk, W.; Minhas, B. Correlations for Prediction of Molecular Diffusivities in Liquids. Can. J. Chem. Eng. 1982, 60, 295–299. [Google Scholar] [CrossRef]
- Scheibel, E.G. Correspondence. Liquid Diffusivities. Viscosity of Gases. Ind. Eng. Chem. 1954, 46, 2007–2008. [Google Scholar] [CrossRef]
- King, C.J.; Hsueh, L.; Mao, K.-W. Liquid Phase Diffusion of Non-Electrolytes at High Dilution. J. Chem. Eng. Data 1965, 10, 348–350. [Google Scholar] [CrossRef]
- Rutten, P.W.M. Diffusion in Liquids. Ph.D. Thesis, Delft Univeristy, Delft, The Netherlands, 1992. [Google Scholar]
- Riazi, M.R.; Whitson, C.H. Estimating Diffusion Coefficients of Dense Fluids. Ind. Eng. Chem. Res. 1993, 32, 3081–3088. [Google Scholar] [CrossRef]
- Wesselingh, J.; Bollen, A. Multicomponent Diffusivities from the Free Volume Theory. Chem. Eng. Res. Des. 1997, 75, 590–602. [Google Scholar] [CrossRef]
- Liu, H.; Silva, C.M.; Macedo, E.A. Generalised Free-Volume Theory for Transport Properties and New Trends about the Relationship between Free Volume and Equations of State. Fluid Phase Equilibria 2002, 202, 89–107. [Google Scholar] [CrossRef]
- Bosma, J.; Wesselingh, J. Estimation of Diffusion Coefficients in Dilute Liquid Mixtures. Chem. Eng. Res. Des. 1999, 4, 325–328. [Google Scholar] [CrossRef]
- Hsu, Y.-D.; Chen, Y.-P. Correlation of the Mutual Diffusion Coefficients of Binary Liquid Mixtures. Fluid Phase Equilibria 1998, 152, 149–168. [Google Scholar] [CrossRef]
- Shapiro, A.A. Thermodynamic Theory of Diffusion and Thermodiffusion Coefficients in Multicomponent Mixtures. J. Non-Equilib. Thermodyn. 2020, 45, 343–372. [Google Scholar] [CrossRef]
- Baghooee, H.; Shapiro, A. Unified Thermodynamic Modelling of Diffusion and Thermodiffusion Coefficients. Fluid Phase Equilibria 2022, 558, 113445. [Google Scholar] [CrossRef]
- Darken, L.S. Diffusion, Mobility and Their Interrelation through Free Energy in Binary Metallic Systems. Trans. Aime 1948, 175, 184–201. [Google Scholar]
- Vignes, A. Diffusion in Binary Solutions. Variation of Diffusion Coefficient with Composition. Ind. Eng. Chem. Fundamen. 1966, 5, 189–199. [Google Scholar] [CrossRef]
- Himmelblau, D. Diffusion of Dissolved Gases in Liquids. Chem. Rev. 1964, 64, 527–550. [Google Scholar] [CrossRef]
- Sigmund, P.M. Prediction of Molecular Diffusion at Reservoir Conditions. Part II-Estimating the Effects of Molecular Diffusion and Convective Mixing in Multicomponent Systems. J. Can. Pet. Technol. 1976, 15, 53–62. [Google Scholar] [CrossRef]
- Berry, V.J., Jr.; Koeller, R. Diffusion in Compressed Binary Gaseous Systems. AIChE J. 1960, 6, 274–280. [Google Scholar] [CrossRef]
- Gover, T.A. Diffusion of Gases: A Physical Chemistry Experiment. J. Chem. Educ. 1967, 44, 409. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Methane in the Liquid Phase of the Methane-Propane System. Ind. Eng. Chem. Chem. Eng. Data Ser. 1958, 3, 54–59. [Google Scholar] [CrossRef]
- Woessner, D.; Snowden, B.S., Jr.; George, R.; Melrose, J. Dense Gas Diffusion Coefficients for the Methane-Propane System. Ind. Eng. Chem. Fundamen. 1969, 8, 779–786. [Google Scholar] [CrossRef]
- Zangi, P.; Rausch, M.H.; Fröba, A.P. Binary Diffusion Coefficients for Gas Mixtures of Propane with Methane and Carbon Dioxide Measured in a Loschmidt Cell Combined with Holographic Interferometry. Int. J. Thermophys. 2019, 40, 18. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Methane-n-Butane-Methane in Liquid Phase. Ind. Eng. Chem. Chem. Eng. Data Ser. 1956, 1, 71–77. [Google Scholar] [CrossRef]
- Christoffersen, K.R. High-Pressure Experiments with Application to Naturally Fractured Chalk Reserviors. Ph.D. Thesis, The Norwegian Institute of Technology University of Trondheim, Trondheim, Norway, 1992. [Google Scholar]
- Riazi, M.R. A New Method for Experimental Measurement of Diffusion Coefficients in Reservoir Fluids. J. Pet. Sci. Eng. 1996, 14, 235–250. [Google Scholar] [CrossRef]
- Imai, M.; Sumikawa, I.; Yamada, T.; Nakano, M. Reservoir Fluid Characterization for Tight Fractured Reservoirs: Effect of Diffusion. In Proceedings of the SPE Asia Pacific Oil and Gas Conference and Exhibition, Perth, Australia, 22 October 2012. [Google Scholar]
- Reamer, H.; Duffy, C.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems: Methane-n-Pentane-Methane in Liuqid Phase. Ind. Eng. Chem. 1956, 48, 282–284. [Google Scholar] [CrossRef]
- Hill, E.; Lacey, W. Rate of Solution of Methane in Quiescent Liquid Hydrocarbons. II. Ind. Eng. Chem. 1934, 26, 1324–1327. [Google Scholar] [CrossRef]
- Wilhelm, E.; Battino, R. Binary Gaseous Diffusion Coefficients. 1. Methane and Carbon Tetrafluroide with n-Hexane, n-Heptane, n-Octane, and 2,2,4-Trimethylpentane at One-Atmosphere Pressure at 10–70. Deg. J. Chem. Eng. Data 1972, 17, 187–189. [Google Scholar] [CrossRef]
- Hayduk, W.; Buckley, W. Effect of Molecular Size and Shape on Diffusivity in Dilute Liquid Solutions. Chem. Eng. Sci. 1972, 27, 1997–2003. [Google Scholar] [CrossRef]
- Kohn, J.P.; Romero, N. Molecular Diffusion Coefficients in Binary Gaseous Systems at One Atmosphere Pressure. N-Hexane-Methane and 3-Methylpentane-Methane Systems. J. Chem. Eng. Data 1965, 10, 125–127. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems: Methane in the Liquid Phase of the Methane–n-Heptane System. AIChE J. 1957, 3, 449–453. [Google Scholar] [CrossRef]
- Taib, M.B.M.; Trusler, J.M. Diffusion Coefficients of Methane in Methylbenzene and Heptane at Temperatures between 323 K and 398 K at Pressures up to 65 MPa. Int. J. Thermophys. 2020, 41, 119. [Google Scholar] [CrossRef]
- Colgate, S.; House, V.; Thieu, V.; Zachery, K.; Hornick, J.; Shalosky, J. High-Pressure Cylindrical Acoustic Resonance Diffusion Measurements of Methane in Liquid Hydrocarbons. Int. J. Thermophys. 1995, 16, 655–662. [Google Scholar] [CrossRef]
- Killie, S.; Hafskjold, B.; Borgen, O.; Ratkje, S.K.; Hovde, E. High-Pressure Diffusion Measurements by Mach-Zehnder Interferometry. AIChE J. 1991, 37, 142–146. [Google Scholar] [CrossRef]
- Erkey, C.; Akgerman, A. Translational-Rotational Coupling Parameters for Mutual Diffusion in N-Octane. AIChE J. 1989, 35, 443–448. [Google Scholar] [CrossRef]
- Reamer, H.; Opfell, J.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Methane-Decane-Methane in Liquid Phase. Ind. Eng. Chem. 1956, 48, 275–282. [Google Scholar] [CrossRef]
- Dysthe, D.K.; Hafskjold, B.; Breer, J.; Cejka, D. Interferometric Technique for Measuring Interdiffusion at High Pressures. J. Phys. Chem. 1995, 99, 11230–11238. [Google Scholar] [CrossRef]
- Dysthe, D.; Hafskjold, B. Inter-and Intradiffusion in Liquid Mixtures of Methane and n-Decane. Int. J. Thermophys. 1995, 16, 1213–1224. [Google Scholar] [CrossRef]
- Jamialahmadi, M.; Emadi, M.; Müller-Steinhagen, H. Diffusion Coefficients of Methane in Liquid Hydrocarbons at High Pressure and Temperature. J. Pet. Sci. Eng. 2006, 53, 47–60. [Google Scholar] [CrossRef]
- Pacheco-Roman, F.J.; Hejazi, S.H.; Maini, B.B. Estimation of Low-Temperature Mass-Transfer Properties of Methane and Carbon Dioxide in n-Decane, Hexadecane, and Bitumen Using the Pressure-Decay Technique. Energy Fuels 2016, 30, 5232–5239. [Google Scholar] [CrossRef]
- Etminan, S.R.; Maini, B.B.; Chen, Z.; Hassanzadeh, H. Constant-Pressure Technique for Gas Diffusivity and Solubility Measurements in Heavy Oil and Bitumen. Energy Fuels 2010, 24, 533–549. [Google Scholar] [CrossRef]
- Ratnakar, R.R.; Dindoruk, B. Measurement of Gas Diffusivity in Heavy Oils and Bitumens by Use of Pressure-Decay Test and Establishment of Minimum Time Criteria for Experiments. SPE J. 2015, 20, SPE-170931-PA. [Google Scholar] [CrossRef]
- Erkey, C.; Akgerman, A. Translational Rotational Coupling Parameters for Mutual Diffusion in Normal Alkanes. AIChE J. 1989, 35, 1907–1911. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Ethane in the Liquid Phase of the Ethane-n-Pentane System. J. Chem. Eng. Data 1961, 6, 481–484. [Google Scholar] [CrossRef]
- Hayduk, W.; Cheng, S. Review of Relation between Diffusivity and Solvent Viscosity in Dilute Liquid Solutions. Chem. Eng. Sci. 1971, 26, 635–646. [Google Scholar] [CrossRef]
- Malik, V.; Hayduk, W. A Steady’state Capillary Cell Method for Measuring Gas-Liquid Diffusion Coefficients. Can. J. Chem. Eng. 1968, 46, 462–466. [Google Scholar] [CrossRef]
- Reamer, H.; Lower, J.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Ethane in the Liquid Phase of the Ethane-n-Decane System. J. Chem. Eng. Data 1964, 9, 54–59. [Google Scholar] [CrossRef]
- Hayduk, W.; Castaneda, R.; Bromfield, H.; Perras, R.R. Diffusivities of Propane in Normal Paraffin, Chlorobenzene, and Butanol Solvents. AIChE J. 1973, 19, 859–861. [Google Scholar] [CrossRef]
- Sabet, N.; Khalifi, M.; Zirrahi, M.; Hassanzadeh, H.; Abedi, J. A New Analytical Model for Estimation of the Molecular Diffusion Coefficient of Gaseous Solvents in Bitumen–Effect of Swelling. Fuel 2018, 231, 342–351. [Google Scholar] [CrossRef]
- Reamer, H.; Lower, J.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. The n-Butane-n-Decane System in the Liquid Phase. J. Chem. Eng. Data 1964, 9, 602–606. [Google Scholar] [CrossRef]
- Bidlack, D.L.; Kett, T.; Kelly, C.; Anderson, D.K. Diffusion in the Solvents Hexane and Carbon Tetrachloride. J. Chem. Eng. Data 1969, 14, 342–343. [Google Scholar] [CrossRef]
- Alizadeh, A.; Wakeham, W. Mutual Diffusion Coefficients for Binary Mixtures of Normal Alkanes. Int. J. Thermophys. 1982, 3, 307–323. [Google Scholar] [CrossRef]
- de Oliveira, C.P.; Fareleira, J.M.; de Castro, C.N. Mutual Diffusivity in Binary Mixtures of N-Heptane with n-Hexane Isomers. Int. J. Thermophys. 1989, 10, 973–982. [Google Scholar] [CrossRef]
- Lopes, M.M.; de Castro, C.N.; de Oliveira, C.P. Mutual Diffusivity in N-Heptane+ n-Hexane Isomers. Fluid Phase Equilibria 1987, 36, 195–205. [Google Scholar] [CrossRef]
- Hayduk, W.; Loakimidis, S. Liquid Diffusivities in Normal Paraffin Solutions. J. Chem. Eng. Data 1976, 21, 255–260. [Google Scholar] [CrossRef]
- Bidlack, D.; Anderson, D. Mutual Diffusion in Nonideal, Nonassociating Liquid Systems. J. Phys. Chem. 1964, 68, 3790–3794. [Google Scholar] [CrossRef]
- Kett, T.K.; Anderson, D.K. Ternary Isothermal Diffusion and the Validity of the Onsager Reciprocal Relations in Nonassociating Systems. J. Phys. Chem. 1969, 73, 1268–1274. [Google Scholar] [CrossRef]
- Shieh, J.J.; Lyons, P. Transport Properties of Liquid N-Alkanes. J. Phys. Chem. 1969, 73, 3258–3264. [Google Scholar] [CrossRef]
- Bidlack, D.; Anderson, D. Mutual Diffusion in the Liquid System Hexane-Hexadecane. J. Phys. Chem. 1964, 68, 206–208. [Google Scholar] [CrossRef]
- Matthews, M.; Akgerman, A. Diffusion Coefficients for Binary Alkane Mixtures to 573 K and 3.5 MPa. AIChE J. 1987, 33, 881–885. [Google Scholar] [CrossRef]
- Lopes, M.L.S.M.; de Castro, C.A.N. Liquid-Phase Diffusivity Measurements of Nalkane Binary Mixtures. In Proceedings of the Ninth European Thermophysical Properties Conference, Manchester, UK, 17 September 1984; Volume 17, pp. 599–606. [Google Scholar]
- Lo, H.Y. Diffusion Coefficients in Binary Liquid N-Alkane Systems. J. Chem. Eng. Data 1974, 19, 236–241. [Google Scholar] [CrossRef]
- Trevoy, D.; Drickamer, H. Diffusion in Binary Liquid Hydrocarbon Mixtures. J. Chem. Phys. 1949, 17, 1117–1120. [Google Scholar] [CrossRef]
- Geet, A.L.V.; Adamson, A.W. Diffusion in Liquid Hydrocarbon Mixtures. J. Phys. Chem. 1964, 68, 238–246. [Google Scholar] [CrossRef]
- Matthews, M.A.; Rodden, J.B.; Akgerman, A. High-Temperature Diffusion of Hydrogen, Carbon Monoxide, and Carbon Dioxide in Liquid n-Heptane, n-Dodecane, and n-Hexadecane. J. Chem. Eng. Data 1987, 32, 319–322. [Google Scholar] [CrossRef]
- Rodden, J.B.; Erkey, C.; Akgerman, A. High-Temperature Diffusion, Viscosity, and Density Measurements in n-Eicosane. J. Chem. Eng. Data 1988, 33, 344–347. [Google Scholar] [CrossRef]
- Rodden, J.B.; Erkey, C.; Akgerman, A. Mutual Diffusion Coefficients for Several Dilute Solutes in N-Octacosane and the Solvent Density at 371–534 K. J. Chem. Eng. Data 1988, 33, 450–453. [Google Scholar] [CrossRef]
- Mueller, C.; Cahill, R. Mass Spectrometric Measurement of Diffusion Coefficients. J. Chem. Phys. 1964, 40, 651–654. [Google Scholar] [CrossRef]
- Pakurar, T.A.; Ferron, J.R. Diffusivities in System: Carbon Dioxide-Nitrogen-Argon. Ind. Eng. Chem. Fundamen. 1966, 5, 553–557. [Google Scholar] [CrossRef]
- Boardman, L.; Wild, N. The Diffusion of Pairs of Gases with Molecules of Equal Mass. Proc. R. Soc. A Math. Phys. Sci. 1937, 162, 511–520. [Google Scholar] [CrossRef]
- Nagasaka, M. Binary Diffusion Coefficients of N-Pentane in Gases. J. Chem. Eng. Data 1973, 18, 388–390. [Google Scholar] [CrossRef]
- Wall, F.; Kidder, G. Mutual Diffusion of Pairs of Gases. J. Phys. Chem. 1946, 50, 235–242. [Google Scholar] [CrossRef]
- Grogan, A.; Pinczewski, V.; Ruskauff, G.J.; Orr, F. Diffusion of CO2 at Reservoir Conditions: Models and Measurements. SPE Res. Eng. 1988, 3, 93–102. [Google Scholar] [CrossRef]
- Umezawa, S.; Nagashima, A. Measurement of the Diffusion Coefficients of Acetone, Benzene, and Alkane in Supercritical CO2 by the Taylor Dispersion Method. J. Supercrit. Fluids 1992, 5, 242–250. [Google Scholar] [CrossRef]
- Takeuchi, H.; Fujine, M.; Sato, T.; Onda, K. Simultaneous Determination of Diffusion Coefficient and Solubility of Gas in Liquid by a Diaphragm Cell. J. Chem. Eng. Jpn. 1975, 8, 252–253. [Google Scholar] [CrossRef]
- Luthjens, L.; De Leng, H.; Warman, J.; Hummel, A. Diffusion Coefficients of Gaseous Scavengers in Organic Liquids Used in Radiation Chemistry. Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1990, 36, 779–784. [Google Scholar] [CrossRef]
- Cadogan, S.P.; Mistry, B.; Wong, Y.; Maitland, G.C.; Trusler, J.M. Diffusion Coefficients of Carbon Dioxide in Eight Hydrocarbon Liquids at Temperatures between (298.15 and 423.15) K at Pressures up to 69 MPa. J. Chem. Eng. Data 2016, 61, 3922–3932. [Google Scholar] [CrossRef]
- Davies, G.; Ponter, A.; Craine, K. The Diffusion of Carbon Dioxide in Organic Liquids. Can. J. Chem. Eng. 1967, 45, 372–376. [Google Scholar] [CrossRef]
- Nikkhou, F.; Keshavarz, P.; Ayatollahi, S.; Jahromi, I.R.; Zolghadr, A. Evaluation of Interfacial Mass Transfer Coefficient as a Function of Temperature and Pressure in Carbon Dioxide/Normal Alkane Systems. Heat Mass Transf. 2015, 51, 477–485. [Google Scholar] [CrossRef]
- Saad, H.; Gulari, E. Diffusion of Carbon Dioxide in Heptane. J. Phys. Chem. 1984, 88, 136–139. [Google Scholar] [CrossRef]
- Saad, H.; Gulari, E. Diffusion of Liquid Hydrocarbons in Supercritical CO2 by Photon Correlation Spectroscopy. Ber. Bunsenges. Phys. Chem. 1984, 88, 834–837. [Google Scholar] [CrossRef]
- Wang, L.-S.; Lang, Z.-X.; Guo, T.-M. Measurement and Correlation of the Diffusion Coefficients of Carbon Dioxide in Liquid Hydrocarbons under Elevated Pressures. Fluid Phase Equilibria 1996, 117, 364–372. [Google Scholar] [CrossRef]
- McManamey, W.; Woollen, J. The Diffusivity of Carbon Dioxide in Some Organic Liquids at 25 ° and 50 °C. AIChE J. 1973, 19, 667–669. [Google Scholar] [CrossRef]
- Du, D.; Zheng, L.; Ma, K.; Wang, F.; Sun, Z.; Li, Y. Determination of Diffusion Coefficient of a Miscible CO2/n-Hexadecane System with Dynamic Pendant Drop Volume Analysis (DPDVA) Technique. Int. J. Heat Mass Transf. 2019, 139, 982–989. [Google Scholar] [CrossRef]
- Robb, W.; Drickamer, H. Diffusion in CO2 up to 150-Atmospheres Pressure. J. Chem. Phys. 1951, 19, 1504–1508. [Google Scholar] [CrossRef]
- Douglass, D.C.; McCall, D.W. Diffusion in Paraffin Hydrocarbons. J. Phys. Chem. 1958, 62, 1102–1107. [Google Scholar] [CrossRef]
- Jeffries, Q.R.; Drickamer, H. Diffusion in CO2–CH4 Mixtures to 225 Atmospheres Pressure. J. Chem. Phys. 1954, 22, 436–437. [Google Scholar] [CrossRef]
- Walker, R.; Westenberg, A. Molecular Diffusion Studies in Gases at High Temperature. I. The “Point Source” Technique. J. Chem. Phys. 1958, 29, 1139–1146. [Google Scholar] [CrossRef]
- Moore, J.W.; Wellek, R.M. Diffusion Coefficients of N-Heptane and n-Decane in n-Alkanes and n-Alcohols at Several Temperatures. J. Chem. Eng. Data 1974, 19, 136–140. [Google Scholar] [CrossRef]
- Chen, B.H.; Chen, S. Diffusion of Slightly Soluble Gases in Liquids: Measurement and Correlation with Implications on Liquid Structures. Chem. Eng. Sci. 1985, 40, 1735–1741. [Google Scholar] [CrossRef]
- Helbaek, M.; Hafskjold, B.; Dysthe, D.; Sørland, G. Self-Diffusion Coefficients of Methane or Ethane Mixtures with Hydrocarbons at High Pressure by NMR. J. Chem. Eng. Data 1996, 41, 598–603. [Google Scholar] [CrossRef]
- Guzman, J.; Garrido, L. Determination of Carbon Dioxide Transport Coefficients in Liquids and Polymers by NMR Spectroscopy. J. Phys. Chem. B 2012, 116, 6050–6058. [Google Scholar] [CrossRef]
- Pomeroy, R.D.; Lacey, W.N.; Scudder, N.F.; Stapp, F.P. Rate of Solution of Methane in Quiescent Liquid Hydrocarbons. Ind. Eng. Chem. 1933, 25, 1014–1019. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Methane in the Liquid Phase of the Methane-Cyclohexane System. J. Chem. Eng. Data 1959, 4, 296–300. [Google Scholar] [CrossRef]
- Renner, T. Measurement and Correlation of Diffusion Coefficients for CO2 and Rich-Gas Applications. SPE Res. Eng. 1988, 3, 517–523. [Google Scholar] [CrossRef]
- Li, Z.; Dong, M. Experimental Study of Carbon Dioxide Diffusion in Oil-Saturated Porous Media under Reservoir Conditions. Ind. Eng. Chem. Res. 2009, 48, 9307–9317. [Google Scholar] [CrossRef]
- Teng, Y.; Liu, Y.; Song, Y.; Jiang, L.; Zhao, Y.; Zhou, X.; Zheng, H.; Chen, J. A Study on CO2 Diffusion Coefficient in n-Decane Saturated Porous Media by MRI. Energy Procedia 2014, 61, 603–606. [Google Scholar] [CrossRef]
- Hao, M.; Song, Y.; Su, B.; Zhao, Y. Diffusion of CO2 in n-Hexadecane Determined from NMR Relaxometry Measurements. Phys. Lett. A 2015, 379, 1197–1201. [Google Scholar] [CrossRef]
- Lv, J.; Chi, Y.; Zhao, C.; Zhang, Y.; Mu, H. Experimental Study of the Supercritical CO2 Diffusion Coefficient in Porous Media under Reservoir Conditions. R. Soc. Open Sci. 2019, 6, 181902. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Tsau, J.-S.; Barati, R. Measurement of CO2 Diffusion Coefficient in the Oil-Saturated Porous Media. J. Pet. Sci. Eng. 2019, 181, 106189. [Google Scholar] [CrossRef]
- Yan, W.; Regueira, T.; Liu, Y.; Stenby, E.H. Density Modeling of High-Pressure Mixtures Using Cubic and Non-Cubic EoS and an Excess Volume Method. Fluid Phase Equilibria 2021, 532, 112884. [Google Scholar] [CrossRef]
- Perez, A.G.; Coquelet, C.; Paricaud, P.; Chapoy, A. Comparative Study of Vapour-Liquid Equilibrium and Density Modelling of Mixtures Related to Carbon Capture and Storage with the SRK, PR, PC-SAFT and SAFT-VR Mie Equations of State for Industrial Uses. Fluid Phase Equilibria 2017, 440, 19–35. [Google Scholar] [CrossRef]
- Barnes, C. Diffusion through a Membrane. Physics 1934, 5, 4–8. [Google Scholar] [CrossRef]
- Robinson, R.A.; Stokes, R.H. Electrolyte Solutions; Butterworth: London, UK, 1960. [Google Scholar]
- Upreti, S.R.; Mehrotra, A.K. Diffusivity of CO2, CH4, C2H6 and N2 in Athabasca Bitumen. Can. J. Chem. Eng. 2010, 80, 116–125. [Google Scholar] [CrossRef]
- Stefan, J. Über Das Gleichgewicht Und Die Bewegung, Insbesondere Die Diffusion von Gasgemengen. Sitz. Akad. Wiss. Math.-Nat. Kl. 1871, 63, 63–124. [Google Scholar]
- Witherspoon, P.; Saraf, D. Diffusion of Methane, Ethane, Propane, and n-Butane in Water from 25 to 43°. J. Phys. Chem. 1965, 69, 3752–3755. [Google Scholar] [CrossRef]
- Kegeles, G.; Gosting, L.J. The Theory of an Interference Method for the Study of Diffusion. J. Am. Chem. Soc. 1947, 69, 2516–2523. [Google Scholar] [CrossRef] [PubMed]
- Hampe, M.J.; Schermuly, W.; Blaß, E. Decrease of Diffusion Coefficients near Binodal States of Liquid-Liquid Systems. Chem. Eng. Technol. 1991, 14, 219–225. [Google Scholar] [CrossRef]
- Yang, C.; Gu, Y. New Experimental Method for Measuring Gas Diffusivity in Heavy Oil by the Dynamic Pendant Drop Volume Analysis (DPDVA). Ind. Eng. Chem. Res. 2005, 44, 4474–4483. [Google Scholar] [CrossRef]
- Ohata, T.; Nakano, M.; Ueda, R. Evaluation of Molecular Diffusion Effect by Using PVT Experimental Data: Impact on Gas Injection to Tight Fractured Gas Condensate/Heavy Oil Reservoirs. In Proceedings of the SPE Asia Pacific Unconventional Resources Conference and Exhibition, Brisbane, Australia, 9 November 2015. [Google Scholar]
- Yang, Y.; Regueira, T.; Rodriguez, H.M.; Shapiro, A.; Stenby, E.H.; Yan, W. Determination of Methane Diffusion Coefficients in Live Oils for Tight Reservoirs at High Pressures. In Proceedings of the SPE Annual Technical Conference and Exhibition, Dubai, United Arab Emirates, 21 September 2021. [Google Scholar]
- Ghasemi, M.; Astutik, W.; Alavian, S.A.; Whitson, C.H.; Sigalas, L.; Olsen, D.; Suicmez, V.S. Determining Diffusion Coefficients for Carbon Dioxide Injection in Oil-Saturated Chalk by Use of a Constant-Volume-Diffusion Method. SPE J. 2017, 22, 505–520. [Google Scholar] [CrossRef]
- Li, S.; Qiao, C.; Li, Z.; Hui, Y. The Effect of Permeability on Supercritical CO2 Diffusion Coefficient and Determination of Diffusive Tortuosity of Porous Media under Reservoir Conditions. J. CO2 Util. 2018, 28, 1–14. [Google Scholar] [CrossRef]
- Li, S.; Qiao, C.; Zhang, C.; Li, Z. Determination of Diffusion Coefficients of Supercritical CO2 under Tight Oil Reservoir Conditions with Pressure-Decay Method. J. CO2 Util. 2018, 24, 430–443. [Google Scholar] [CrossRef]
- Zhang, C.; Qiao, C.; Li, S.; Li, Z. The Effect of Oil Properties on the Supercritical CO2 Diffusion Coefficient under Tight Reservoir Conditions. Energies 2018, 11, 1495. [Google Scholar] [CrossRef]
- Zhang, X.; Shaw, J. Liquid-Phase Mutual Diffusion Coefficients for Heavy Oil+ Light Hydrocarbon Mixtures. Pet. Sci. Technol. 2007, 25, 773–790. [Google Scholar] [CrossRef]
- Zhang, X.; Fulem, M.; Shaw, J.M. Liquid-Phase Mutual Diffusion Coefficients for Athabasca Bitumen+ Pentane Mixtures. J. Chem. Eng. Data 2007, 52, 691–694. [Google Scholar] [CrossRef]
- Sadighian, A.; Becerra, M.; Bazyleva, A.; Shaw, J.M. Forced and Diffusive Mass Transfer between Pentane and Athabasca Bitumen Fractions. Energy Fuels 2011, 25, 782–790. [Google Scholar] [CrossRef]
- Guerrero Aconcha, U.E.; Kantzas, A. Diffusion of Hydrocarbon Gases in Heavy Oil and Bitumen. In Proceedings of the Latin American and Caribbean Petroleum Engineering Conference, Cartagena, Colombia, 31 May 2009. [Google Scholar]
- Imai, M.; Mikami, K.; Suganuma, T.; Tsuchiya, Y.; Nakagawa, K.; Takahashi, S. Determination of Binary Diffusion Coefficients between Hot Liquid Solvents and Bitumen with X-ray CT. J. Pet. Sci. Eng. 2019, 177, 496–507. [Google Scholar] [CrossRef]
- Fadaei, H.; Scarff, B.; Sinton, D. Rapid Microfluidics-Based Measurement of CO2 Diffusivity in Bitumen. Energy Fuels 2011, 25, 4829–4835. [Google Scholar] [CrossRef]
- Talebi, S.; Abedini, A.; Lele, P.; Guerrero, A.; Sinton, D. Microfluidics-Based Measurement of Solubility and Diffusion Coefficient of Propane in Bitumen. Fuel 2017, 210, 23–31. [Google Scholar] [CrossRef]
- Unatrakarn, D.; Asghari, K.; Condor, J. Experimental Studies of CO2 and CH4 Diffusion Coefficient in Bulk Oil and Porous Media. Energy Procedia 2011, 4, 2170–2177. [Google Scholar] [CrossRef]
- Guo, P.; Wang, Z.; Xu, Y.; Du, J. Research on Molecular Diffusion Coefficient of Gas-Oil System Under High Temperature and High Pressure. In Mass Transfer in Chemical Engineering Processes; Markoš, J., Ed.; IntechOpen: London, UK, 2011. [Google Scholar]
- Guo, P.; Tu, H.; Ye, A.; Wang, Z. Predicted Dependence of Gas—Liquid Diffusion Coefficient on Capillary Pressure in Porous Media. Chem. Technol. Fuels Oils 2017, 53, 54–67. [Google Scholar] [CrossRef]
- Wang, S.; Hou, J.; Liu, B.; Zhao, F.; Yuan, G.; Liu, G. The Pressure-Decay Method for Nature Convection Accelerated Diffusion of CO2 in Oil and Water under Elevated Pressures. Energy Sources Part A Recovery Util. Environ. Eff. 2013, 35, 538–545. [Google Scholar] [CrossRef]
- Li, B.; Zhang, Q.; Cao, A.; Bai, H.; Xu, J. Experimental and Numerical Studies on the Diffusion of CO2 from Oil to Water. J. Therm. Sci. 2020, 29, 268–278. [Google Scholar] [CrossRef]
- Etminan, S.R.; Maini, B.B.; Chen, Z. Determination of Mass Transfer Parameters in Solvent-Based Oil Recovery Techniques Using a Non-Equilibrium Boundary Condition at the Interface. Fuel 2014, 120, 218–232. [Google Scholar] [CrossRef]
- Tharanivasan, A.K.; Yang, C.; Gu, Y. Measurements of Molecular Diffusion Coefficients of Carbon Dioxide, Methane, and Propane in Heavy Oil under Reservoir Conditions. Energy Fuels 2006, 20, 2509–2517. [Google Scholar] [CrossRef]
- Yang, C.; Gu, Y. Diffusion Coefficients and Oil Swelling Factors of Carbon Dioxide, Methane, Ethane, Propane, and Their Mixtures in Heavy Oil. Fluid Phase Equilibria 2006, 243, 64–73. [Google Scholar] [CrossRef]
- Zhang, Y.; Hyndman, C.; Maini, B. Measurement of Gas Diffusivity in Heavy Oils. J. Pet. Sci. Eng. 2000, 25, 37–47. [Google Scholar] [CrossRef]
- Yang, Z.; Dong, M.; Gong, H.; Li, Y. Determination of Mass Transfer Coefficient of Methane in Heavy Oil-Saturated Unconsolidated Porous Media Using Constant-Pressure Technique. Ind. Eng. Chem. Res. 2017, 56, 7390–7400. [Google Scholar] [CrossRef]
- Zheng, S.; Yang, D. Determination of Individual Diffusion Coefficients of C3H8/n-C4H10/CO2/Heavy-Oil Systems at High Pressures and Elevated Temperatures by Dynamic Volume Analysis. SPE J. 2017, 22, 799–816. [Google Scholar] [CrossRef]
- Shi, Y.; Zheng, S.; Yang, D. Determination of Individual Diffusion Coefficients of Alkane Solvent (s)–CO2–Heavy Oil Systems with Consideration of Natural Convection Induced by Swelling Effect. Int. J. Heat Mass Transf. 2017, 107, 572–585. [Google Scholar] [CrossRef]
- Li, H.A.; Sun, H.; Yang, D. Effective Diffusion Coefficients of Gas Mixture in Heavy Oil under Constant-Pressure Conditions. Heat Mass Transf. 2017, 53, 1527–1540. [Google Scholar] [CrossRef]
- Li, H.; Yang, D. Determination of Individual Diffusion Coefficients of Solvent/CO2 Mixture in Heavy Oil with Pressure-Decay Method. SPE J. 2016, 21, 131–143. [Google Scholar] [CrossRef]
- Wen, Y.; Kantzas, A. Monitoring Bitumen-Solvent Interactions with Low-Field Nuclear Magnetic Resonance and X-ray Computer-Assisted Tomography. Energy Fuels 2005, 19, 1319–1326. [Google Scholar] [CrossRef]
- Guerrero-Aconcha, U.E.; Salama, D.; Kantzas, A. Diffusion Coefficient of N-Alkanes in Heavy Oil. In Proceedings of the SPE Annual Technical Conference and Exhibition, Denver, CO, USA, 21 September 2008. [Google Scholar]
- Ratnakar, R.; Dindoruk, B. On the Exact Representation of Pressure Decay Tests: New Modeling and Experimental Data for Diffusivity Measurement in Gas-Oil/Bitumen Systems. In Proceedings of the SPE Annual Technical Conference and Exhibition, Dubai, United Arab Emirates, 26 September 2016. [Google Scholar]
- Behzadfar, E.; Hatzikiriakos, S.G. Diffusivity of CO2 in Bitumen: Pressure–Decay Measurements Coupled with Rheometry. Energy Fuels 2014, 28, 1304–1311. [Google Scholar] [CrossRef]
- Upreti, S.R.; Mehrotra, A.K. Experimental Measurement of Gas Diffusivity in Bitumen: Results for Carbon Dioxide. Ind. Eng. Chem. Res. 2000, 39, 1080–1087. [Google Scholar] [CrossRef]
- Nguyen, T.; Ali, S. Effect of Nitrogen on the Solubility and Diffusivity of Carbon Dioxide into Oil and Oil Recovery by the Immiscible WAG Process. J. Can. Pet. Technol. 1998, 37, PETSOC-98-02-02. [Google Scholar] [CrossRef]
- Zheng, S.; Yang, D. Experimental and Theoretical Determination of Diffusion Coefficients of CO2-Heavy Oil Systems by Coupling Heat and Mass Transfer. J. Energy Resour. Technol. 2017, 139, 022901. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, Q.; Yuan, Q.; Zhang, L.; Feng, J.; Chu, B.; Zeng, F.; Zhu, G. Determining CO2 Diffusion Coefficient in Heavy Oil in Bulk Phase and in Porous Media Using Experimental and Mathematical Modeling Methods. Fuel 2020, 263, 116205. [Google Scholar] [CrossRef]
- Yang, C.; Gu, Y. A New Method for Measuring Solvent Diffusivity in Heavy Oil by Dynamic Pendant Drop Shape Analysis (DPDSA). SPE J. 2006, 11, 48–57. [Google Scholar] [CrossRef]
- Kavousi, A.; Torabi, F.; Chan, C.W.; Shirif, E. Experimental Measurement and Parametric Study of CO2 Solubility and Molecular Diffusivity in Heavy Crude Oil Systems. Fluid Phase Equilibria 2014, 371, 57–66. [Google Scholar] [CrossRef]
- Lou, X.; Chakraborty, N.; Karpyn, Z.T.; Ayala, L.F.; Nagarajan, N.; Wijaya, Z. Experimental Study of Gas/Liquid Diffusion in Porous Rocks and Bulk Fluids to Investigate the Effect of Rock-Matrix Hindrance. SPE J. 2021, 26, 1174–1188. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients in Hydrocarbon Systems. Methane in the Liquid Phase of the Methane-Santa Fe Springs Crude Oil System. J. Chem. Eng. Data 1959, 4, 15–21. [Google Scholar] [CrossRef]
- Reamer, H.; Duffy, C.; Sage, B. Methane-White Oil-Methane in Liquid Phase. Ind. Eng. Chem. 1956, 48, 285–288. [Google Scholar] [CrossRef]
- Ratnakar, R.R.; Dindoruk, B.; Odikpo, G.; Lewis, E.J. Measurement and Quantification of Diffusion-Induced Compositional Variations in Absence of Convective Mixing at Reservoir Conditions. Transp. Porous Media 2019, 128, 29–43. [Google Scholar] [CrossRef]
- Reamer, H.; Sage, B. Diffusion Coefficients of Ethane in the Liquid Phase of the Ethane-White Oil System. J. Chem. Eng. Data 1961, 6, 180–184. [Google Scholar] [CrossRef]
- Hill, E.; Lacey, W. Rate of Solution of Propane in Quiescent Liquid Hydrocarbons. Ind. Eng. Chem. 1934, 26, 1327–1331. [Google Scholar] [CrossRef]
- Dong, X.; Shi, Y.; Yang, D. Quantification of Mutual Mass Transfer of CO2/N2–Light Oil Systems by Dynamic Volume Analysis. Ind. Eng. Chem. Res. 2018, 57, 16495–16507. [Google Scholar] [CrossRef]
- Yang, D.; Gu, Y. Determination of Diffusion Coefficients and Interface Mass-Transfer Coefficients of the Crude Oil- CO2 System by Analysis of the Dynamic and Equilibrium Interfacial Tensions. Ind. Eng. Chem. Res. 2008, 47, 5447–5455. [Google Scholar] [CrossRef]
- Rajan, S.M.; Goren, S.L. Gas Absorption by Drops Traveling on a Vertical Wire. AIChE J. 1967, 13, 91–96. [Google Scholar] [CrossRef]
- Shu, G.; Dong, M.; Hassanzadeh, H.; Chen, S. Effects of Operational Parameters on Diffusion Coefficients of CO2 in a Carbonated Water–Oil System. Ind. Eng. Chem. Res. 2017, 56, 12799–12810. [Google Scholar] [CrossRef]
- Feng, S.; Li, C.; Peng, X.; Shao, L.; Liu, W. Digital Holography Interferometry for Measuring the Mass Diffusion Coefficients of N2 in RP-3 and RP-5 Jet Fuels. Aircr. Eng. Aerosp. Technol. 2019, 91, 1093–1099. [Google Scholar] [CrossRef]
- Tyn, M.; Calus, W. Estimating Liquid Molal Volume. Processing 1975, 21, 16–17. [Google Scholar]
- Hirschfelder, J.O.; Curtiss, C.F.; Bird, R.B.; Mayer, M.G. Molecular Theory of Gases and Liquids; Wiley: New York, NY, USA, 1954. [Google Scholar]
- Fairbanks, D.; Wilke, C. Diffusion Coefficients in Multicomponent Gas Mixtures. Ind. Eng. Chem. 1950, 42, 471–475. [Google Scholar] [CrossRef]
- Stiel, L.I.; Thodos, G. The Viscosity of Nonpolar Gases at Normal Pressures. AIChE J. 1961, 7, 611–615. [Google Scholar] [CrossRef]
- Jossi, J.A.; Stiel, L.I.; Thodos, G. The Viscosity of Pure Substances in the Dense Gaseous and Liquid Phases. AIChE J. 1962, 8, 59–63. [Google Scholar] [CrossRef]
- Fuller, E.N.; Schettler, P.D.; Giddings, J.C. New Method for Prediction of Binary Gas-Phase Diffusion Coefficients. Ind. Eng. Chem. 1966, 58, 18–27. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Fredenslund, A. An Improved Corresponding States Model for the Prediction of Oil and Gas Viscosities and Thermal Conductivities. Chem. Eng. Sci. 1987, 42, 182–186. [Google Scholar] [CrossRef]
- Gross, J.; Sadowski, G. Perturbed-Chain SAFT: An Equation of State Based on a Perturbation Theory for Chain Molecules. Ind. Eng. Chem. Res. 2001, 40, 1244–1260. [Google Scholar] [CrossRef]
- von Solms, N.; Michelsen, M.L.; Kontogeorgis, G.M. Computational and Physical Performance of a Modified PC-SAFT Equation of State for Highly Asymmetric and Associating Mixtures. Ind. Eng. Chem. Res. 2003, 42, 1098–1105. [Google Scholar] [CrossRef]
- Kunz, O.; Wagner, W. The GERG-2008 Wide-Range Equation of State for Natural Gases and Other Mixtures: An Expansion of GERG-2004. J. Chem. Eng. Data 2012, 57, 3032–3091. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Fredenslund, A.; Thomassen, P. Properties of Oils and Natural Gases; Gulf Publishing Company: Houston, TX, USA, 1989. [Google Scholar]
- Yan, W.; Varzandeh, F.; Stenby, E.H. PVT Modeling of Reservoir Fluids Using PC-SAFT EoS and Soave-BWR EoS. Fluid Phase Equilibria 2015, 386, 96–124. [Google Scholar] [CrossRef]
- Quiñones-Cisneros, S.E.; Zéberg-Mikkelsen, C.K.; Stenby, E.H. The Friction Theory (f-Theory) for Viscosity Modeling. Fluid Phase Equilibria 2000, 169, 249–276. [Google Scholar] [CrossRef]
- Quiñones-Cisneros, S.E.; Zéberg-Mikkelsen, C.K.; Stenby, E.H. One Parameter Friction Theory Models for Viscosity. Fluid Phase Equilibria 2001, 178, 1–16. [Google Scholar] [CrossRef]
- Aasberg-Petersen, K.; Knudsen, K.; Fredenslund, A. Prediction of Viscosities of Hydrocarbon Mixtures. Fluid Phase Equilibria 1991, 70, 293–308. [Google Scholar] [CrossRef]



















| N2 | 1 | 1 | ||||||||||||||||||||||||||
| CO2 | 2 | 2 | ||||||||||||||||||||||||||
| H2S | 3 | 3 | ||||||||||||||||||||||||||
| C1 | 4 | 4 | ||||||||||||||||||||||||||
| C2 | 5 | N | 5 | |||||||||||||||||||||||||
| C3 | 6 | N | 6 | |||||||||||||||||||||||||
| iC4 | 7 | 7 | ||||||||||||||||||||||||||
| C4 | 8 | 8 | ||||||||||||||||||||||||||
| iC5 | 9 | 9 | ||||||||||||||||||||||||||
| C5 | 10 | N | 10 | |||||||||||||||||||||||||
| C6 | 11 | 11 | ||||||||||||||||||||||||||
| C7 | 12 | 12 | ||||||||||||||||||||||||||
| C8 | 13 | 13 | ||||||||||||||||||||||||||
| C9 | 14 | N | 14 | |||||||||||||||||||||||||
| C10 | 15 | 15 | ||||||||||||||||||||||||||
| C11 | 16 | 16 | ||||||||||||||||||||||||||
| C12 | 17 | N | 17 | |||||||||||||||||||||||||
| C13 | 18 | 18 | ||||||||||||||||||||||||||
| C14 | 19 | 19 | ||||||||||||||||||||||||||
| C15 | 20 | 20 | ||||||||||||||||||||||||||
| C16 | 21 | N | 21 | |||||||||||||||||||||||||
| C17 | 22 | 22 | ||||||||||||||||||||||||||
| C18 | 23 | 23 | ||||||||||||||||||||||||||
| C20 | 24 | 24 | ||||||||||||||||||||||||||
| C24 | 25 | 25 | ||||||||||||||||||||||||||
| C28 | 26 | 26 | ||||||||||||||||||||||||||
| C32 | 27 | 27 |
| Systems | Phase | Method | Np | P (bar) | T (K) | x1 | Source |
|---|---|---|---|---|---|---|---|
| C1-C2 | G | LDC | 15 | 68.9–551.6 | 313–350 | 0.8 | [58] |
| G | CVD | 1 | 1.0 | 298 | - | [59] | |
| C1-C3 | G | LDC | 57 | 13.7–206.8 | 311–378 | 0.09–0.9 | [39] |
| L | CP | 22 | 24.9–66.6 | 278–344 | 0.07–0.33 | [60] | |
| G | NMR | 36 | 121.7–173.4 | 298–364 | 0.3–0.7 | [61] | |
| G | IF | 37 | 0.5–5.0 | 293–314 | 0.17–0.84 | [62] | |
| C1-C4 | G | LDC | 26 | 13.8–173.7 | 311–378 | 0.11–0.97 | [39] |
| L | CP | 19 | 19.6–100.0 | 278–378 | 0.1–0.5 | [63] | |
| C1-C5 | G/L | CVD | 8 | 94.3–149.5 | 295 | - | [64] |
| L | CVD | 1 | 71.0 | 311 | - | [65] | |
| G/L | CVD | 2 | 102.0 | 311 | - | [66] | |
| L | CP | 41 | 17.2–155.1 | 278–411 | 0.02–0.69 | [67] | |
| L | CP | 1 | 20.7 | 303 | 0.1 | [68] | |
| C1-C6 | G | Stefan | 4 | 1.0 | 283–328 | - | [69] |
| L | CC | 1 | 1.0 | 298 | 0 | [70] | |
| L | CP | 1 | 20.7 | 303 | 0.11 | [68] | |
| G | Stefan | 5 | 1.0 | 298–333 | - | [71] | |
| C1-C7 | G | Stefan | 5 | 1.0 | 283–343 | - | [69] |
| L | CP | 36 | 23.1–210.3 | 278–444 | 0.08–0.75 | [72] | |
| L | TD | 19 | 10.2–629.9 | 323–398 | 0 | [73] | |
| L | CC | 1 | 1.0 | 298 | 0 | [70] | |
| L | CP | 1 | 20.7 | 303 | 0.1 | [68] | |
| C1-C8 | G | Stefan | 5 | 1.0 | 283–343 | - | [69] |
| L | CC | 1 | 1.0 | 298 | 0 | [70] | |
| L | Acoustic | 4 | 17.2 | 281–312 | - | [74] | |
| L | IF | 6 | 17.2 | 301–424 | 0.03 | [75] | |
| G/L | CVD | 12 | 93.9–183.7 | 295 | - | [64] | |
| L | TD | 6 | 17.2 | 304–435 | 0 | [76] | |
| C1-C9 | L | Acoustic | 4 | 17.2 | 281–312 | - | [74] |
| C1-C10 | L | CP | 40 | 34.5–275.8 | 278–411 | 0.13–0.7 | [77] |
| L | Acoustic | 4 | 17.2 | 281–312 | - | [74] | |
| G/L | CVD | 4 | 97.2–180.5 | 295 | - | [64] | |
| L | IF | 9 | 200.6- 598.0 | 303–423 | 0.1 | [78] | |
| L | IF | 24 | 250.0- 600.0 | 303–304 | 0 | [79] | |
| C1-C12 | L | CC | 3 | 1.0 | 273–323 | 0.11–0.97 | [70] |
| L | CP | 33 | 40.0–350.0 | 318–354 | 0 | [80] | |
| L | CVD | 3 | 35.0 | 273–298 | - | [81] | |
| L | CP | 2 | 34.5–34.6 | 318–338 | - | [82] | |
| C1-C16 | L | CC | 1 | 1.0 | 298 | - | [70] |
| L | CVD | 2 | 60.1–63.8 | 298–353 | 0 | [83] | |
| L | TD | 8 | 8.6 | 302–541 | 0 | [84] | |
| G/L | CVD | 8 | 95.6–179.6 | 299 | - | [64] | |
| L | CVD | 2 | 35.1–35.4 | 293–298 | 0 | [81] | |
| C2-C3 | G | CVD | 1 | 1.0 | 298 | - | [59] |
| C2-C5 | L | CP | 26 | 3.2–46.4 | 278–411 | - | [85] |
| C2-C6 | L | CC | 1 | 1.0 | 298 | - | [86] |
| L | CC | 1 | 1.0 | 303 | 0.06–0.8 | [87] | |
| C2-C7 | L | CC | 1 | 1.0 | 298 | 0.03 | [86] |
| L | CC | 2 | 1.0 | 303–313 | 0.03 | [87] | |
| C2-C8 | L | TD | 6 | 17.2 | 304–435 | 0.03 | [76] |
| L | CC | 1 | 1.0 | 298 | 0.03 | [86] | |
| C2-C10 | L | CP | 30 | 3.3–73.8 | 278–478 | 0 | [88] |
| C2-C12 | L | CC | 1 | 1.0 | 298 | 0.03 | [86] |
| C2-C16 | L | TD | 8 | 8.6 | 301–540 | 0.04–0.54 | [84] |
| L | CC | 1 | 1.0 | 298 | 0.04 | [86] | |
| C3-C6 | L | CC | 1 | 1.0 | 298 | 0 | [89] |
| C3-C7 | L | CC | 1 | 1.0 | 298 | 0.04 | [89] |
| C3-C8 | L | TD | 6 | 17.2 | 304–435 | 0.12 | [76] |
| L | CC | 1 | 1.0 | 298 | 0.12 | [89] | |
| C3-C16 | L | CC | 1 | 1.0 | 298 | 0 | [89] |
| L | TD | 8 | 8.6 | 299–540 | 0.12 | [84] | |
| C4-C10 | L | CP | 2 | 6.9 | 377–411 | 0.14 | [90] |
| L | CP | 8 | 6.8–20.8 | 378–444 | 0 | [91] | |
| C4-C16 | L | TD | 8 | 8.6 | 298–542 | - | [84] |
| C5-C6 | L | IF | 1 | 1.0 | 298 | 0.25–0.71 | [92] |
| C5-C8 | L | TD | 6 | 17.2 | 304–435 | 0 | [76] |
| C5-C16 | L | TD | 8 | 8.6 | 300–541 | 0 | [84] |
| C6-C7 | L | IF | 1 | 1.0 | 298 | 0 | [92] |
| L | TD | 28 | 1.0 | 300–333 | 0 | [93] | |
| L | TD | 10 | 1.0 | 283–298 | 1 | [94] | |
| L | TD | 1 | 1.0 | 297 | 0–1 | [95] | |
| C6-C8 | L | IF | 1 | 1.0 | 298 | 0–1 | [92] |
| L | TD | 30 | 1.0 | 295–333 | 1 | [93] | |
| C6-C10 | L | IF | 1 | 1.0 | 298 | 1 | [92] |
| C6-C12 | L | IF | 1 | 1.0 | 298 | 0–1 | [92] |
| L | CDM | 2 | 1.0 | 298 | 1 | [96] | |
| L | IF | 7 | 1.0 | 298 | 1 | [97] | |
| L | IF | 2 | 1.0 | 298 | 0.98–1 | [98] | |
| L | IF | 27 | 1.0 | 298–308 | 0.02–0.99 | [99] | |
| C6-C16 | L | IF | 1 | 1.0 | 298 | 0–1 | [92] |
| L | IF | 8 | 1.0 | 298 | 0–1 | [100] | |
| L | IF | 2 | 1.0 | 298 | 1 | [98] | |
| C6-C18 | L | IF | 1 | 1.0 | 298 | 0.01–1 | [92] |
| C6-C24 | L | CDM | 1 | 1.0 | 298 | 0–1 | [96] |
| C6-C32 | L | CDM | 1 | 1.0 | 298 | 1 | [96] |
| C7-C8 | L | TD | 4 | 1.0–34.8 | 299–477 | 1 | [101] |
| L | TD | 10 | 1.0 | 298–308 | 1 | [102] | |
| L | TD | 25 | 1.0 | 293–343 | 1 | [93] | |
| L | TD | 6 | 17.2 | 304–435 | 1 | [76] | |
| C7-C10 | L | TD | 5 | 1.0–34.8 | 299–477 | 0–1 | [101] |
| L | DC | 21 | 1.0 | 298 | 0 | [103] | |
| C7-C12 | L | TD | 5 | 1.0–35.5 | 299–477 | 1 | [101] |
| L | DC | 20 | 1.0 | 298 | 0.04–0.96 | [103] | |
| L | DC | 3 | 1.0 | 298–338 | 1 | [104] | |
| L | TD | 5 | 1.0 | 298 | 0.08–0.97 | [102] | |
| C7-C14 | L | TD | 5 | 1.0–35.5 | 299–477 | 0.5 | [101] |
| L | DC | 23 | 1.0 | 298 | 0–1 | [103] | |
| L | DC | 1 | 1.0 | 298 | 1 | [104] | |
| C7-C16 | L | TD | 8 | 1.0–35.5 | 299–477 | 0.05–0.97 | [101] |
| L | DC | 3 | 1.0 | 298–338 | 0.5 | [104] | |
| L | IF | 7 | 1.0 | 298 | 1 | [97] | |
| C7-C18 | L | DC | 1 | 1.0 | 298 | 0.5 | [104] |
| C8-C10 | L | TD | 6 | 17.2 | 304–435 | 0.02–0.99 | [76] |
| C8-C12 | L | TD | 9 | 14.1–34.4 | 304–566 | 0.5 | [101] |
| L | DC | 21 | 1.0 | 298–333 | 1 | [105] | |
| L | TD | 6 | 17.2 | 304–435 | 0 | [76] | |
| C8-C14 | L | DC | 24 | 1.0 | 298 | 0.14–0.93 | [103] |
| L | TD | 6 | 17.2 | 304–435 | 1 | [76] | |
| C8-C16 | L | TD | 10 | 14.2–35.1 | 323–564 | 0.04–0.98 | [106] |
| L | TD | 4 | 8.6 | 420–542 | 1 | [84] | |
| L | IF | 2 | 1.0 | 298 | 0 | [99] | |
| C8-C20 | L | TD | 5 | 13.8 | 375–534 | 0 | [107] |
| C8-C24 | L | CDM | 1 | 1.0 | 298 | 0–1 | [96] |
| C8-C28 | L | TD | 5 | 13.8 | 373–534 | 0 | [108] |
| C8-C32 | L | CDM | 1 | 1.0 | 298 | 0.99 | [96] |
| C10-C12 | L | TD | 5 | 14.1–14.5 | 304–566 | 0 | [101] |
| C10-C16 | L | TD | 5 | 14.2–14.4 | 323–564 | 1 | [106] |
| L | TD | 4 | 8.6 | 420–540 | 0 | [84] | |
| L | IF | 11 | 1.0 | 298 | 0 | [99] | |
| C10-C20 | L | TD | 2 | 13.8 | 413–495 | 0 | [107] |
| C12-C14 | L | TD | 5 | 14.1–14.5 | 304–566 | 0–1 | [101] |
| C12-C16 | L | TD | 5 | 14.1–14.5 | 304–566 | 0 | [101] |
| L | TD | 5 | 14.2–14.4 | 323–564 | 1 | [106] | |
| L | IF | 2 | 1.0 | 298 | 1 | [98] | |
| L | TD | 4 | 8.6 | 421–541 | 0 | [84] | |
| C12-C20 | L | TD | 5 | 13.8 | 375–534 | 0–1 | [107] |
| C12-C24 | L | CDM | 1 | 1.0 | 298 | 0 | [96] |
| C12-C28 | L | TD | 5 | 13.8 | 373–534 | 0 | [108] |
| C14-C16 | L | TD | 5 | 14.2–14.4 | 323–564 | 0.56 | [106] |
| L | TD | 4 | 8.6 | 419–541 | 0 | [84] | |
| C14-C20 | L | TD | 2 | 1.0–629.9 | 273–566 | 0 | [107] |
| C16-C20 | L | TD | 5 | 13.8 | 375–534 | 0 | [107] |
| C16-C28 | L | TD | 5 | 13.8 | 373–534 | 0–1 | [108] |
| C1-N2 | G | LDC | 5 | 34.5–172.4 | 313 | 0 | [58] |
| G | Other | 3 | 13.8 | 371–534 | 0 | [109] | |
| G | LDC | 26 | 13.9–139.3 | 313–366 | 0.8 | [39] | |
| C2-N2 | G | CP | 5 | 34.5–172.4 | 313 | - | [58] |
| C2-CO2 | G | CVD | 1 | 1.0 | 298 | 0.5 | [59] |
| N2-CO2 | G | Other | 34 | 1.0 | 1081–1810 | 0.8 | [110] |
| G | CVD | 6 | 1.0 | 288–290 | - | [111] | |
| N2-C5 | G | Stefan | 9 | 1.0 | 258–298 | 0.1–0.7 | [112] |
| G/L | CVD | 8 | 94.3–149.5 | 295 | - | [64] | |
| N2-C8 | L | IF | 5 | 150.0 | 303–423 | - | [75] |
| G/L | CVD | 4 | 98.7–179.4 | 295 | - | [64] | |
| N2-C10 | L | IF | 12 | 75.0–150.0 | 291–422 | 0.06 | [75] |
| G/L | CVD | 4 | 97.2–184.6 | 295 | - | [64] | |
| N2-C16 | G/L | CVD | 6 | 93.6–177.8 | 299 | 0.08 | [64] |
| CO2-C3 | G | CVD | 2 | 1.0 | 298 | - | [113] |
| G | IF | 35 | 0.5–5.0 | 293–313 | - | [62] | |
| CO2-C5 | L | HCT | 9 | 19.2–38.4 | 298 | - | [114] |
| L | TD | 5 | 90.0–105.0 | 299–308 | 0.17–0.83 | [115] | |
| CO2-C6 | L | DC | 1 | 1.0 | 298 | 0.28–0.69 | [116] |
| L | DC | 1 | 1.0 | 298 | 1 | [117] | |
| L | TD | 5 | 90.0–105.0 | 299–308 | - | [115] | |
| L | TD | 16 | 12.0–660.0 | 298–423 | - | [118] | |
| CO2-C7 | L | Other | 1 | 1.0 | 298 | 1 | [119] |
| L | DC | 1 | 1.0 | 298 | 0 | [116] | |
| L | TD | 5 | 90.0–105.0 | 299–308 | - | [115] | |
| L | TD | 30 | 10.0–680.0 | 298–423 | - | [118] | |
| L | Other | 1 | 34.5 | 333 | 1 | [120] | |
| L | Other | 33 | 17.2–73.1 | 283–323 | 0 | [121] | |
| L | Other | 17 | 78.1–108.6 | 310–341 | - | [122] | |
| CO2-C8 | L | CP | 10 | 7.5–36.7 | 290–311 | 0.3–0.89 | [123] |
| L | TD | 5 | 90.0–105.0 | 299–308 | 0.96–0.99 | [115] | |
| L | TD | 30 | 9.0–690.0 | 298–423 | 0.13–0.62 | [118] | |
| CO2-C9 | L | TD | 5 | 90.0–105.0 | 299–308 | 1 | [115] |
| CO2-C10 | L | HCT | 15 | 16.5–56.4 | 298 | 0 | [114] |
| L | TD | 5 | 90.0–105.0 | 299–308 | 1 | [115] | |
| L | TD | 16 | 11.0–680.0 | 298–423 | 0.2–0.82 | [118] | |
| CO2-C11 | L | TD | 5 | 90.0–105.0 | 299–308 | 1 | [115] |
| CO2-C12 | L | TD | 9 | 13.9–34.5 | 304–567 | 0 | [106] |
| L | TD | 5 | 90.0–105.0 | 299–308 | 1 | [115] | |
| L | TD | 15 | 12.0–650.0 | 298–423 | 0 | [118] | |
| L | CVD | 3 | 35.4–36.0 | 273–298 | 1 | [81] | |
| CO2-C14 | L | CP | 24 | 9.1–40.4 | 290–331 | 0 | [123] |
| L | TD | 5 | 90.0–105.0 | 299–308 | - | [115] | |
| L | CM | 2 | 1.0 | 298–323 | 0.18–0.59 | [124] | |
| CO2-C16 | L | HCT | 11 | 31.0–52.9 | 298 | 1 | [114] |
| L | PDM | 23 | 70.0–190.0 | 300–353 | - | [125] | |
| L | TD | 10 | 14.0–34.6 | 323–564 | 0.34–0.58 | [106] | |
| L | TD | 27 | 10.0–690.0 | 298–423 | 0.69–0.84 | [118] | |
| L | CC | 2 | 1.0 | 298–323 | 0 | [86] | |
| L | Other | 1 | 69.0 | 313 | 0 | [120] | |
| L | CVD | 2 | 35.6–36.1 | 293–298 | 0.01 | [81] | |
| CO2-C20 | L | TD | 5 | 13.8 | 374–533 | - | [107] |
| CO2-C28 | L | TD | 5 | 13.8 | 371–534 | - | [108] |
| Parameters | Models |
|---|---|
| Density | PR, SRK, SAFT, GERG *, PR-VT, SRK-VT |
| Viscosity | FT-PR *, FT-SRK, LBC-PR, LBC-PRVT, CS2 |
| Low-pressure density-diffusivity product | Chapman–Enskog *, Fuller |
| Composition derivatives of fugacity coefficients | GERG, PR*, SRK, SAFT |
| Type | Systems | Np | AAD% |
|---|---|---|---|
| Refined oil products | CO2 + kerosine [119] | 1 | 37.3 |
| CO2 + white oil [197] | 2 | 60.8 | |
| C1 + white oil [191] | 5 | 25.0 | |
| C2 + white oil [193] | 4 | 55.6 | |
| C3 + kerosene [194] | 19 | 60.3 | |
| C3 + spray oil [194] | 12 | 31.1 | |
| Average | 45.0 | ||
| Ordinary crude oil | CO2-crude oil [119,169,196,198] | 17 | 600.0 |
| Heavy oil and bitumen | N2 [146] | 4 | 70.2 |
| CO2 [82,146,151,163,165,170,171,173,176,177,178,183,184,186,188] | 31 | 89.9 | |
| C1 [82,146,165,170,171,172,173,174] | 20 | 90.5 | |
| C2 [146,172] | 7 | 96.1 | |
| C3 [161,164,171,172,177,178] | 15 | 94.6 | |
| C4 [176,178] | 2 | 99.8 | |
| C5 [158,159,160,179] | 48 | 99.1 | |
| C6 [179] | 1 | 100.0 | |
| C7 [179] | 8 | 99.9 | |
| Average | 93.3 | ||
| Total | 196 | 106.9 |
| Systems | ES | RW | WC | HM | Viscosity |
|---|---|---|---|---|---|
| Light oils | |||||
| C1 in light oil [189] | −83.5 | 119.1 | 33.1 | 135.7 | LBC |
| CO2 in live oil [154] | −0.5 | 769.8 | 1065.7 | 1623.1 | LBC |
| CO2 in STO [154] | −57.7 | −38.4 | −73.7 | −28.9 | LBC |
| C1 in live oil [152] | −56.0 | −2.7 | −50.1 | −29.4 | LBC |
| Heavy oils | |||||
| CO2 in heavy oil [176] | 2709.5 | −72.0 | −98.2 | −67.9 | Paper |
| CO2 in heavy oil [176] | 1077.2 | −88.2 | −99.3 | −86.7 | Paper |
| CO2 in heavy oil [176] | 2794.4 | −96.9 | −99.9 | −94.1 | Paper |
| CO2 in heavy oil [185] | 2677.7 | −93.8 | −99.9 | −92.0 | Paper |
| CO2 in heavy oil [185] | 1145.7 | −93.4 | −99.7 | −91.1 | Paper |
| CO2 in heavy oil [185] | 516.8 | −94.0 | −99.5 | −91.5 | Paper |
| AAD | |||||
| Light oils | 49.4 | 232.5 | 305.7 | 454.3 | |
| Heavy oils | 1820.2 | 89.7 | 99.4 | 87.2 | |
| All | 1111.9 | 146.8 | 181.9 | 234.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Stenby, E.H.; Shapiro, A.A.; Yan, W. Diffusion Coefficients in Systems Related to Reservoir Fluids: Available Data and Evaluation of Correlations. Processes 2022, 10, 1554. https://doi.org/10.3390/pr10081554
Yang Y, Stenby EH, Shapiro AA, Yan W. Diffusion Coefficients in Systems Related to Reservoir Fluids: Available Data and Evaluation of Correlations. Processes. 2022; 10(8):1554. https://doi.org/10.3390/pr10081554
Chicago/Turabian StyleYang, Yibo, Erling H. Stenby, Alexander A. Shapiro, and Wei Yan. 2022. "Diffusion Coefficients in Systems Related to Reservoir Fluids: Available Data and Evaluation of Correlations" Processes 10, no. 8: 1554. https://doi.org/10.3390/pr10081554