
The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
Rationale
2. Results and Discussion
2.1. In Silico Studies
2.1.1. Docking Study
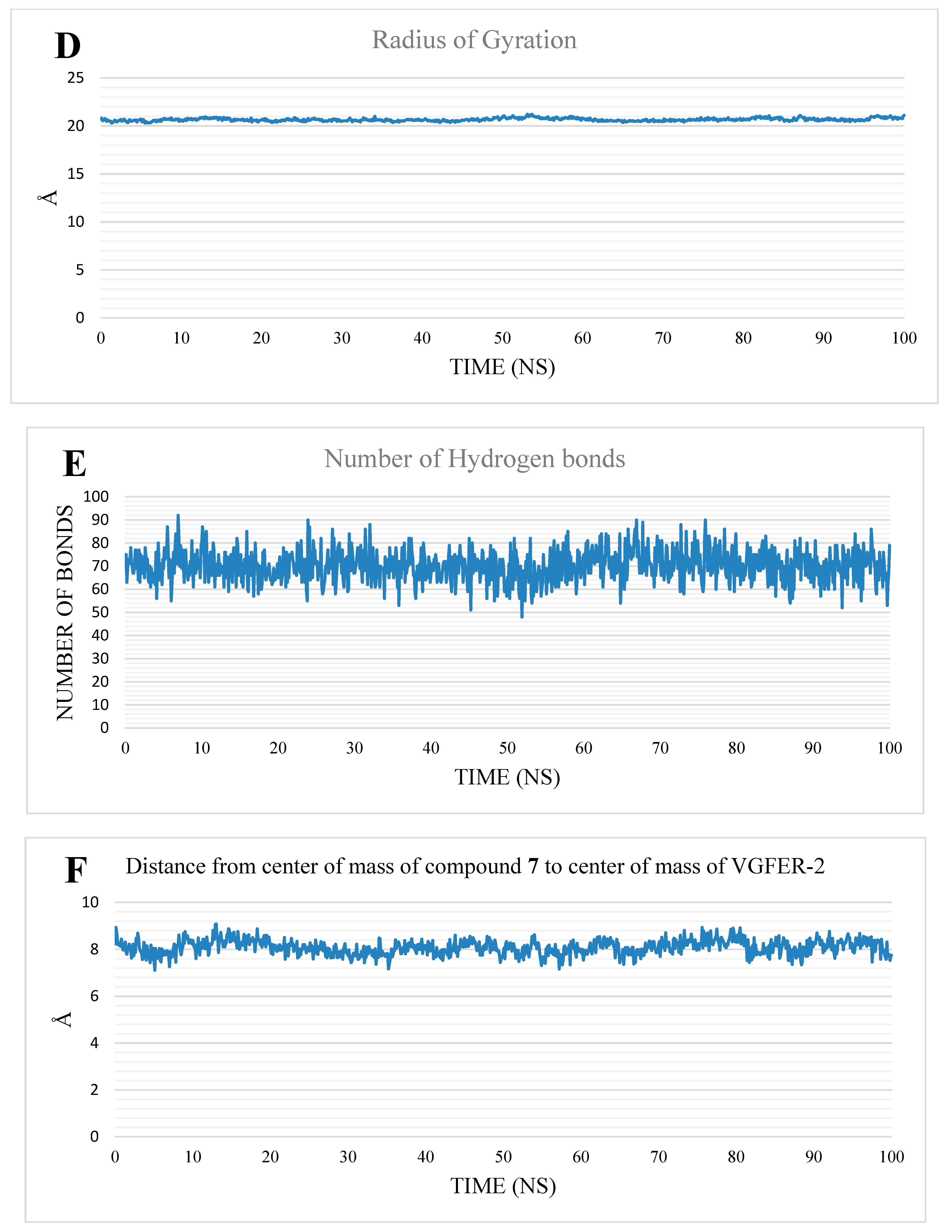
2.1.2. MD Studies
2.1.3. MM-GBSA
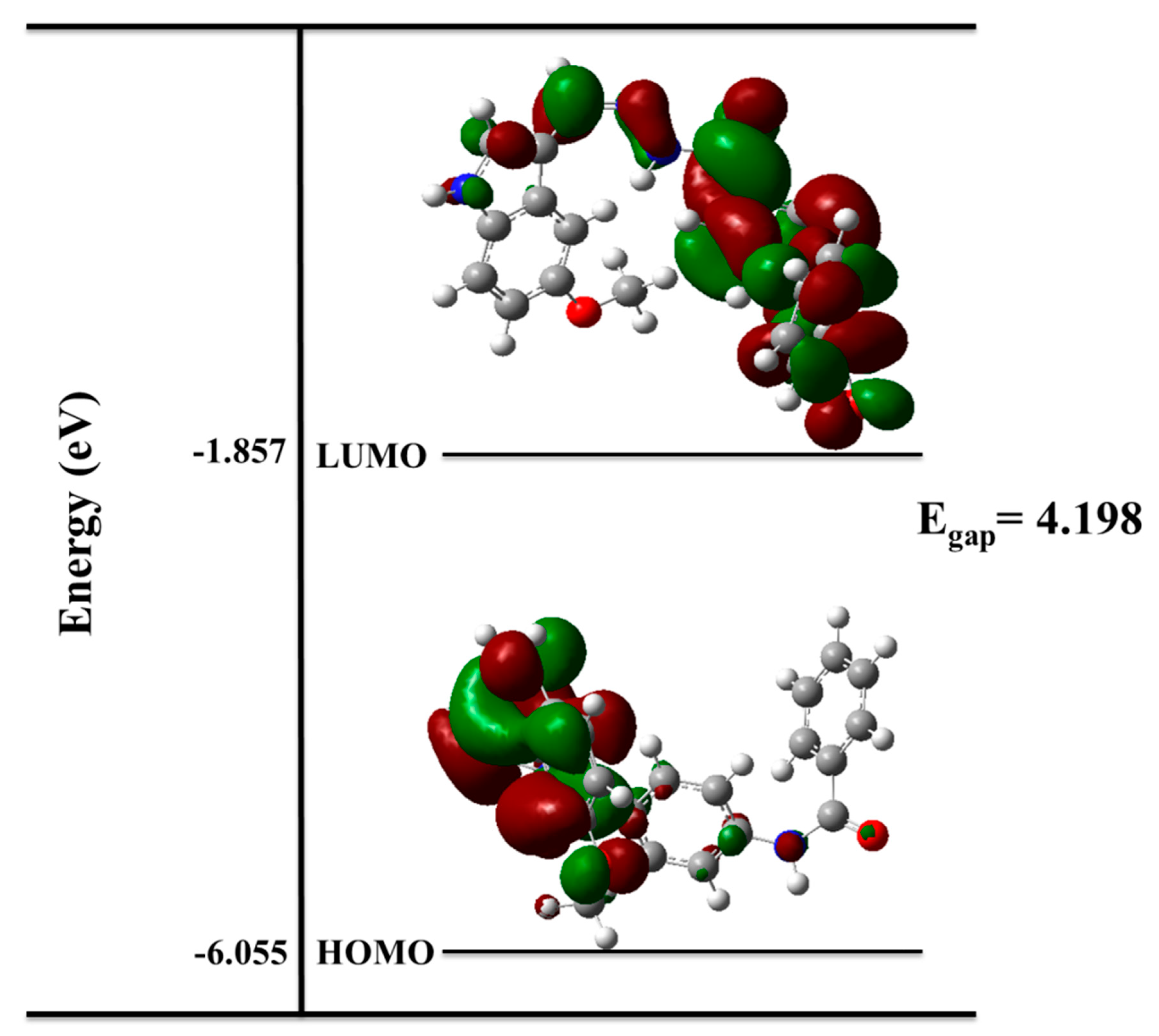
2.1.4. Density Functional Theory (DFT)
Molecular Structure Optimization
Quantum Chemistry Calculations
The Electron Density Maps
2.1.5. ADMET Profiling Study
2.1.6. In Silico Toxicity Studies
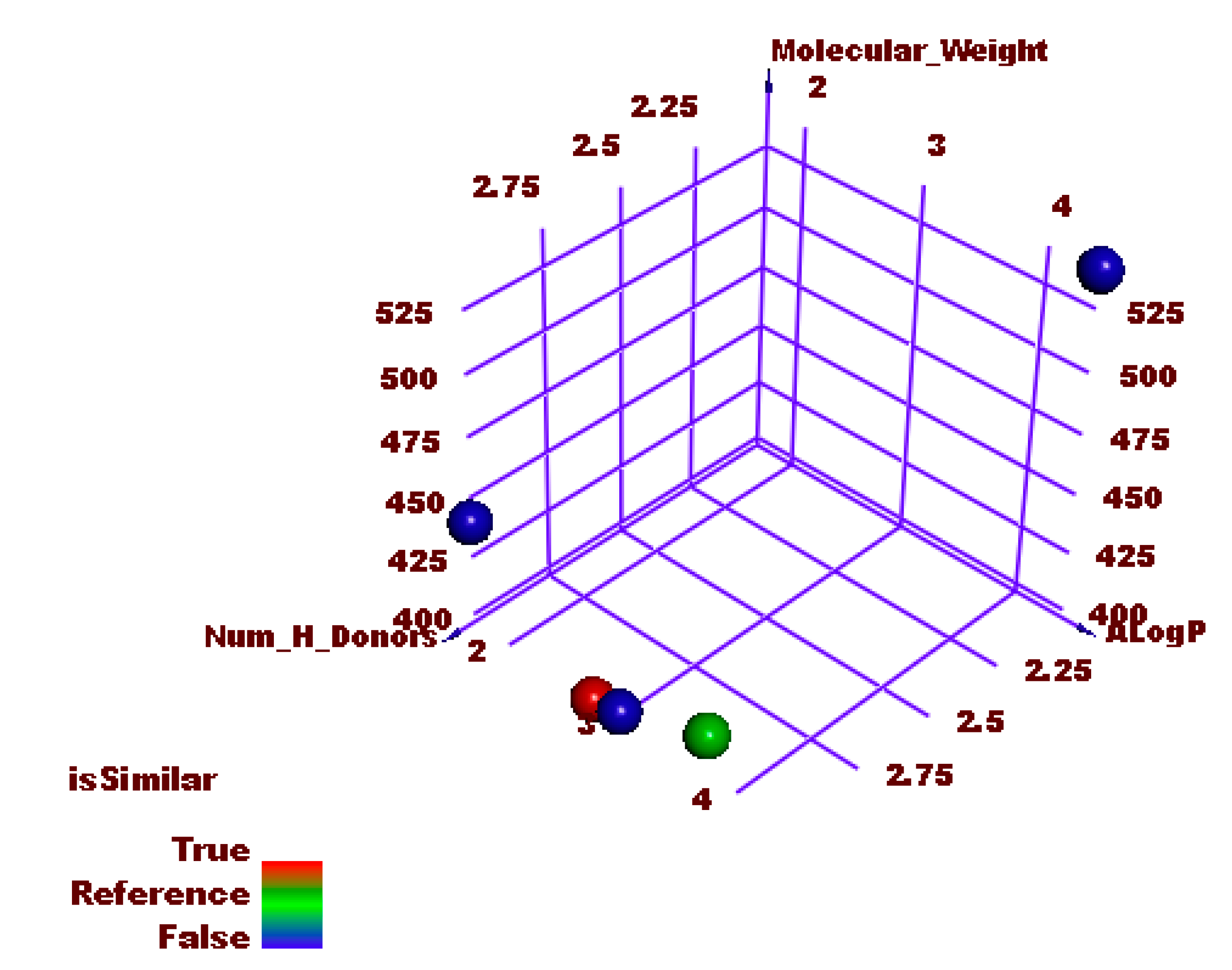
2.1.7. Molecular Similarity
Similarity Check Using Physical Properties
Flexible Alignment
2.2. Chemistry
2.3. Biological Examinations
2.3.1. VGFER-2 Inhibition
2.3.2. Cytotoxicity
2.3.3. Safety Evaluation
3. Conclusions
4. Experimental
4.1. In Silico Studies
4.1.1. Docking Studies
4.1.2. MD Simulation
4.1.3. MM-GBSA
4.1.4. DFT
4.1.5. ADMET Studies
4.1.6. Toxicity Studies
4.2. Chemistry
4.2.1. General Procedure for the Synthesis of Compound 5

4.2.2. General Procedure for the Synthesis of Compound 7

4.3. Biological Studies
4.3.1. In Vitro VEGFR-2 Inhibition
4.3.2. In Vitro Antiproliferative Activity
4.3.3. Safety Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Cancer, Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 13 June 2022).
- El-Dash, Y.; Elzayat, E.; Abdou, A.M.; Hassan, R.A. Novel thienopyrimidine-aminothiazole hybrids: Design, synthesis, antimicrobial screening, anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis and VEGFR-2 inhibition. Bioorg. Chem. 2021, 114, 105137. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.R.; Muñoz-Chápuli, R.; Medina, M.A. Anti-angiogenic drugs: From bench to clinical trials. Med. Res. Rev. 2006, 26, 483–530. [Google Scholar] [CrossRef] [PubMed]
- El-Zahabi, M.A.; Sakr, H.; El-Adl, K.; Zayed, M.; Abdelraheem, A.S.; Eissa, S.I.; Elkady, H.; Eissa, I.H. Design, synthesis, and biological evaluation of new challenging thalidomide analogs as potential anticancer immunomodulatory agents. Bioorg. Chem. 2020, 104, 104218. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.; Albassam, H.; Dahab, M.A.; Mahdy, H.A. Identification of new [1, 2, 4] triazolo [4, 3-a] quinoxalines as potent VEGFR-2 tyrosine kinase inhibitors: Design, synthesis, anticancer evaluation, and in silico studies. Bioorg. Med. Chem. 2021, 46, 116384. [Google Scholar] [CrossRef]
- Elrazaz, E.Z.; Serya, R.A.; Ismail, N.S.; Albohy, A.; Abou El Ella, D.A.; Abouzid, K.A. Discovery of Potent Thieno [2, 3-d] pyrimidine VEGFR-2 Inhibitors: Design, Synthesis and Enzyme Inhibitory Evaluation Supported by Molecular Dynamics Simulations. Bioorg. Chem. 2021, 113, 105019. [Google Scholar] [CrossRef]
- Sana, S.; Reddy, V.G.; Bhandari, S.; Reddy, T.S.; Tokala, R.; Sakla, A.P.; Bhargava, S.K.; Shankaraiah, N. Exploration of carbamide derived pyrimidine-thioindole conjugates as potential VEGFR-2 inhibitors with anti-angiogenesis effect. Eur. J. Med. Chem. 2020, 200, 112457. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Aucar, M.G.; Adler, N.S. Computational chemistry in drug lead discovery and design. Int. J. Quantum Chem. 2019, 119, e25678. [Google Scholar] [CrossRef] [Green Version]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; El-Gamal, K.M.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; Elsohly, M.A. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4 (3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorg. Chem. 2020, 94, 103422. [Google Scholar] [CrossRef]
- Eissa, I.H.; El-Helby, A.-G.A.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A. Discovery of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2020, 105, 104380. [Google Scholar] [CrossRef]
- Eissa, I.H.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4 (3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg. Chem. 2021, 107, 104532. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4 (3H)-ones as potential VEGFR-2 inhibitors. Bioorg. Med. Chem. 2021, 29, 115872. [Google Scholar] [CrossRef] [PubMed]
- Yousef, R.G.; Ibrahim, A.; Khalifa, M.M.; Eldehna, W.M.; Gobaara, I.M.; Mehany, A.B.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I.H. Discovery of new nicotinamides as apoptotic VEGFR-2 inhibitors: Virtual screening, synthesis, anti-proliferative, immunomodulatory, ADMET, toxicity, and molecular dynamic simulation studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- El-Helby, A.G.A.; Sakr, H.; Eissa, I.H.; Abulkhair, H.; Al-Karmalawy, A.A.; El-Adl, K. Design, synthesis, molecular docking, and anticancer activity of benzoxazole derivatives as VEGFR-2 inhibitors. Arch. Der Pharm. 2019, 352, 1900113. [Google Scholar] [CrossRef] [PubMed]
- Saleh, N.M.; Abdel-Rahman, A.A.H.; Omar, A.M.; Khalifa, M.M.; El-Adl, K. Pyridine-derived VEGFR-2 inhibitors: Rational design, synthesis, anticancer evaluations, in silico ADMET profile, and molecular docking. Arch. Der Pharm. 2021, 354, 2100085. [Google Scholar] [CrossRef]
- Taghour, M.S.; Elkady, H.; Eldehna, W.M.; El-Deeb, N.M.; Kenawy, A.M.; Elkaeed, E.B.; Alsfouk, A.A.; Alesawy, M.S.; Metwaly, A.M.; Eissa, I.H. Design and synthesis of thiazolidine-2, 4-diones hybrids with 1, 2-dihydroquinolones and 2-oxindoles as potential VEGFR-2 inhibitors: In-vitro anticancer evaluation and in-silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 1903–1917. [Google Scholar] [CrossRef]
- Parmar, D.R.; Soni, J.Y.; Guduru, R.; Rayani, R.H.; Kusurkar, R.V.; Vala, A.G.; Talukdar, S.N.; Eissa, I.H.; Metwaly, A.M.; Khalil, A. Discovery of new anticancer thiourea-azetidine hybrids: Design, synthesis, in vitro antiproliferative, SAR, in silico molecular docking against VEGFR-2, ADMET, toxicity, and DFT studies. Bioorg. Chem. 2021, 115, 105206. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; El-Adl, K.; El-Hddad, S.S.; Elhady, M.M.; Saleh, N.M.; Khalifa, M.M.; Khedr, F.; Alswah, M.; Nayl, A.A.; Ghoneim, M.M. Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2, 4-diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPARγ Agonists. Pharmaceuticals 2022, 15, 226. [Google Scholar] [CrossRef]
- El-Adl, K.; Sakr, H.M.; Yousef, R.G.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; Eissa, I.H. Discovery of new quinoxaline-2 (1H)-one-based anticancer agents targeting VEGFR-2 as inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2021, 114, 105105. [Google Scholar] [CrossRef]
- Yousef, R.G.; Sakr, H.M.; Eissa, I.H.; Mehany, A.B.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Abulkhair, H.S.; El-Adl, K. New quinoxaline-2 (1 H)-ones as potential VEGFR-2 inhibitors: Design, synthesis, molecular docking, ADMET profile and anti-proliferative evaluations. New J. Chem. 2021, 45, 16949–16964. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Mahdy, H.A.; Alsaif, N.A.; Obaidullah, A.J.; Alkahtani, H.M.; Al-Mehizia, A.A.; Alsubaie, S.M.; Dahab, M.A.; Eissa, I.H. New bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, in silico studies, and anticancer evaluation. Bioorg. Chem. 2021, 112, 104949. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Eissa, I.H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Alasmari, A.F.; Albassam, H.; Elkady, H.; Elwan, A. Design, synthesis, docking, ADMET studies, and anticancer evaluation of new 3-methylquinoxaline derivatives as VEGFR-2 inhibitors and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1760–1782. [Google Scholar] [CrossRef]
- Alanazi, M.M.; Elkady, H.; Alsaif, N.A.; Obaidullah, A.J.; Alanazi, W.A.; Al-Hossaini, A.M.; Alharbi, M.A.; Eissa, I.H.; Dahab, M.A. Discovery of new quinoxaline-based derivatives as anticancer agents and potent VEGFR-2 inhibitors: Design, synthesis, and in silico study. J. Mol. Struct. 2021, 1253, 132220. [Google Scholar] [CrossRef]
- El-Metwally, S.A.; Abou-El-Regal, M.M.; Eissa, I.H.; Mehany, A.B.; Mahdy, H.A.; Elkady, H.; Elwan, A.; Elkaeed, E.B. Discovery of thieno [2, 3-d] pyrimidine-based derivatives as potent VEGFR-2 kinase inhibitors and anti-cancer agents. Bioorg. Chem. 2021, 112, 104947. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; Alesawy, M.S.; Saleh, A.M.; Elkaeed, E.B.; Alsfouk, B.A.; El-Attar, A.-A.M.; Metwaly, A.M. Ligand and structure-based in silico determination of the most promising SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase complex inhibitors among 3009 FDA approved drugs. Molecules 2022, 27, 2287. [Google Scholar] [CrossRef] [PubMed]
- Alsaif, N.A.; Dahab, M.A.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Mahdy, H.A.; Elkady, H. New quinoxaline derivatives as VEGFR-2 inhibitors with anticancer and apoptotic activity: Design, molecular modeling, and synthesis. Bioorg. Chem. 2021, 110, 104807. [Google Scholar] [CrossRef] [PubMed]
- Elkaeed, E.B.; Elkady, H.; Belal, A.; Alsfouk, B.A.; Ibrahim, T.H.; Abdelmoaty, M.; Arafa, R.K.; Metwaly, A.M.; Eissa, I.H. Multi-Phase In Silico Discovery of Potential SARS-CoV-2 RNA-Dependent RNA Polymerase Inhibitors among 3009 Clinical and FDA-Approved Related Drugs. Processes 2022, 10, 530. [Google Scholar] [CrossRef]
- Mohammed, S.O.; El Ashry, E.S.H.; Khalid, A.; Amer, M.R.; Metwaly, A.M.; Eissa, I.H.; Elkaeed, E.B.; Elshobaky, A.; Hafez, E.E. Expression, Purification, and Comparative Inhibition of Helicobacter pylori Urease by Regio-Selectively Alkylated Benzimidazole 2-Thione Derivatives. Molecules 2022, 27, 865. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Arenz, C.; Ishmuratova, M.Y.; Toppet, S.; Dehaen, W.; Alsfouk, B.A.; Elkaeed, E.B.; Eissa, I.H. Jusanin, a New Flavonoid from Artemisia commutata with an In Silico Inhibitory Potential against the SARS-CoV-2 Main Protease. Molecules 2022, 27, 1636. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Ishmuratova, M.Y.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H.; Metwaly, A.M. Isolation and In Silico SARS-CoV-2 Main Protease Inhibition Potential of Jusan Coumarin, a New Dicoumarin from Artemisia glauca. Molecules 2022, 27, 2281. [Google Scholar] [CrossRef]
- Eissa, I.H.; Khalifa, M.M.; Elkaeed, E.B.; Hafez, E.E.; Alsfouk, A.A.; Metwaly, A.M. In silico exploration of potential natural inhibitors against SARS-CoV-2 nsp10. Molecules 2021, 26, 6151. [Google Scholar] [CrossRef]
- Suleimen, Y.M.; Jose, R.A.; Suleimen, R.N.; Arenz, C.; Ishmuratova, M.; Toppet, S.; Dehaen, W.; Alsfouk, A.A.; Elkaeed, E.B.; Eissa, I.H. Isolation and In Silico Anti-SARS-CoV-2 Papain-Like Protease Potentialities of Two Rare 2-Phenoxychromone Derivatives from Artemisia spp. Molecules 2022, 27, 1216. [Google Scholar] [CrossRef] [PubMed]
- Alesawy, M.S.; Elkaeed, E.B.; Alsfouk, A.A.; Metwaly, A.M.; Eissa, I. In Silico Screening of Semi-Synthesized Compounds as Potential Inhibitors for SARS-CoV-2 Papain-like Protease: Pharmacophoric Features, Molecular Docking, ADMET, Toxicity and DFT Studies. Molecules 2021, 26, 6593. [Google Scholar] [CrossRef]
- Adnane, L.; Trail, P.A.; Taylor, I.; Wilhelm, S.M. Sorafenib (BAY 43-9006, Nexavar®), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006, 407, 597–612. [Google Scholar] [PubMed]
- Peng, F.-W.; Liu, D.-K.; Zhang, Q.-W.; Xu, Y.-G.; Shi, L. VEGFR-2 inhibitors and the therapeutic applications thereof: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 987–1004. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.S.; Grierson, P.M.; Picus, J.; Lockhart, A.C.; Roth, B.J.; Liu, J.; Morton, A.; Chan, E.; Huffman, J.; Liang, C. Vorolanib (X-82), an oral anti-VEGFR/PDGFR/CSF1R tyrosine kinase inhibitor, with everolimus in solid tumors: Results of a phase I study. Investig. New Drugs 2021, 39, 1298–1305. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Latiano, T.P.; Parente, P.; Chiarazzo, C.; Limosani, F.; Di Maggio, G.; Maiello, E. The potential role of nintedanib in treating colorectal cancer. Expert Opin. Pharmacother. 2017, 18, 1153–1162. [Google Scholar] [CrossRef]
- Papich, M.G. Papich Handbook of Veterinary Drugs-E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2020. [Google Scholar]
- El-Helby, A.-G.A.; Ayyad, R.R.; El-Adl, K.; Elkady, H. Phthalazine-1, 4-dione derivatives as non-competitive AMPA receptor antagonists: Design, synthesis, anticonvulsant evaluation, ADMET profile and molecular docking. Mol. Divers. 2019, 23, 283–298. [Google Scholar] [CrossRef] [PubMed]
- El-Helby, A.G.A.; Ayyad, R.R.; Zayed, M.F.; Abulkhair, H.S.; Elkady, H.; El-Adl, K. Design, synthesis, in silico ADMET profile and GABA-A docking of novel phthalazines as potent anticonvulsants. Arch. Der Pharm. 2019, 352, 1800387. [Google Scholar] [CrossRef]
- Wei, K.; Louis, H.; Emori, W.; Idante, P.S.; Agwamba, E.C.; Cheng, C.-R.; Eno, E.A.; Unimuke, T.O. Antispasmodic activity of carnosic acid extracted from rosmarinus officinalis: Isolation, spectroscopic characterization, DFT studies, and in silico molecular docking investigations. J. Mol. Struct. 2022, 1260, 132795. [Google Scholar] [CrossRef]
- Eno, E.A.; Mbonu, J.I.; Louis, H.; Patrick-Inezi, F.S.; Gber, T.E.; Unimuke, T.O.; Okon, E.E.; Benjamin, I.; Offiong, O.E. Antimicrobial activities of 1-phenyl-3-methyl-4-trichloroacetyl-pyrazolone: Experimental, DFT studies, and molecular docking investigation. J. Indian Chem. Soc. 2022, 99, 100524. [Google Scholar] [CrossRef]
- Benjamin, I.; Udoikono, A.D.; Louis, H.; Agwamba, E.C.; Unimuke, T.O.; Owen, A.E.; Adeyinka, A.S. Antimalarial potential of naphthalene-sulfonic acid derivatives: Molecular electronic properties, vibrational assignments, and in-silico molecular docking studies. J. Mol. Struct. 2022, 1264, 133298. [Google Scholar] [CrossRef]
- Asogwa, F.C.; Agwamba, E.C.; Louis, H.; Muozie, M.C.; Benjamin, I.; Gber, T.E.; Mathias, G.E.; Adeyinka, A.S.; Ikeuba, A.I. Structural Benchmarking, Density Functional Theory Simulation, Spectroscopic Investigation and Molecular Docking of N-(1H-pyrrol-2-yl) methylene)-4-methylaniline as Castration-Resistant Prostate Cancer Chemotherapeutic Agent. Chem. Phys. Impact 2022, 5, 100091. [Google Scholar] [CrossRef]
- Louis, H.; Mathias, G.E.; Unimuke, T.O.; Emori, W.; Ling, L.; Owen, A.E.; Adeyinka, A.S.; Ntui, T.N.; Cheng, C.-R. Isolation, characterization, molecular electronic structure investigation, and in-silico modeling of the anti-inflammatory potency of trihydroxystilbene. J. Mol. Struct. 2022, 1266, 133418. [Google Scholar] [CrossRef]
- Udoikono, A.D.; Louis, H.; Eno, E.A.; Agwamba, E.C.; Unimuke, T.O.; Igbalagh, A.T.; Edet, H.O.; Odey, J.O.; Adeyinka, A.S. Reactive azo compounds as a potential chemotherapy drugs in the treatment of malignant glioblastoma (GBM): Experimental and theoretical studies. J. Photochem. Photobiol. 2022, 10, 100116. [Google Scholar] [CrossRef]
- Husein, D.Z.; Hassanien, R.; Khamis, M. Cadmium oxide nanoparticles/graphene composite: Synthesis, theoretical insights into reactivity and adsorption study. RSC Adv. 2021, 11, 27027–27041. [Google Scholar] [CrossRef]
- Wang, T.; Husein, D.Z. Novel synthesis of multicomponent porous nano-hybrid composite, theoretical investigation using DFT and dye adsorption applications: Disposing of waste with waste. Environ. Sci. Pollut. Res. 2022, 1–28. [Google Scholar] [CrossRef]
- Turchi, M.; Cai, Q.; Lian, G. An evaluation of in-silico methods for predicting solute partition in multiphase complex fluids–A case study of octanol/water partition coefficient. Chem. Eng. Sci. 2019, 197, 150–158. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Enoch, S.J.; Ezendam, J.; Sewald, K.; Roggen, E.L.; Cochrane, S. An adverse outcome pathway for sensitization of the respiratory tract by low-molecular-weight chemicals: Building evidence to support the utility of in vitro and in silico methods in a regulatory context. Appl. Vitr. Toxicol. 2017, 3, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Altamash, T.; Amhamed, A.; Aparicio, S.; Atilhan, M. Effect of hydrogen bond donors and acceptors on CO2 absorption by deep eutectic solvents. Processes 2020, 8, 1533. [Google Scholar] [CrossRef]
- Wan, Y.; Tian, Y.; Wang, W.; Gu, S.; Ju, X.; Liu, G. In silico studies of diarylpyridine derivatives as novel HIV-1 NNRTIs using docking-based 3D-QSAR, molecular dynamics, and pharmacophore modeling approaches. RSC Adv. 2018, 8, 40529–40543. [Google Scholar] [CrossRef] [Green Version]
- Escamilla-Gutiérrez, A.; Ribas-Aparicio, R.M.; Córdova-Espinoza, M.G.; Castelán-Vega, J.A. In silico strategies for modeling RNA aptamers and predicting binding sites of their molecular targets. Nucleosides Nucleotides Nucleic Acids 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Morphological similarity: A 3D molecular similarity method correlated with protein-ligand recognition. J. Comput.-Aided Mol. Des. 2000, 14, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ren, J.-X.; Ma, J.-X.; Ding, L. Development of an in silico prediction model for chemical-induced urinary tract toxicity by using naïve Bayes classifier. Mol. Divers. 2019, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Elkady, H.; Elwan, A.; El-Mahdy, H.A.; Doghish, A.S.; Ismail, A.; Taghour, M.S.; Elkaeed, E.B.; Eissa, I.H.; Dahab, M.A.; Mahdy, H.A. New benzoxazole derivatives as potential VEGFR-2 inhibitors and apoptosis inducers: Design, synthesis, anti-proliferative evaluation, flowcytometric analysis, and in silico studies. J. Enzym. Inhib. Med. Chem. 2022, 37, 397–410. [Google Scholar] [CrossRef]
- Alsaif, N.A.; Taghour, M.S.; Alanazi, M.M.; Obaidullah, A.J.; Al-Mehizia, A.A.; Alanazi, M.M.; Aldawas, S.; Elwan, A.; Elkady, H. Discovery of new VEGFR-2 inhibitors based on bis ([1, 2, 4] triazolo)[4, 3-a: 3′, 4′-c] quinoxaline derivatives as anticancer agents and apoptosis inducers. J. Enzym. Inhib. Med. Chem. 2021, 36, 1093–1114. [Google Scholar] [CrossRef]
- Elkaeed, E.B.; Youssef, F.S.; Eissa, I.H.; Elkady, H.; Alsfouk, A.A.; Ashour, M.L.; El Hassab, M.A.; Abou-Seri, S.M.; Metwaly, A.M. Multi-Step In Silico Discovery of Natural Drugs against COVID-19 Targeting Main Protease. Int. J. Mol. Sci. 2022, 23, 6912. [Google Scholar] [CrossRef]
- Belal, A.; Abdel Gawad, N.M.; Mehany, A.B.; Abourehab, M.A.; Elkady, H.; Al-Karmalawy, A.A.; Ismael, A.S. Design, synthesis and molecular docking of new fused 1 H-pyrroles, pyrrolo [3, 2-d] pyrimidines and pyrrolo [3, 2-e][1, 4] diazepine derivatives as potent EGFR/CDK2 inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 1884–1902. [Google Scholar] [CrossRef]
- NIH Methyl 4-Aminobenzoate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/12082 (accessed on 1 May 2022).
- NIH Methyl 4-Benzamidobenzoate. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/727255 (accessed on 1 May 2022).
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Number | No. of Hydrogen Bonds | Amino Acids in the Receptor | No. of Hydrophobic Interactions | Amino Acids in the Receptor |
|---|---|---|---|---|
| C1 | 3 | K866–E915–D1044 | 6 | I886–L887–I890–L1017–L1033–F1045 |
| C2 | 3 | K866–E915–D1044 | 4 | I886–I890–L1017–L1033 |
| C3 | 3 | K866–E915–D1044 | 7 | I886–I890 (2)–F916–L1017–D1044–F1045 |
| IP | EA | μ (eV) | χ (eV) | η (eV) | σ (eV) | ω (eV) | Dm (Debye) | TE (eV) | ∆Nmax | ∆E (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| −6.055 | −1.857 | −3.956 | 3.956 | 2.099 | 0.476 | 16.421 | 6.472 | −37,323.8 | 1.885 | −16.421 |
| Comp. | RF-FDA-C | TD50-M (mg/kg/day) | MD-R (g/kg) | R-O-LD50 (g/kg) | LOAEL-C (g/kg) | Skin Irritancy | Eye Irritancy |
|---|---|---|---|---|---|---|---|
| Compound 7 | Non-Carcinogen | 15.480 | 0.486 | 0.874 | 0.136 | Non-Irritant | Mild |
| Sorafenib | 19.236 | 0.089 | 0.823 | 0.005 | Non-Irritant | Mild |
| Comp. | ALog p | M. Wt | HBA | HBD | Rotatable Bonds | Rings | Aromatic Rings | MFPSA | Minimum Distance |
|---|---|---|---|---|---|---|---|---|---|
| Toceranib V | 2.761 | 396.458 | 3 | 3 | 5 | 4 | 2 | 0.191 | 1.12 |
| Compound 7 | 3.745 | 412.441 | 4 | 3 | 6 | 4 | 4 | 0.233 | 0.00 |
| Sunitinib II | 2.997 | 398.474 | 3 | 3 | 7 | 3 | 2 | 0.179 | 1.35 |
| Nintedanib IV | 4.392 | 539.625 | 7 | 2 | 8 | 5 | 4 | 0.178 | 1.83 |
| Vorolanib III | 1.671 | 439.483 | 3 | 3 | 3 | 4 | 2 | 0.213 | 1.31 |
| MCF-7 IC50 (μM) | HCT 116 IC50 (μM) | VEGFR IC50 (nM) | |
|---|---|---|---|
| Compound 7 | 12.93 ± 0.54 | 11.52 ± 0.70 | 25 ± 1.29 |
| Sorafenib | 4.32 ± 0.33 | 7.28 ± 0.53 | 35 ± 1.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkaeed, E.B.; Yousef, R.G.; Elkady, H.; Gobaara, I.M.M.; Alsfouk, A.A.; Husein, D.Z.; Ibrahim, I.M.; Metwaly, A.M.; Eissa, I.H. The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches. Processes 2022, 10, 1391. https://doi.org/10.3390/pr10071391
Elkaeed EB, Yousef RG, Elkady H, Gobaara IMM, Alsfouk AA, Husein DZ, Ibrahim IM, Metwaly AM, Eissa IH. The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches. Processes. 2022; 10(7):1391. https://doi.org/10.3390/pr10071391
Chicago/Turabian StyleElkaeed, Eslam B., Reda G. Yousef, Hazem Elkady, Ibraheem M. M. Gobaara, Aisha A. Alsfouk, Dalal Z. Husein, Ibrahim M. Ibrahim, Ahmed M. Metwaly, and Ibrahim H. Eissa. 2022. "The Assessment of Anticancer and VEGFR-2 Inhibitory Activities of a New 1H-Indole Derivative: In Silico and In Vitro Approaches" Processes 10, no. 7: 1391. https://doi.org/10.3390/pr10071391