
Extended Overview of Ocular Phenotype with Recent Advances in Hypohidrotic Ectodermal Dysplasia
 , , ,
, , ,  ,
, {kind=link}
Abstract
:1. Ectodermal Dysplasia
Definition and Classification
2. Hypohidrotic Ectodermal Dysplasia
2.1. Definition
2.2. Genetics and Molecular Basis
2.3. Clinical Manifestations
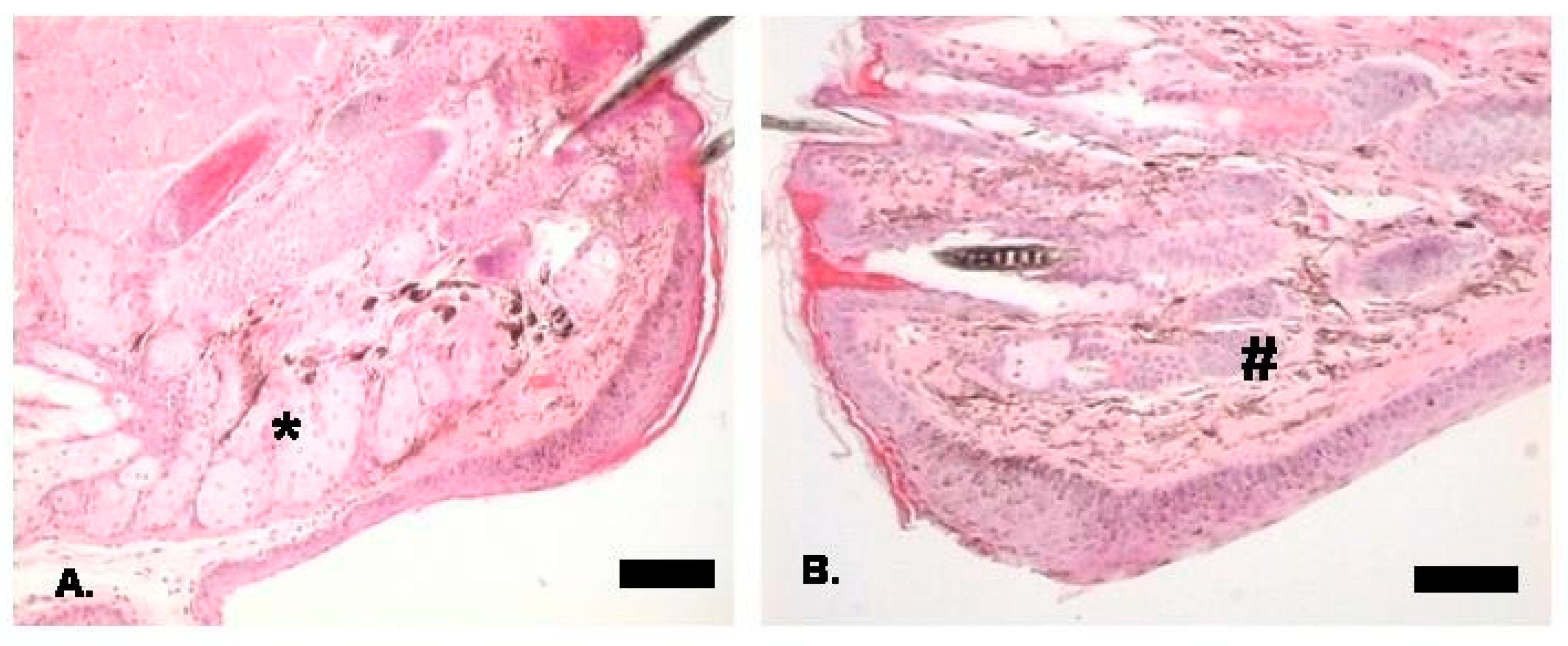
3. Ocular Manifestations of HED
3.1. Dry Eye Disease
3.2. Anterior Segment and Eyelids
3.3. Lacrimal Drainage System
3.4. Other Ocular Manifestations
3.5. Molecular Basis of Ocular Phenotype in HED
4. Management of Ocular Manifestations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Freire-Maia, N. Ectodermal dysplasias. Hum. Hered. 1971, 21, 309–312. [Google Scholar] [CrossRef]
- Freire-Maia, N. Ectodermal dysplasias revisited. Acta Genet. Med. Gemellol. 1977, 26, 121–131. [Google Scholar] [CrossRef]
- Irvine, A.D. Ectodermal Dysplasias. In Pediatric Dermatology, 2nd ed.; Harper, J.I., Oranje, O.P., Prose, N., Eds.; Blackwell Scientific: Oxford, UK, 2005; pp. 1412–1466. [Google Scholar]
- Kaercher, T. Ocular symptoms and signs in patients with ectodermal dysplasia syndromes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2004, 242, 495–500. [Google Scholar] [CrossRef]
- Freire-Maia, N.; Pinheiro, M. Ectodermal Dysplasias: A Clinical and Genetic Study; Alan R. Liss: New York, NY, USA, 1984; p. 251. [Google Scholar]
- Wright, J.T.; Fete, M.; Schneider, H.; Zinser, M.; Koster, M.I.; Clarke, A.J.; Hadj-Rabia, S.; Tadini, G.; Pagnan, N.; Visinoni, A.F.; et al. Ectodermal dysplasias: Classification and organization by phenotype, genotype and molecular pathway. Am. J. Med. Genet. A 2019, 179, 442–447. [Google Scholar] [CrossRef]
- Kere, J.; Srivastava, A.K.; Montonen, O.; Zonana, J.; Thomas, N.; Ferguson, B.; Munoz, F.; Morgan, D.; Clarke, A.; Baybayan, P.; et al. X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat. Genet. 1996, 13, 409–416. [Google Scholar]
- Headon, D.J.; Emmal, S.A.; Ferguson, B.M.; Tucker, A.S.; Justice, M.J.; Sharpe, P.T.; Zonana, J.; Overbeek, P.A. Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 414, 913–916. [Google Scholar] [CrossRef]
- Wisniewski, S.A.; Trzeciak, W.H. A new mutation resulting in the truncation of the TRAF6-interacting domain of XEDAR: A possible novel cause of hypohidrotic ectodermal dysplasia. J. Med. Genet. 2012, 49, 499–501. [Google Scholar] [CrossRef]
- Nguyen-Nielsen, M.; Skovbo, S.; Svaneby, D.; Pedersen, L.; Fryzek, J. The prevalence of X-linked hypohidrotic ectodermal dysplasia (XLHED) in Denmark, 1995–2010. Eur. J. Med. Genet. 2013, 56, 236–242. [Google Scholar] [CrossRef]
- Gökdere, S.; Schneider, H.; Hehr, U.; Willen, L.; Schneider, P.; Maier-Wohlfart, S. Functional and clinical analysis of five EDA variants associated with ectodermal dysplasia but with a hard-to-predict significance. Front. Genet. 2022, 13, 934395. [Google Scholar] [CrossRef]
- Li, L.; Zhou, Y.; Tian, R.; Zhang, C. Prenatal ultrasound findings of ectodermal dysplasia: A case report. BMC Pregnancy Childbirth 2022, 22, 100. [Google Scholar] [CrossRef]
- Huttner, K. Future developments in XLHED treatment approaches. Am. J. Med. Genet. Part A 2014, 164, 2433–2436. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Faschingbauer, F.; Schuepbach-Mallepell, S.; Körber, I.; Wohlfart, S.; Dick, A.; Wahlbuhl, M.; Kowalczyk-Quintas, C.; Vigolo, M.; Kirby, N.; et al. Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. N. Engl. J. Med. 2018, 378, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Tyagi, V.; Hashim, A.A. Ocular and non-ocular manifestations of hypohidrotic ectodermal dysplasia. BMJ Case Rep. 2011, 2011, bcr0120113731. [Google Scholar] [CrossRef]
- Lind, L.K.; Stecksén-Blicks, C.; Lejon, K.; Schmitt-Egenolf, M. EDAR mutation in autosomal dominant hypohidrotic ectodermal dysplasia in two Swedish families. BMC Med. Genet. 2006, 7, 80. [Google Scholar] [CrossRef]
- Priolo, M.; Laganà, C. Ectodermal dysplasias: A new clinical-genetic classification. J. Med. Genet. 2001, 38, 579–585. [Google Scholar] [CrossRef]
- Bayés, M.; Hartung, A.J.; Ezer, S.; Pispa, J.; Thesleff, I.; Srivastava, A.K.; Kere, J. The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin-A with deletion mutations in collagenous repeats. Hum. Mol. Genet. 1998, 7, 1661–1669. [Google Scholar] [CrossRef]
- Monreal, A.; Zonana, J.; Ferguson, B. Identification of a new splice of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am. J. Hum. Genet. 1998, 63, 380–389. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pispa, J.; Hartung, A.J.; Du, Y.; Ezer, S.; Jenks, T.; Shimada, T.; Pekkanen, M.; Mikkola, M.L.; Ko, M.S.; et al. The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc. Natl. Acad. Sci. USA 1997, 94, 13069–13074. [Google Scholar] [CrossRef]
- Ferguson, M.; Naunheim, K. Resection for Barrett’s mucosa with high-grade dysplasia: Implications for prophylactic photodynamic therapy. J. Thorac. Cardiovasc. Surg. 1997, 114, 824–829. [Google Scholar] [CrossRef]
- Callea, M.; Nieminen, P.; Willoughby, C.; Clarich, G.; Yavuz, I.; Vinciguerra, A.; Di Stazio, M.; Giglio, S.; Sani, I.; Maglione, M.; et al. A novel INDEL mutation in the EDA gene resulting in a distinct X-linked hypohidrotic ectodermal dysplasia phenotype in an Italian family. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 341–343. [Google Scholar] [CrossRef]
- Huang, S.X.; Liang, J.L.; Sui, W.G.; Lin, H.; Xue, W.; Chen, J.J.; Zhang, Y.; Gong, W.W.; Dai, Y.; Ou, M.L. EDA mutation as a cause of hypohidrotic ectodermal dysplasia: A case report and review of the literature. Genet. Mol. Res. 2015, 14, 10344–10351. [Google Scholar] [CrossRef]
- Clarke, A.; Phillips, D.I.; Brown, R.; Harper, P.S. Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch. Dis Child. 1987, 62, 989–996. [Google Scholar] [CrossRef]
- Inazawa-Terada, M.; Namiki, T.; Omigawa, C.; Fujimoto, T.; Munetsugu, T.; Ugajin, T.; Shimomura, Y.; Ohshima, Y.; Yoshida, K.; Niizeki, H.; et al. An epidemiological survey of anhidrotic/hypohidrotic ectodermal dysplasia in Japan: High prevalence of allergic diseases. J. Dermatol. 2022, 49, 422–431. [Google Scholar] [CrossRef]
- Pozo-Molina, G.; Reyes-Reali, J.; Mendoza-Ramos, M.I.; Villalobos-Molina, R.; Garrido-Guerrero, E.; Méndez-Cruz, A.R. Novel missense mutation in the EDA1 gene identified in a family with hypohidrotic ectodermal dysplasia. Int. J. Dermatol. 2015, 54, 790–794. [Google Scholar] [CrossRef]
- Wright, J.T.; Grange, D.K.; Fete, M.; Adam, M.P.; Everman, D.B.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.; Gripp, K.W.; et al. Hypohidrotic Ectodermal Dysplasia [Updated 1 June 2017]. In GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2003; pp. 1993–2020. [Google Scholar]
- Callea, M.; Teggi, R.; Yavuz, I.; Tadini, G.; Priolo, M.; Crovella, S.; Clarich, G.; Grasso, D.L. Ear nose throat manifestations in hypoidrotic ectodermal dysplasia. Int. J. Pediatr. Otorhinolaryngol. 2013, 77, 1801–1804. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Gholse, A.; Nagarajaram, H.A.; Dalal, A.B.; Gupta, N.; Dutta, A.K.; Danda, S.; Gupta, R.; Sankar, H.V.; Bhavani, G.S.; et al. Ectodysplasin pathogenic variants affecting the furin-cleavage site and unusual clinical features define X-linked hypohidrotic ectodermal dysplasia in India. Am. J. Med. Genet. A 2022, 188, 788–805. [Google Scholar] [CrossRef]
- Cambiaghi, S.; Restano, L.; Pääkkönen, K.; Caputo, R.; Kere, J. Clinical findings in mosaic carriers of hypohidrotic ectodermal dysplasia. Arch. Dermatol. 2000, 136, 217–224. [Google Scholar] [CrossRef]
- Clarke, A.; Burn, J. Sweat testing to identify female carriers of X linked hypohidrotic ectodermal dysplasia. J. Med. Genet. 1991, 28, 330–333. [Google Scholar] [CrossRef]
- Clarke, A. Hypohidrotic ectodermal dysplasia. J. Med. Genet. 1987, 24, 659–663. [Google Scholar] [CrossRef]
- Gipson, I.K. The ocular surface: The challenge to enable and protect vision: The Friedenwald lecture. Investig. Ophthalmol Vis. Sci. 2007, 48, 4390–4398. [Google Scholar] [CrossRef]
- Ekins, M.B.; Waring, G.O., III. Absent Meibomian glands and reduced corneal sensation in hypohidrotic ectodermal dysplasia. J. Pediatr. Ophthalmol. Strabismus 1981, 18, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Khaled, A.; Kharfi, M.; Bouzgarrou, A.; M’halla, H.; Jones, M.; Fazaa, B.; Kamoun, M.R. Anhidrotic/hypohidrotic ectodermal dysplasia: Ten cases. Tunis Med. 2009, 87, 805–809. [Google Scholar]
- Piccione, M.; Serra, G.; Sanfilippo, C.; Andreucci, E.; Sani, I.; Corsello, G. A new mutation in EDA gene in X-linked hypohidrotic ectodermal dysplasia associated with keratoconus. Minerva Pediatr. 2012, 64, 59–64. [Google Scholar] [PubMed]
- Dietz, J.; Kaercher, T.; Schneider, A.T.; Zimmermann, T.; Huttner, K.; Johnson, R.; Schneider, H. Early respiratory and ocular involvement in X-linked hypohidrotic ectodermal dysplasia. Eur. J. Pediatr. 2013, 172, 1023–1031. [Google Scholar] [CrossRef]
- Di Iorio, E.; Kaye, S.B.; Ponzin, D.; Barbaro, V.; Ferrari, S.; Böhm, E.; Nardiello, P.; Castaldo, G.; McGrath, J.A.; Willoughby, C.E. Limbal stem cell deficiency and ocular phenotype in ectrodactyly-ectodermal dysplasia-clefting syndrome caused by p63 mutations. Oftalmología 2012, 119, 74–83. [Google Scholar] [CrossRef]
- Kaercher, T.; Dietz, J.; Jacobi, C.; Berz, R.; Schneider, H. Diagnosis of X-linked hypohidrotic ectodermal dysplasia by meibography and infrared thermography of the eye. Curr. Eye Res. 2015, 40, 884–890. [Google Scholar] [CrossRef]
- Saw, V.P.J.; Dart, J.K.G.; Sitaru, C.; Zillikens, D. Cicatrising conjunctivitis with anti-basement membrane autoantibodies in ectodermal dysplasia. Br. J. Ophthalmol. 2008, 92, 1403–1410. [Google Scholar] [CrossRef]
- Beckerman, B.L. Lacrimal anomalies in anhidrotic ectodermal dysplasia. Am. J. Ophthalmol. 1973, 75, 728–730. [Google Scholar] [CrossRef]
- Wilson, F.M., II; Grayson, M.; Pieroni, D. Corneal changes in ectodermal dysplasia. Case report, histopathology, and differential diagnosis. Am. J. Ophthalmol. 1973, 75, 17–27. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, L.; Li, X.; Li, C.; Zhang, H. Lower lid ectropion in hypohidrotic ectodermal dysplasia. Case Rep. Ophthalmol. Med. 2015, 2015, 952834. [Google Scholar] [CrossRef]
- Chen, X.; Zeng, W.X.; Duan, B.Y.; Lin, Y.Y.; Liu, J.; Zhang, Z.D. Clinical features, surgical outcomes and genetic analysis of ectodermal dysplasia with ocular diseases. Int. J. Ophthalmol. 2022, 15, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Viljoen, D.L.; Winship, W.S. A new form of hypohidrotic ectodermal dysplasia. Am. J. Med. Genet. 1988, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, M.; Pascual-Castrovejo, I.; Di Rocco, C. Hypohidroticectodermal Dysplasia (HED). In Neurocutaneous Disorders: Phakomatoses and Hamartoneoplastic Syndromes, 1st ed.; Springer: Vienna, Austria, 2008; pp. 957–965. [Google Scholar]
- Liakos, G.M. Anhidrotic ectodermal dysplasia with lacrimal anomalies. Br. J. Ophthalmol. 1979, 63, 520–522. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Bhoot, M.; Jain, K. Hypohidrotic Ectodermal Dysplasia: A Rare Disorder with Bilateral Infantile Glaucoma. J. Glaucoma 2019, 28, e58–e60. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, N.; Eliott, D.; Garcia-Valenzuela, E.; Baker, J. Bilateral panuveitis in a child with hypohidrotic ectodermal dysplasia. Am. J. Ophthalmol. 2002, 134, 443–445. [Google Scholar] [CrossRef]
- Callea, M.; Vinciguerra, A.; Willoughby, C.E.; Deroma, L.; Clarich, G. Infantile bilateral glaucoma in a child with ectodermal dysplasia. Ophthalmic Genet. 2013, 34, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Li, S.; Chen, X.; Ma, B.; He, H.; Liu, T.; Yu, J.; Zhang, L.; Chen, Y.; Liu, Z.; et al. Meibomian Gland Absence Related Dry Eye in Ectodysplasin a Mutant Mice. Am. J. Pathol. 2016, 186, 32–42. [Google Scholar] [CrossRef]
- Cui, C.Y.; Smith, J.A.; Schlessinger, D.; Chan, C.C. X-linked anhidrotic ectodermal dysplasia disruption yields a mouse model for ocular surface disease and resultant blindness. Am. J. Pathol. 2005, 167, 89–95. [Google Scholar] [CrossRef]
- Kuony, A.; Ikkala, K.; Kalha, S.; Magalhães, A.C.; Pirttiniemi, A.; Michon, F. Ectodysplasin-A signaling is a key integrator in the lacrimal gland-cornea feedback loop. Development 2019, 146, dev176693. [Google Scholar] [CrossRef]
- Li, S.; Zhou, J.; Bu, J.; Ning, K.; Zhang, L.; Li, J.; Guo, Y.; He, X.; He, H.; Cai, X.; et al. Ectodysplasin A protein promotes corneal epithelial cell proliferation. J. Biol. Chem. 2017, 292, 13391–13401. [Google Scholar] [CrossRef]
- Vargas, G.A.; Fantino, E.; George-Nascimento, C.; Gargus, J.J.; Haigler, H.T. Reduced epidermal growth factor receptor expression in hypohidrotic ectodermal dysplasia and Tabby mice. J. Clin. Investig. 1996, 97, 2426–2432. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, J.; Zhang, L.; Li, J.; Yu, J.; Ning, K.; Qu, Y.; He, H.; Chen, Y.; Reinach, P.S.; et al. Ectodysplasin A regulates epithelial barrier function through sonic hedgehog signalling pathway. J. Cell. Mol. Med. 2018, 22, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.Y.; Wang, L.X.; Deng, Y.P. Transgenic dry eye mouse models: Powerful tools to study dry eye disease. Int. J. Ophthalmol. 2022, 15, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Moshirfar, M.; Williams, D.J.; Ronquillo, Y.C.; Ply, B.K. Potential Risks of Corneal Refractive Surgery in Patients with Ectodermal Dysplasia. Ophthalmol. Ther. 2022, 11, 1281–1289. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callea, M.; Bignotti, S.; Semeraro, F.; Cammarata-Scalisi, F.; El-Feghaly, J.; Morabito, A.; Romano, V.; Willoughby, C.E. Extended Overview of Ocular Phenotype with Recent Advances in Hypohidrotic Ectodermal Dysplasia. Children 2022, 9, 1357. https://doi.org/10.3390/children9091357
Callea M, Bignotti S, Semeraro F, Cammarata-Scalisi F, El-Feghaly J, Morabito A, Romano V, Willoughby CE. Extended Overview of Ocular Phenotype with Recent Advances in Hypohidrotic Ectodermal Dysplasia. Children. 2022; 9(9):1357. https://doi.org/10.3390/children9091357
Chicago/Turabian StyleCallea, Michele, Stefano Bignotti, Francesco Semeraro, Francisco Cammarata-Scalisi, Jinia El-Feghaly, Antonino Morabito, Vito Romano, and Colin E. Willoughby. 2022. "Extended Overview of Ocular Phenotype with Recent Advances in Hypohidrotic Ectodermal Dysplasia" Children 9, no. 9: 1357. https://doi.org/10.3390/children9091357