
New Directions in Understanding Atopic March Starting from Atopic Dermatitis

 , ,
, , {kind=link}
Abstract
:1. Introduction
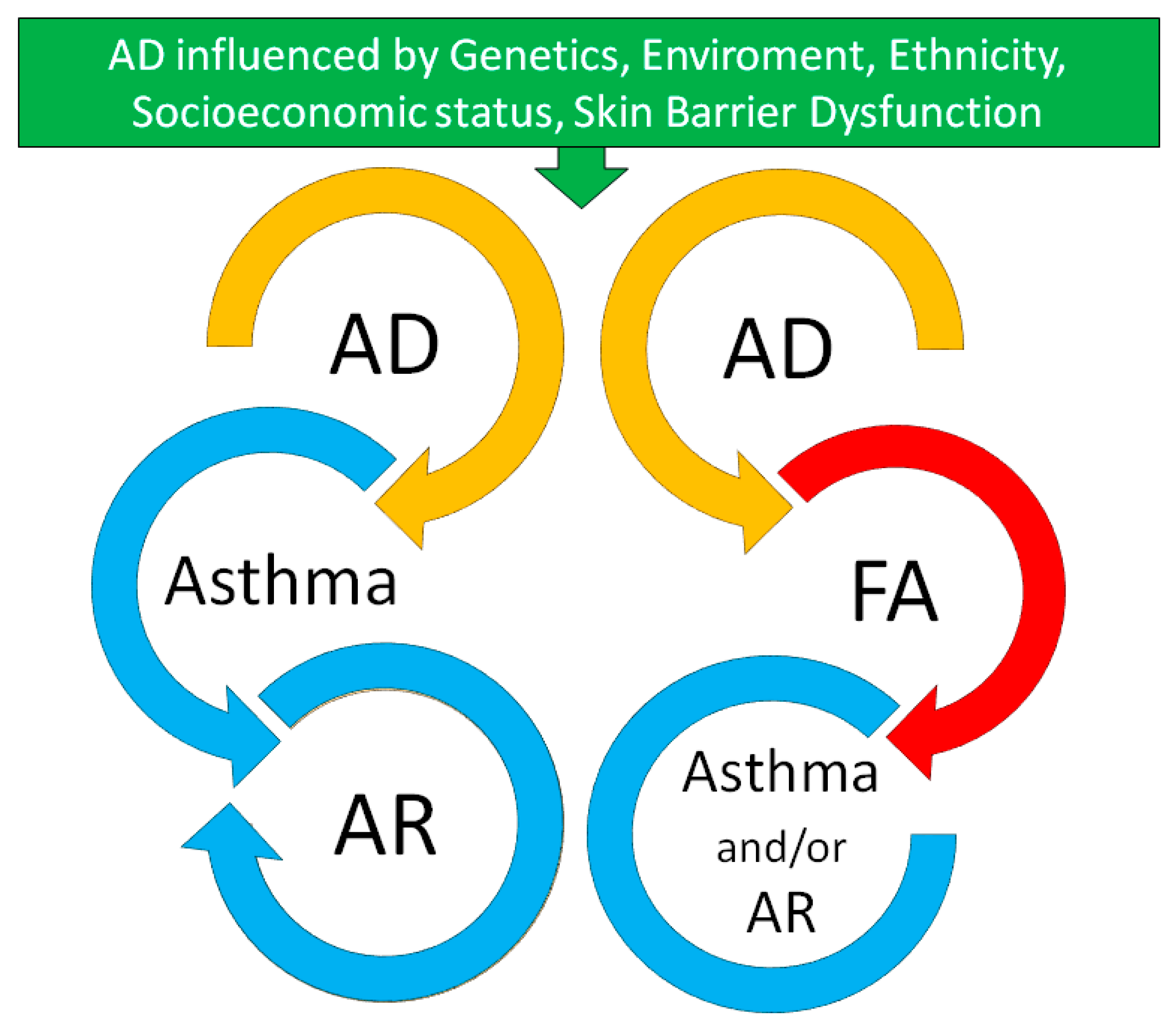
2. The Atopic March: Myth or Reality?
3. Atopic March Risk Factors
3.1. Age of Onset and Severity of Atopic Dermatitis
3.2. Allergic Polysensitization
3.3. Family History for Atopy
3.4. Genetic Factors
4. Pathogenic Mechanisms Underlying Atopic Dermatitis and Possibly Atopic March
4.1. Outside-In vs. Inside-Out Hypotheses
4.2. The Exposome Hypothesis
4.3. The Dysbiosis Hypothesis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hill, D.A.; Spergel, J.M. The atopic march: Critical evidence and clinical relevance. Ann. Allergy Asthma Immunol. 2018, 120, 131–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punekar, Y.S.; Sheikh, A. Establishing the sequential progression of multiple allergic diagnoses in a UK birth cohort using the General Practice Research Database. Clin. Exp. Allergy 2009, 39, 1889–1895. [Google Scholar] [CrossRef]
- Paller, A.S.; Spergel, J.M.; Mina-Osorio, P.; Irvine, A.D. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J. Allergy Clin. Immunol. 2019, 143, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Belgrave, D.C.; Granell, R.; Simpson, A.; Guiver, J.; Bishop, C.; Buchan, I.; Henderson, A.J.; Custovic, A. Developmental profiles of eczema, wheeze, and rhinitis: Two population-based birth cohort studies. PLoS Med. 2014, 11, e1001748. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W. The atopic march: Fact or folklore? Ann. Allergy Asthma Immunol. 2018, 120, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.D.; Wright, A.L.; Taussig, L.M.; Holberg, C.J.; Halonen, M.; Morgan, W.J. Asthma and wheezing in the first six years of life. The Group Health Medical Associates. N. Engl. J. Med. 1995, 332, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Ravnborg, N.; Ambikaibalan, D.; Agnihotri, G.; Price, S.; Rastogi, S.; Patel, K.R.; Singam, V.; Andersen, Y.; Halling, A.S.; Silverberg, J.I.; et al. Prevalence of asthma in patients with atopic dermatitis: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2021, 84, 471–478. [Google Scholar] [CrossRef]
- Abo-Zaid, G.; Sharpe, R.A.; Fleming, L.E.; Depledge, M.; Osborne, N.J. Association of Infant Eczema with Childhood and Adult Asthma: Analysis of Data from the 1958 Birth Cohort Study. Int. J. Environ. Res. Public Health 2018, 7, 1415. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, N.; Sánchez, J.; Zakzuk, J.; Bornacelly, A.; Quiróz, C.; Alvarez, Á.; Puello, M.; Mendoza, K.; Martínez, D.; Mercado, D.; et al. Particular characteristics of allergic symptoms in tropical environments: Follow up to 24 months in the FRAAT birth cohort study. BMC Pulm. Med. 2012, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Illi, S.; von Mutius, E.; Lau, S.; Nickel, R.; Grüber, C.; Niggemann, B.; Wahn, U.; Multicenter Allergy Study Group. The natural course of atopic dermatitis from birth to age 7 years and the association with asthma. J. Allergy Clin. Immunol. 2004, 113, 925–931. [Google Scholar] [CrossRef]
- Lowe, A.J.; Angelica, B.; Su, J.; Lodge, C.J.; Hill, D.J.; Erbas, B.; Bennett, C.M.; Gurrin, L.C.; Axelrad, C.; Abramson, M.J.; et al. Age at onset and persistence of eczema are related to subsequent risk of asthma and hay fever from birth to 18 years of age. Pediatr. Allergy Immunol. 2017, 28, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, D.; Sjöberg, O.; Foucard, T. Development of allergies and asthma in infants and young children with atopic dermatitis—A prospective follow-up to 7 years of age. Allergy 2000, 55, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.P.; Butland, B.K.; Anderson, H.R. Incidence and prognosis of asthma and wheezing illness from early childhood to age 33 in a national British cohort. BMJ 1996, 312, 1195–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roduit, C.; Frei, R.; Depner, M.; Karvonen, A.M.; Renz, H.; Braun-Fahrländer, C.; Schmausser-Hechfellner, E.; Pekkanen, J.; Riedler, J.; Dalphin, J.C.; et al. Phenotypes of Atopic Dermatitis Depending on the Timing of Onset and Progression in Childhood. JAMA Pediatr. 2017, 171, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Margolis, J.S.; Abuabara, K.; Bilker, W.; Hoffstad, O.; Margolis, D.J. Persistence of mild to moderate atopic dermatitis. JAMA Dermatol. 2014, 150, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Ballardini, N.; Kull, I.; Söderhäll, C.; Lilja, G.; Wickman, M.; Wahlgren, C.F. Eczema severity in preadolescent children and its relation to sex, filaggrin mutations, asthma, rhinitis, aggravating factors and topical treatment: A report from the BAMSE birth cohort. Br. J. Dermatol. 2013, 168, 588–594. [Google Scholar] [CrossRef]
- Ricci, G.; Patrizi, A.; Baldi, E.; Menna, G.; Tabanelli, M.; Masi, M. Long-term follow-up of atopic dermatitis: Retrospective analysis of related risk factors and association with concomitant allergic diseases. J. Am. Acad. Dermatol. 2006, 55, 765–771. [Google Scholar] [CrossRef]
- Paternoster, L.; Savenije, O.; Heron, J.; Evans, D.M.; Vonk, J.M.; Brunekreef, B.; Wijga, A.H.; Henderson, A.J.; Koppelman, G.H.; Brown, S.J. Identification of atopic dermatitis subgroups in children from 2 longitudinal birth cohorts. J. Allergy Clin. Immunol. 2018, 141, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Tsakok, T.; Marrs, T.; Mohsin, M.; Baron, S.; du Toit, G.; Till, S.; Flohr, C. Does atopic dermatitis cause food allergy? A systematic review. J. Allergy Clin. Immunol. 2016, 137, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.; Hanifin, J.; Boguniewicz, M.; Eichenfield, L.F.; Spergel, J.M.; Dakovic, R.; Paller, A.S. Study of the Atopic March: Development of Atopic Comorbidities. Pediatr. Dermatol. 2016, 33, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Du Toit, G.; Sayre, P.H.; Roberts, G.; Lawson, K.; Sever, M.L.; Bahnson, H.T.; Fisher, H.R.; Feeney, M.; Radulovic, S.; Basting, M.; et al. Allergen specificity of early peanut consumption and effect on development of allergic disease in the Learning Early About Peanut Allergy study cohort. J. Allergy Clin. Immunol. 2018, 141, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anto, J.M.; Bousquet, J.; Akdis, M.; Auffray, C.; Keil, T.; Momas, I.; Postma, D.S.; Valenta, R.; Wickman, M.; Cambon-Thomsen, A.; et al. Mechanisms of the Development of Allergy (MeDALL): Introducing novel concepts in allergy phenotypes. J. Allergy Clin. Immunol. 2017, 139, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickman, M.; Asarnoj, A.; Tillander, H.; Andersson, N.; Bergström, A.; Kull, I.; Melén, E.; Pershagen, G.; Ahlstedt, S.; Lilja, G.; et al. Childhood-to-adolescence evolution of IgE antibodies to pollens and plant foods in the BAMSE cohort. J. Allergy Clin. Immunol. 2014, 133, 580–582. [Google Scholar] [CrossRef] [PubMed]
- Gabet, S.; Just, J.; Couderc, R.; Bousquet, J.; Seta, N.; Momas, I. Early polysensitization is associated with allergic multimorbidity in PARIS birth cohort infants. Pediatr. Allergy Immunol. 2016, 27, 831–837. [Google Scholar] [CrossRef]
- Alduraywish, S.A.; Standl, M.; Lodge, C.J.; Abramson, M.J.; Allen, K.J.; Erbas, B.; von Berg, A.; Heinrich, J.; Lowe, A.J.; Dharmage, S.C. Is there a march from early food sensitization to later childhood allergic airway disease? Results from two prospective birth cohort studies. Pediatr. Allergy Immunol. 2017, 28, 30–37. [Google Scholar] [CrossRef]
- Alduraywish, S.A.; Lodge, C.J.; Campbell, B.; Allen, K.J.; Erbas, B.; Lowe, A.J.; Dharmage, S.C. The march from early life food sensitization to allergic disease: A systematic review and meta-analyses of birth cohort studies. Allergy 2016, 71, 77–89. [Google Scholar] [CrossRef]
- Hill, D.A.; Grundmeier, R.W.; Ram, G.; Spergel, J.M. The epidemiologic characteristics of healthcare provider-diagnosed eczema, asthma, allergic rhinitis, and food allergy in children: A retrospective cohort study. BMC Pediatr. 2016, 16, 133. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.M.; Lefebvre, D.L.; Dharma, C.; Dai, D.; Lou, W.; Subbarao, P.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; Sears, M.R.; et al. Predicting the atopic march: Results from the Canadian Healthy Infant Longitudinal Development Study. J. Allergy Clin. Immunol. 2018, 141, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Havstad, S.L.; Sitarik, A.; Kim, H.; Zoratti, E.M.; Ownby, D.; Johnson, C.C.; Wegienka, G. Increased risk of asthma at age 10 years for children sensitized to multiple allergens. Ann. Allergy Asthma Immunol. 2021, 127, 441–445. [Google Scholar] [CrossRef]
- Bousquet, J.; Vignola, A.M.; Demoly, P. Links between rhinitis and asthma. Allergy 2003, 58, 691–706. [Google Scholar] [CrossRef] [Green Version]
- Depner, M.; Fuchs, O.; Genuneit, J.; Karvonen, A.M.; Hyvärinen, A.; Kaulek, V.; Roduit, C.; Weber, J.; Schaub, B.; Lauener, R.; et al. Clinical and epidemiologic phenotypes of childhood asthma. Am. J. Respir. Crit. Care Med. 2014, 189, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Grabenhenrich, L.B.; Gough, H.; Reich, A.; Eckers, N.; Zepp, F.; Nitsche, O.; Forster, J.; Schuster, A.; Schramm, D.; Bauer, C.P.; et al. Early-life determinants of asthma from birth to age 20 years: A German birth cohort study. J. Allergy Clin. Immunol. 2014, 133, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Gabet, S.; Just, J.; Couderc, R.; Seta, N.; Momas, I. Asthma and allergic rhinitis risk depends on house dust mite specific IgE levels in PARIS birth cohort children. Int. J. Hyg. Environ. Health. 2016, 219, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Herr, M.; Just, J.; Nikasinovic, L.; Foucault, C.; Le Marec, A.M.; Giordanella, J.P.; Momas, I. Risk factors and characteristics of respiratory and allergic phenotypes in early childhood. J. Allergy Clin. Immunol. 2012, 130, 389–396. [Google Scholar] [CrossRef]
- Hudson, T.J. Skin barrier function and allergic risk. Nat. Genet. 2006, 38, 399–400. [Google Scholar] [CrossRef]
- Mischke, D.; Korge, B.P.; Marenholz, I.; Volz, A.; Ziegler, A. Genes encoding structural proteins of epidermal cornification and S100 calcium-binding proteins form a gene complex (“epidermal differentiation complex”) on human chromosome 1q21. J. Investig. Dermatol. 1996, 106, 989–992. [Google Scholar] [CrossRef] [Green Version]
- Irvine, A.D.; McLean, W.H.; Leung, D.Y. Filaggrin mutations associated with skin and allergic diseases. N. Engl. J. Med. 2011, 365, 1315–1327. [Google Scholar] [CrossRef] [Green Version]
- Weidinger, S.; O’Sullivan, M.; Illig, T.; Baurecht, H.; Depner, M.; Rodriguez, E.; Ruether, A.; Klopp, N.; Vogelberg, C.; Weiland, S.K.; et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J. Allergy Clin. Immunol. 2008, 121, 1203–1209. [Google Scholar] [CrossRef]
- McAleer, M.A.; Irvine, A.D. The multifunctional role of filaggrin in allergic skin disease. J. Allergy Clin. Immunol. 2013, 131, 280–291. [Google Scholar] [CrossRef]
- Rodríguez, E.; Baurecht, H.; Herberich, E.; Wagenpfeil, S.; Brown, S.J.; Cordell, H.J.; Irvine, A.D.; Weidinger, S. Meta-analysis of filaggrin polymorphisms in eczema and asthma: Robust risk factors in atopic disease. J. Allergy Clin. Immunol. 2009, 123, 1361–1370. [Google Scholar] [CrossRef]
- Marenholz, I.; Rivera, V.A.; Esparza-Gordillo, J.; Bauerfeind, A.; Lee-Kirsch, M.A.; Ciechanowicz, A.; Kurek, M.; Piskackova, T.; Macek, M.; Lee, Y.A. Association screening in the Epidermal Differentiation Complex (EDC) identifies an SPRR3 repeat number variant as a risk factor for eczema. J. Investig. Dermatol. 2011, 131, 1644–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, S.P.; Goh, C.S.; Brown, S.J.; Palmer, C.N.; Porter, R.M.; Cole, C.; Campbell, L.E.; Gierlinski, M.; Barton, G.J.; Schneider, G.; et al. Tmem79/Matt is the matted mouse gene and is a predisposing gene for atopic dermatitis in human subjects. J. Allergy Clin. Immunol. 2013, 132, 1121–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.A.; Vonk, J.M.; Baurecht, H.; Marenholz, I.; Tian, C.; Hoffman, J.D.; Helmer, Q.; Tillander, A.; Ullemar, V.; van Dongen, J.; et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 2017, 49, 1752–1757. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Willis-Owen, S.A.; Kamatani, Y.; Baurecht, H.; Morar, N.; Liang, L.; Edser, P.; Street, T.; Rodriguez, E.; O’Regan, G.M.; et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum. Mol. Genet. 2013, 22, 4841–4856. [Google Scholar] [CrossRef] [Green Version]
- Marenholz, I.; Esparza-Gordillo, J.; Rüschendorf, F.; Bauerfeind, A.; Strachan, D.P.; Spycher, B.D.; Baurecht, H.; Margaritte-Jeannin, P.; Sääf, A.; Kerkhof, M.; et al. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat. Commun. 2015, 6, 8804. [Google Scholar] [CrossRef] [Green Version]
- Johansson, E.; Biagini Myers, J.M.; Martin, L.J.; He, H.; Pilipenko, V.; Mersha, T.; Weirauch, M.; Salomonis, N.; Ryan, P.; LeMasters, G.K.; et al. KIF3A genetic variation is associated with pediatric asthma in the presence of eczema independent of allergic rhinitis. J. Allergy Clin. Immunol. 2017, 140, 595–598. [Google Scholar] [CrossRef] [Green Version]
- Ezratty, E.J.; Stokes, N.; Chai, S.; Shah, A.S.; Williams, S.E.; Fuchs, E. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 2011, 145, 1129–1141. [Google Scholar] [CrossRef] [Green Version]
- Croyle, M.J.; Lehman, J.M.; O’Connor, A.K.; Wong, S.Y.; Malarkey, E.B.; Iribarne, D.; Dowdle, W.E.; Schoeb, T.R.; Verney, Z.M.; Athar, M.; et al. Role of epidermal primary cilia in the homeostasis of skin and hair follicles. Development 2011, 138, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Johansson, E.; Biagini Myers, J.M.; Martin, L.J.; He, H.; Ryan, P.; LeMasters, G.K.; Bernstein, D.I.; Lockey, J.; Khurana Hershey, G.K. Identification of two early life eczema and non-eczema phenotypes with high risk for asthma development. Clin. Exp. Allergy 2019, 49, 829–837. [Google Scholar] [CrossRef]
- Ferreira, M.; Vonk, J.M.; Baurecht, H.; Marenholz, I.; Tian, C.; Hoffman, J.D.; Helmer, Q.; Tillander, A.; Ullemar, V.; Lu, Y.; et al. Eleven loci with new reproducible genetic associations with allergic disease risk. J. Allergy Clin. Immunol. 2019, 143, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Czarnowicki, T.; Krueger, J.G.; Guttman-Yassky, E. Skin barrier and immune dysregulation in atopic dermatitis: An evolving story with important clinical implications. J. Allergy Clin. Immunol. Pract. 2014, 2, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Looman, K.I.M.; van Meel, E.R.; Grosserichter-Wagener, C.; Vissers, F.J.M.; Klingenberg, J.H.; de Jong, N.W.; de Jongste, J.C.; Pasmans, S.G.M.A.; Duijts, L.; van Zelm, M.C.; et al. Associations of Th2, Th17, Treg cells, and IgA+ memory B cells with atopic disease in children: The Generation R Study. Allergy 2020, 75, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Hatano, Y.; Williams, M.L. Basis for the barrier abnormality in atopic dermatitis: Outside-inside-outside pathogenic mechanisms. J. Allergy Clin. Immunol. 2008, 121, 1337–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleimer, R.P.; Berdnikovs, S. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J. Allergy Clin. Immunol. 2017, 139, 1752–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biagini Myers, J.M.; Sherenian, M.G.; Baatyrbek Kyzy, A.; Alarcon, R.; An, A.; Flege, Z.; Morgan, D.; Gonzalez, T.; Stevens, M.L.; He, H.; et al. Events in Normal Skin Promote Early-Life Atopic Dermatitis-The MPAACH Cohort. J. Allergy Clin. Immunol. Pract. 2020, 8, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, K.; Haruna, M. Short-Term skin problems in infants aged 0-3 months affect food allergies or atopic dermatitis until 2 years of age, among infants of the general population. Allergy Asthma Clin. Immunol. 2019, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Berdyshev, E.; Goleva, E. Cutaneous barrier dysfunction in allergic diseases. J. Allergy Clin. Immunol. 2020, 145, 1485–1497. [Google Scholar] [CrossRef]
- Leung, D.; Calatroni, A.; Zaramela, L.S.; LeBeau, P.K.; Dyjack, N.; Brar, K.; David, G.; Johnson, K.; Leung, S.; Ramirez-Gama, M.; et al. The nonlesional skin surface distinguishes atopic dermatitis with food allergy as a unique endotype. Sci. Transl. Med. 2019, 11, eaav2685. [Google Scholar] [CrossRef]
- Kelleher, M.M.; Cro, S.; Cornelius, V.; Lodrup Carlsen, K.C.; Skjerven, H.O.; Rehbinder, E.M.; Lowe, A.J.; Dissanayake, E.; Shimojo, N.; Yonezawa, K.; et al. Skin care interventions in infants for preventing eczema and food allergy. Cochrane Database Syst. Rev. 2021, 52, CD013534. [Google Scholar]
- Akdis, C.A. The epithelial barrier hypothesis proposes a comprehensive understanding of the origins of allergic and other chronic noncommunicable diseases. J. Allergy Clin. Immunol. 2022, 149, 41–44. [Google Scholar] [CrossRef]
- Fackelmann, G.; Sommer, S. Microplastics and the gut microbiome: How chronically exposed species may suffer from gut dysbiosis. Mar. Pollut. Bull. 2019, 143, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Choi, D.; Han, S.; Choi, J.; Hong, J. An assessment of the toxicity of polypropylene microplastics in human derived cells. Sci. Total Environ. 2019, 684, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, M.; Tian, D.; Qiu, L.; Li, T. Effects of polystyrene microbeads on cytotoxicity and transcriptomic profiles in human Caco-2 cells. Environ. Toxicol. 2020, 35, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.D.; Chen, C.W.; Chen, Y.C.; Chen, H.H.; Lee, J.S.; Lin, C.H. Polystyrene microplastic particles: In vitro pulmonary toxicity assessment. J. Hazard. Mater. 2020, 385, 121575. [Google Scholar] [CrossRef]
- Krempski, J.W.; Dant, C.; Nadeau, K.C. The origins of allergy from a systems approach. Ann. Allergy. Asthma Immunol. 2020, 125, 507–516. [Google Scholar] [CrossRef]
- Reid, C.E.; Brauer, M.; Johnston, F.H.; Jerrett, M.; Balmes, J.R.; Elliott, C.T. Critical Review of Health Impacts of Wildfire Smoke Exposure. Environ. Health Perspect. 2016, 124, 1334–1343. [Google Scholar] [CrossRef] [Green Version]
- Prunicki, M.; Stell, L.; Dinakarpandian, D.; de Planell-Saguer, M.; Lucas, R.W.; Hammond, S.K.; Balmes, J.R.; Zhou, X.; Paglino, T.; Sabatti, C.; et al. Exposure to NO2, CO, and PM2.5 is linked to regional DNA methylation differences in asthma. Clin. Epigenet. 2018, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.V.; Lozupone, C.A.; Schwartz, D.A. The environment, epigenome, and asthma. J. Allergy Clin. Immunol. 2017, 140, 14e23. [Google Scholar] [CrossRef] [Green Version]
- Botha, M.; Basera, W.; Facey-Thomas, H.E.; Gaunt, B.; Genuneit, J.; Gray, C.L.; Kiragu, W.; Ramjith, J.; Watkins, A.; Levin, M.E. Nutrition and allergic diseases in urban and rural communities from the South African Food Allergy cohort. Pediatr. Allergy Immunol. 2019, 30, 511–521. [Google Scholar] [CrossRef]
- Lunjani, N.; Tan, G.; Dreher, A.; Sokolowska, M.; Groeger, D.; Warwyzniak, M.; Altunbulakli, C.; Westermann, P.; Basera, W.; Hobane, L.; et al. Environment-dependent alterations of immune mediators in urban and rural South African children with atopic dermatitis. Allergy 2022, 77, 569–581. [Google Scholar] [CrossRef]
- Luger, T.; Amagai, M.; Dreno, B.; Dagnelie, M.A.; Liao, W.; Kabashima, K.; Schikowski, T.; Proksch, E.; Elias, P.M.; Simon, M.; et al. Atopic dermatitis: Role of the skin barrier, environment, microbiome, and therapeutic agents. J. Dermatol. Sci. 2021, 102, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, B.E.; Ahn, K.; Leung, D. Interactions Between Atopic Dermatitis and Staphylococcus aureus Infection: Clinical Implications. Allergy Asthma Immunol. Res. 2019, 11, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Silverberg, J.I. Public health burden and epidemiology of atopic dermatitis. Dermatol. Clin. 2017, 35, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; Balica, S.; Hsu, C.Y.; Jean-Decoster, C.; Lauze, C.; Redoules, D.; Viodé, C.; Schmitt, A.M.; Serre, G.; Simon, M.; et al. Staphylococcus aureus density on lesional and nonlesional skin is strongly associated with disease severity in atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 1272–1274. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis toTrigger Cytokine Expression. J. Invest. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef] [Green Version]
- Forbes-Blom, E.; Camberis, M.; Prout, M.; Tang, S.C.; Le Gros, G. Staphylococcal-derived superantigen enhances peanut induced Th2 responses in the skin. Clin. Exp. Allergy 2012, 42, 305–314. [Google Scholar] [CrossRef]
- Jones, A.L.; Curran-Everett, D.; Leung, D. Food allergy is associated with Staphylococcus aureus colonization in children with atopic dermatitis. J. Allergy Clin. Immunol. 2016, 137, 1247–1248. [Google Scholar] [CrossRef]
- Patrick, G.J.; Liu, H.; Alphonse, M.P.; Dikeman, D.A.; Youn, C.; Otterson, J.C.; Wang, Y.; Ravipati, A.; Mazhar, M.; Denny, G.; et al. Epicutaneous Staphylococcus aureus induces IL-36 to enhance IgE production and ensuing allergic disease. J. Clin. Investig. 2021, 131, e143334. [Google Scholar] [CrossRef]
- Jang, H.; Matsuda, A.; Jung, K.; Karasawa, K.; Matsuda, K.; Oida, K.; Ishizaka, S.; Ahn, G.; Amagai, Y.; Moon, C.; et al. Skin pH Is the Master Switch of Kallikrein 5-Mediated Skin Barrier Destruction in a Murine Atopic Dermatitis Model. J. Investig. Dermatol. 2016, 136, 127–135. [Google Scholar] [CrossRef]
- Krysko, O.; Teufelberger, A.; van Nevel, S.; Krysko, D.V.; Bachert, C. Protease/antiprotease network in allergy: The role of Staphylococcus aureus protease-like proteins. Allergy 2019, 74, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Oh, M.H.; Park, J.U.; Myers, A.C.; Dong, C.; Zhu, Z.; Zheng, T. Epicutaneous exposure to staphylococcal superantigen enterotoxin B enhances allergic lung inflammation via an IL-17A dependent mechanism. PLoS ONE 2012, 7, e39032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsilochristou, O.; du Toit, G.; Sayre, P.H.; Roberts, G.; Lawson, K.; Sever, M.L.; Bahnson, H.T.; Radulovic, S.; Basting, M.; Plaut, M.; et al. Association of Staphylococcus aureus colonization with food allergy occurs independently of eczema severity. J. Allergy Clin. Immunol. 2019, 144, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Kaplan, M.H.; Turner, M.J.; Kloepfer, K.M.; Kumar, R. Exposure: Staphylococcus aureus skin colonization predisposes to food allergy in the Learning Early about Allergy to Peanut (LEAP) and LEAP-On studies. J. Allergy Clin. Immunol. 2019, 144, 404–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peroni, D.G.; Nuzzi, G.; Trambusti, I.; Di Cicco, M.E.; Comberiati, P. Microbiome Composition and Its Impact on the Development of Allergic Diseases. Front. Immunol. 2020, 11, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.A.; Connolly, J.; Hourihane, J.O.; Fallon, P.G.; McLean, W.; Murray, D.; Jo, J.H.; Segre, J.A.; Kong, H.H.; Irvine, A.D. Skin microbiome before development of atopic dermatitis: Early colonization with commensal staphylococci at 2 months is associated with a lower risk of atopic dermatitis at 1 year. J. Allergy Clin. Immunol. 2017, 139, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuji, T.; Hata, T.R.; Tong, Y.; Cheng, J.Y.; Shafiq, F.; Butcher, A.M.; Salem, S.S.; Brinton, S.L.; Rudman Spergel, A.K.; Johnson, K.; et al. Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nat. Med. 2021, 27, 700–709. [Google Scholar] [CrossRef]
- Hu, T.; Dong, Y.; Yang, C.; Zhao, M.; He, Q. Pathogenesis of Children’s Allergic Diseases: Refocusing the Role of the Gut Microbiota. Front. Physiol. 2021, 14, 749544. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiello, N.; Comberiati, P.; Giannetti, A.; Ricci, G.; Carello, R.; Galli, E. New Directions in Understanding Atopic March Starting from Atopic Dermatitis. Children 2022, 9, 450. https://doi.org/10.3390/children9040450
Maiello N, Comberiati P, Giannetti A, Ricci G, Carello R, Galli E. New Directions in Understanding Atopic March Starting from Atopic Dermatitis. Children. 2022; 9(4):450. https://doi.org/10.3390/children9040450
Chicago/Turabian StyleMaiello, Nunzia, Pasquale Comberiati, Arianna Giannetti, Giampaolo Ricci, Rossella Carello, and Elena Galli. 2022. "New Directions in Understanding Atopic March Starting from Atopic Dermatitis" Children 9, no. 4: 450. https://doi.org/10.3390/children9040450