
Identification of the Rare Ala871Glu Mutation in the Androgen Receptor Gene Leading to Complete Androgen Insensitivity Syndrome in an Adolescent Girl with Primary Amenorrhea
 , , ,
, , ,
Abstract
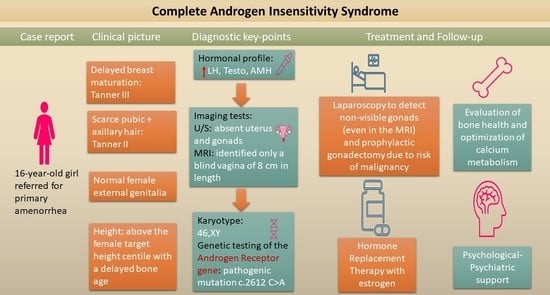
:
1. Introduction
2. Case Report
2.1. Patient Information
2.2. Clinical Findings and Diagnostic Assessment
2.3. Therapeutic Intervention and Outcome
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hughes, I.A.; Deeb, A. Androgen Resistance. Best Pract. Res. Clin. Endocrinol. Metab. 2006, 20, 577–598. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The Nuclear Receptor Superfamily: The Second Decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, A.O.; Faber, P.W.; Van Rooij, H.C.J.; Kuiper, G.G.J.M.; Ris, C.; Klaassen, P.; Van der Korput, J.A.G.M.; Voorhorst, M.M.; Van Laar, J.H.; Mulder, E.; et al. The human androgen receptor: Domain structure, genomic organization and regulation of expression. J. Steroid Biochem. 1989, 34, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Batista, R.L.; Craveiro, F.L.; Ramos, R.M.; Mendonca, B.B. Mild Androgen Insensitivity Syndrome: The Current Landscape. Endocr. Pract. 2022, 28, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, D.T.; Linglart, A.; Morel, Y.; Chaussain, J.-L. Puberty in Subjects with Complete Androgen Insensitivity Syndrome. Horm. Res. Paediatr. 2006, 65, 126–131. [Google Scholar] [CrossRef]
- Hornig, N.C.; Holterhus, P.M. Molecular Basis of Androgen Insensitivity Syndromes. Mol. Cell. Endocrinol. 2021, 523, 111146. [Google Scholar] [CrossRef]
- Chen, F.; Chen, X.; Jiang, F.; Leng, F.; Liu, W.; Gui, Y.; Yu, J. Computational Analysis of Androgen Receptor (AR) Variants to Decipher the Relationship between Protein Stability and Related-Diseases. Sci. Rep. 2020, 10, 12101. [Google Scholar] [CrossRef]
- Hashmi, A.; Hanif, F.; Hanif, S.M.; Abdullah, F.E.; Shamim, M.S. Complete Androgen Insensitivity Syndrome. J. Coll. Physicians Surg. Pak. 2008, 18, 442–444. [Google Scholar]
- Boehmer, A.L.M.; Brüggenwirth, H.; Van Assendelft, C.; Otten, B.J.; Verleun-Mooijman, M.C.T.; Niermeijer, M.F.; Brunner, H.G.; Rouwé, C.W.; Waelkens, J.J.; Oostdijk, W.; et al. Genotype Versus Phenotype in Families with Androgen Insensitivity Syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 4151–4160. [Google Scholar] [CrossRef]
- Audi, L.; Fernández-Cancio, M.; Carrascosa, A.; Andaluz, P.; Torán, N.; Piró, C.; Vilaró, E.; Vicens-Calvet, E.; Gussinyé, M.; Albisu, M.A.; et al. Novel (60%) and Recurrent (40%) Androgen Receptor Gene Mutations in a Series of 59 Patients with a 46, XY Disorder of Sex Development. J. Clin. Endocrinol. Metab. 2010, 95, 1876–1888. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, M.R.; Gubbala, S.P.; Delphine Silvia, C.R.W.; Amanchy, R. Molecular Diagnostics of Disorders of Sexual Development: An Indian Survey and Systems Biology Perspective. Syst. Biol. Reprod. Med. 2019, 65, 105–120. [Google Scholar] [CrossRef]
- Parisi, M.A.; Ramsdell, L.A.; Burns, M.W.; Carr, M.C.; Grady, R.E.; Gunther, D.F.; Kletter, G.B.; McCauley, E.; Mitchell, M.E.; Opheim, K.E.; et al. A Gender Assessment Team: Experience with 250 Patients over a Period of 25 Years. Genet. Med. 2007, 9, 348–357. [Google Scholar] [CrossRef] [Green Version]
- Wisniewski, A.B.; Batista, R.L.; Costa, E.M.F.; Finlayson, C.; Sircili, M.H.P.; Dénes, F.T.; Domenice, S.; Mendonca, B.B. Management of 46, XY Differences/Disorders of Sex Development (DSD) Throughout Life. Endocr. Rev. 2019, 40, 1547–1572. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Berrevoets, C.A.; Doesburg, P.; Steketee, K.; Trapman, J.; Brinkmann, A.O. Functional Interactions of the AF-2 Activation Domain Core Region of the Human Androgen Receptor with the Amino-Terminal Domain and with the Transcriptional Coactivator TIF2 (Transcriptional Intermediary Factor 2). Mol. Endocrinol. 1998, 12, 1172–1183. [Google Scholar] [CrossRef]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural Evidence for Ligand Specificity in the Binding Domain of the Human Androgen Receptor. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, D.; Dermitzaki, E.; Kleanthous, K.; Papadimitriou, A.; Mastorakos, G. MON-541 Successful Treatment of Normocalcemic Hyperparathyroidism in Children. J. Endocr. Soc. 2019, 3, MON-541. [Google Scholar] [CrossRef]
- Kallali, W.; Messiaen, C.; Saïdi, R.; Lessim, S.; Viaud, M.; Dulon, J.; Nedelcu, M.; Samara, D.; Houang, M.; Donadille, B.; et al. Age at Diagnosis in Patients with Chronic Congenital Endocrine Conditions: A Regional Cohort Study from a Reference Center for Rare Diseases. Orphanet. J. Rare Dis. 2021, 16, 469. [Google Scholar] [CrossRef]
- Melo, K.F.S.; Mendonca, B.B.; Billerbeck, A.E.C.; Costa, E.M.F.; Inácio, M.; Silva, F.A.Q.; Leal, A.M.O.; Latronico, A.C.; Arnhold, I.J.P. Clinical, Hormonal, Behavioral, and Genetic Characteristics of Androgen Insensitivity Syndrome in a Brazilian Cohort: Five Novel Mutations in the Androgen Receptor Gene. J. Clin. Endocrinol. Metab. 2003, 88, 3241–3250. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Yao, F.; Tian, T.; Deng, S.; Luo, M.; Tian, Q. Clinical Characteristics and Molecular Genetics of Complete Androgen Insensitivity Syndrome Patients: A Series Study of 30 Cases from a Chinese Tertiary Medical Center. Fertil. Steril. 2021, 115, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, G.; Cosci o di Coscio, M.; Masini, B.; Baldinotti, F.; Caligo, M.A.; Tyutyusheva, N.; Sessa, M.R.; Peroni, D.; Bertelloni, S. Disorders of Sexual Development with XY Karyotype and Female Phenotype: Clinical Findings and Genetic Background in a Cohort from a Single Centre. J. Endocrinol. Investig. 2021, 44, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yin, X.; Li, P. Clinical, Hormonal and Genetic Characteristics of Androgen Insensitivity Syndrome in 39 Chinese Patients. Reprod. Biol. Endocrinol. 2020, 18, 34. [Google Scholar] [CrossRef] [PubMed]
- Cheikhelard, A.; Morel, Y.; Thibaud, E.; Lortat-Jacob, S.; Jaubert, F.; Polak, M.; Nihoul-Fekete, C. Long-Term Followup and Comparison Between Genotype and Phenotype in 29 Cases of Complete Androgen Insensitivity Syndrome. J. Urol. 2008, 180, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Hannema, S.; Scott, I.; Rajpert-De Meyts, E.; Skakkebæk, N.; Coleman, N.; Hughes, I. Testicular Development in the Complete Androgen Insensitivity Syndrome. J. Pathol. 2006, 208, 518–527. [Google Scholar] [CrossRef]
- Van, Y.H.; Lin, J.L.; Huang, S.F.; Luo, C.C.; Hwang, C.S.; Lo, F.S. Novel Point Mutations in Complete Androgen Insensitivity Syndrome with Incomplete Müllerian Regression: Two Taiwanese Patients. Eur. J. Pediatr. 2003, 162, 781–784. [Google Scholar] [CrossRef]
- Güven, A.; Dursun, F.; Özkanli, S.; Güçlüer, B.; Kuru, L.I. Complete Androgen Insensitivity Syndrome and Discordant Müllerian Remnants: Two Cases with Novel Mutation in the Androgen Receptor. J. Pediatr. Endocrinol. Metab. 2013, 26, 909–914. [Google Scholar] [CrossRef]
- Nichols, J.L.; Bieber, E.J.; Gell, J.S. Case of Sisters with Complete Androgen Insensitivity Syndrome and Discordant Müllerian Remnants. Fertil. Steril. 2009, 91, 932.e15–932.e18. [Google Scholar] [CrossRef]
- Cools, M.; Looijenga, L. Update on the Pathophysiology and Risk Factors for the Development of Malignant Testicular Germ Cell Tumors in Complete Androgen Insensitivity Syndrome. Sex. Dev. 2017, 11, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Barros, B.A.; de Oliveira, L.R.; Surur, C.R.C.; Barros-Filho, A.D.A.; Maciel-Guerra, A.T.; Guerra-Junior, G. Complete Androgen Insensitivity Syndrome and Risk of Gonadal Malignancy: Systematic Review. Ann. Pediatr. Endocrinol. Metab. 2021, 26, 19–23. [Google Scholar] [CrossRef]
- Ouyang, Y.; Tan, S.; Yu, Y.; Luo, B.; Yin, W.; Luo, L. Gonadal Tumor and Malignancy in 118 Patients with Disorders of Sex Development with Y Chromosome. Int. J. Gynecol. Obstet. 2021, 158, 285–288. [Google Scholar] [CrossRef]
- Sandberg, D.; Gardner, M.; Cohen-Kettenis, P. Psychological Aspects of the Treatment of Patients with Disorders of Sex Development. Semin. Reprod. Med. 2012, 30, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Danilovic, D.L.S.; Correa, P.H.S.; Costa, E.M.F.; Melo, K.F.S.; Mendonca, B.B.; Arnhold, I.J.P. Height and Bone Mineral Density in Androgen Insensitivity Syndrome with Mutations in the Androgen Receptor Gene. Osteoporos. Int. 2007, 18, 369–374. [Google Scholar] [CrossRef]
- King, T.F.J.; Wat, W.Z.M.; Creighton, S.M.; Conway, G.S. Bone Mineral Density in Complete Androgen Insensitivity Syndrome and the Timing of Gonadectomy. Clin. Endocrinol. 2017, 87, 136–140. [Google Scholar] [CrossRef]
- Bertelloni, S.; Baroncelli, G.I.; Federico, G.; Cappa, M.; Lala, R.; Saggese, G. Altered Bone Mineral Density in Patients with Complete Androgen Insensitivity Syndrome. Horm. Res. 1998, 50, 309–314. [Google Scholar] [CrossRef]
- Han, T.S.; Goswami, D.; Trikudanathan, S.; Creighton, S.M.; Conway, G.S. Comparison of Bone Mineral Density and Body Proportions between Women with Complete Androgen Insensitivity Syndrome and Women with Gonadal Dysgenesis. Eur. J. Endocrinol. 2008, 159, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Gava, G.; Mancini, I.; Orsili, I.; Bertelloni, S.; Alvisi, S.; Seracchioli, R.; Meriggiola, M.C. Bone Mineral Density, Body Composition and Metabolic Profiles in Adult Women with Complete Androgen Insensitivity Syndrome and Removed Gonads Using Oral or Transdermal Estrogens. Eur. J. Endocrinol. 2019, 181, 711–718. [Google Scholar] [CrossRef]
- Muñoz-Torres, M.; Jódar, E.; Quesada, M.; Escobar-Jiménez, F. Bone Mass in Androgen-Insensitivity Syndrome: Response to Hormonal Replacement Therapy. Calcif. Tissue Int. 1995, 57, 94–96. [Google Scholar] [CrossRef]
- Misakian, A.; McLoughlin, M.; Pyle, L.C.; Kolon, T.F.; Kelly, A.; Vogiatzi, M.G. Case Report: Low Bone and Normal Lean Mass in Adolescents with Complete Androgen Insensitivity Syndrome. Front. Endocrinol. 2021, 12, 727131. [Google Scholar] [CrossRef]
- Papadimitriou, D.T.; Chrysis, D.; Nyktari, G.; Zoupanos, G.; Liakou, E.; Papadimitriou, A.; Mastorakos, G. Replacement of Male Mini-Puberty. J. Endocr. Soc. 2019, 3, 1275–1282. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.F.; Cheng, A.; Hughes, I.A. Assessment of the Gonadotrophin-Gonadal Axis in Androgen Insensitivity Syndrome. Arch. Dis. Child. 1999, 80, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.R.; Foley, T.P.; Corvol, P.; Park, I.-J.; Kowarski, A.; Blizzard, R.M.; Jones, H.W.; Migeon, C.J. Plasma concentration of testosterone, dihydrotestosterone, testosterone-oestradiol binding globulin, and pituitary gonadotrophins in the syndrome of male pseudo-hermaphroditism with testicular feminization. Acta Endocrinol. 1972, 70, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Rey, R.; Mebarki, F.; Forest, M.G.; Mowszowicz, I.; Cate, R.L.; Morel, Y.; Chaussain, J.L.; Josso, N. Anti-Müllerian Hormone in Children with Androgen Insensitivity. J. Clin. Endocrinol. Metab. 1994, 79, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Rey, R.A.; Belville, C.; Nihoul-Fékété, C.; Michel-Calemard, L.; Forest, M.G.; Lahlou, N.; Jaubert, F.; Mowszowicz, I.; David, M.; Saka, N.; et al. Evaluation of Gonadal Function in 107 Intersex Patients by Means of Serum Antimüllerian Hormone Measurement. J. Clin. Endocrinol. Metab. 1999, 84, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Troiano, R.N.; McCarthy, S.M. Müllerian Duct Anomalies: Imaging and Clinical Issues. Radiology 2004, 233, 19–34. [Google Scholar] [CrossRef]
- Hederström, E.; Forsberg, L.; Kullendorff, C.-M. Ultrasonography of the Undescended Testis. Acta Radiol. Diagn. 1985, 26, 453–456. [Google Scholar] [CrossRef]
- Kanemoto, K.; Hayashi, Y.; Kojima, Y.; Maruyama, T.; Ito, M.; Kohri, K. Accuracy of Ultrasonography and Magnetic Resonance Imaging in the Diagnosis of Non-Palpable Testis. Int. J. Urol. 2005, 12, 668–672. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Achermann, J.; Alderson, J.; Crouch, N.S.; Elford, S.; Hughes, I.A.; Krone, N.; McGowan, R.; Mushtaq, T.; O’Toole, S.; et al. Society for Endocrinology UK Guidance on the Initial Evaluation of a Suspected Difference or Disorder of Sex Development (Revised 2021). Clin. Endocrinol. 2021, 95, 818–840. [Google Scholar] [CrossRef]
- Seppä, S.; Kuiri-Hänninen, T.; Holopainen, E.; Voutilainen, R. MANAGEMENT OF ENDOCRINE DISEASE: Diagnosis and Management of Primary Amenorrhea and Female Delayed Puberty. Eur. J. Endocrinol. 2021, 184, R225–R242. [Google Scholar] [CrossRef]
- Akcay, T.; Fernandez-Cancio, M.; Turan, S.; Güran, T.; Audi, L.; Bereket, A. AR and SRD5A2 Gene Mutations in a Series of 51 Turkish 46, XY DSD Children with a Clinical Diagnosis of Androgen Insensitivity. Andrology 2014, 2, 572–578. [Google Scholar] [CrossRef]
- Hiort, O.; Klauber, G.; Cendron, M.; Sinnecker, G.H.G.; Keim, L.; Schwinger, E.; Wolfe, H.J.; Yandell, D.W. Molecular Characterization of the Androgen Receptor Gene in Boys with Hypospadias. Eur. J. Pediatr. 1994, 153, 317–321. [Google Scholar] [CrossRef]
- Hiort, O.; Sinnecker, G.H.G.; Holterbus, P.M.; Nitsche, E.M.; Kruse, K. Inherited and de Novo Androgen Receptor Gene Mutations: Investigation of Single-Case Families. J. Pediatr. 1998, 132, 939–943. [Google Scholar] [CrossRef]
- Albers, N.; Ulrichs, C.; Gluer, S.; Hiort, O.; Sinnecken, G.H.G.; Mildenberger, H.; Brodehl, J. Etiologic Classification of Severe Hypospadias: Implications for Prognosis and Management. J. Pediatr. 1997, 131, 386–392. [Google Scholar] [CrossRef]
- Zenteno, J.C.; Chávez, B.; Vilchis, F.; Kofman-Alfaro, S. Phenotypic Heterogeneity Associated with Identical Mutations in Residue 870 of the Androgen Receptor. Horm. Res. 2002, 57, 90–93. [Google Scholar] [CrossRef]
- Bhangoo, A.; Paris, F.; Philibert, P.; Audran, F.; Ten, S.; Sultan, C. Isolated Micropenis Reveals Partial Androgen Insensitivity Syndrome Confirmed by Molecular Analysis. Asian J. Androl. 2010, 12, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Cheng, J.; Yin, X.; Liu, G.; Lu, Z.; Sheng, H.; Cai, Y.; Shi, Q.; Liu, L. Clinical and Molecular Characteristics in 15 Patients with Androgen Receptor Gene Mutations from South China. Andrologia 2017, 49, e12763. [Google Scholar] [CrossRef]
- Knoke, I.; Jakubiczka, S.; Ottersen, T.; Göppinger, A.; Wieacker, P. A (870) E Mutation of the Androgen Receptor Gene in a Patient with Complete Androgen Insensitivity Syndrome and Sertoli Cell Tumor. Cancer Genet. Cytogenet. 1997, 98, 139–141. [Google Scholar] [CrossRef]
- Lee, P.A.; Nordenström, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.-M.; Lin-Su, K.; Looijenga, L.H.J., 3rd; et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef]
- Kopper, N.W.; Gudeman, J.; Thompson, D.J. Transdermal Hormone Therapy in Postmenopausal Women: A Review of Metabolic Effects and Drug Delivery Technologies. Drug Des. Devel. Ther. 2008, 2, 193. [Google Scholar] [CrossRef] [Green Version]
- Rohr, U.D.; Volko, C.D.; Schindler, A.E. Comparison of Steady State Development and Reduction of Menopausal Symptoms after Oral or Transdermal Delivery of 17-β-Estradiol in Young Healthy Symptomatic Menopausal Women. Horm. Mol. Biol. Clin. Investig. 2014, 18, 123–136. [Google Scholar] [CrossRef]
- Birnbaum, W.; Marshall, L.; Werner, R.; Kulle, A.; Holterhus, P.M.; Rall, K.; Köhler, B.; Richter-Unruh, A.; Hartmann, M.F.; Wudy, S.A.; et al. Oestrogen versus Androgen in Hormone-Replacement Therapy for Complete Androgen Insensitivity Syndrome: A Multicentre, Randomised, Double-Dummy, Double-Blind Crossover Trial. Lancet Diabetes Endocrinol. 2018, 6, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Engberg, H.; Strandqvist, A.; Nordenström, A.; Butwicka, A.; Nordenskjöld, A.; Hirschberg, A.L.; Frisén, L. Increased Psychiatric Morbidity in Women with Complete Androgen Insensitivity Syndrome or Complete Gonadal Dysgenesis. J. Psychosom. Res. 2017, 101, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Khorashad, B.S.; Aghili, Z.; Kreukels, B.P.C.; Reid, A.G.; Roshan, G.M.; Hiradfar, M.; Talaei, A.; Cohen Kettenis, P.T. Mental Health and Disorders of Sex Development/Intersex Conditions in Iranian Culture: Congenital Adrenal Hyperplasia, 5-α Reductase Deficiency-Type 2, and Complete Androgen Insensitivity Syndrome. Arch. Sex. Behav. 2018, 47, 931–942. [Google Scholar] [CrossRef]
- D’Alberton, F.; Assante, M.T.; Foresti, M.; Balsamo, A.; Bertelloni, S.; Dati, E.; Nardi, L.; Bacchi, M.L.; Mazzanti, L. Quality of Life and Psychological Adjustment of Women Living with 46, XY Differences of Sex Development. J. Sex. Med. 2015, 12, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Dati, E.; Baroncelli, G.I.; Mora, S.; Russo, G.; Baldinotti, F.; Parrini, D.; Erba, P.; Simi, P.; Bertelloni, S. Body Composition and Metabolic Profile in Women with Complete Androgen Insensitivity Syndrome. Sex. Dev. 2009, 3, 188–193. [Google Scholar] [CrossRef]




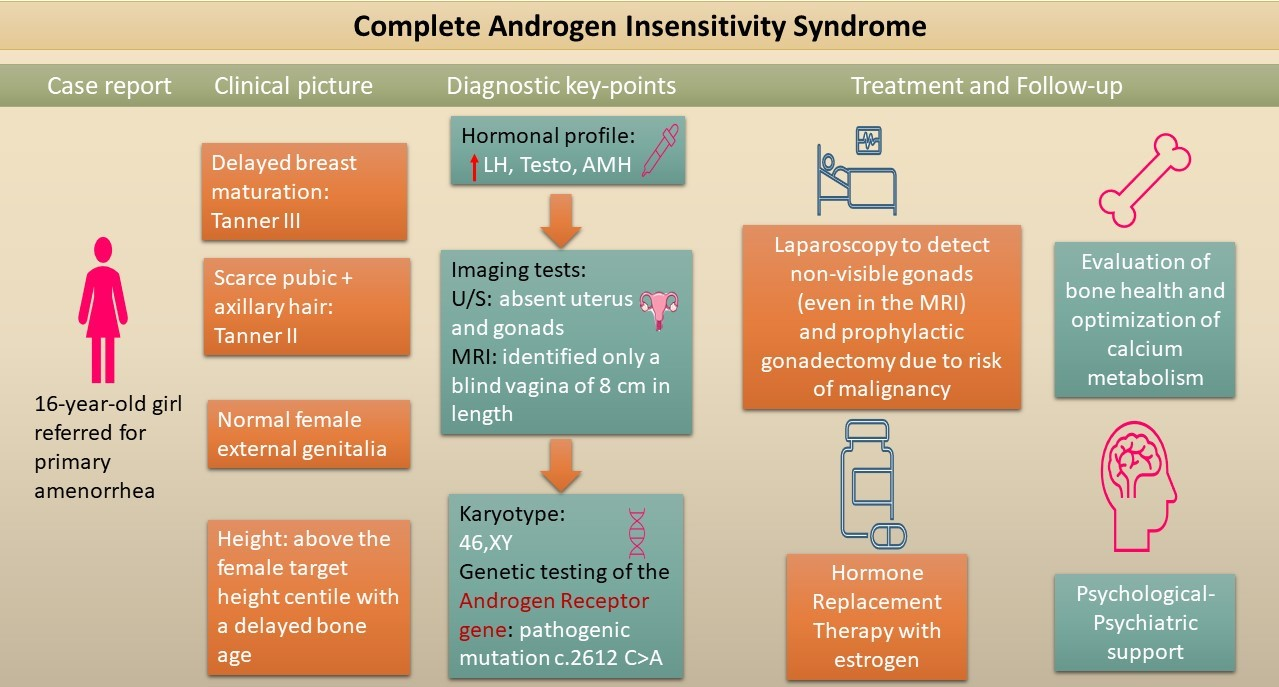{kind=link}
{kind=link}
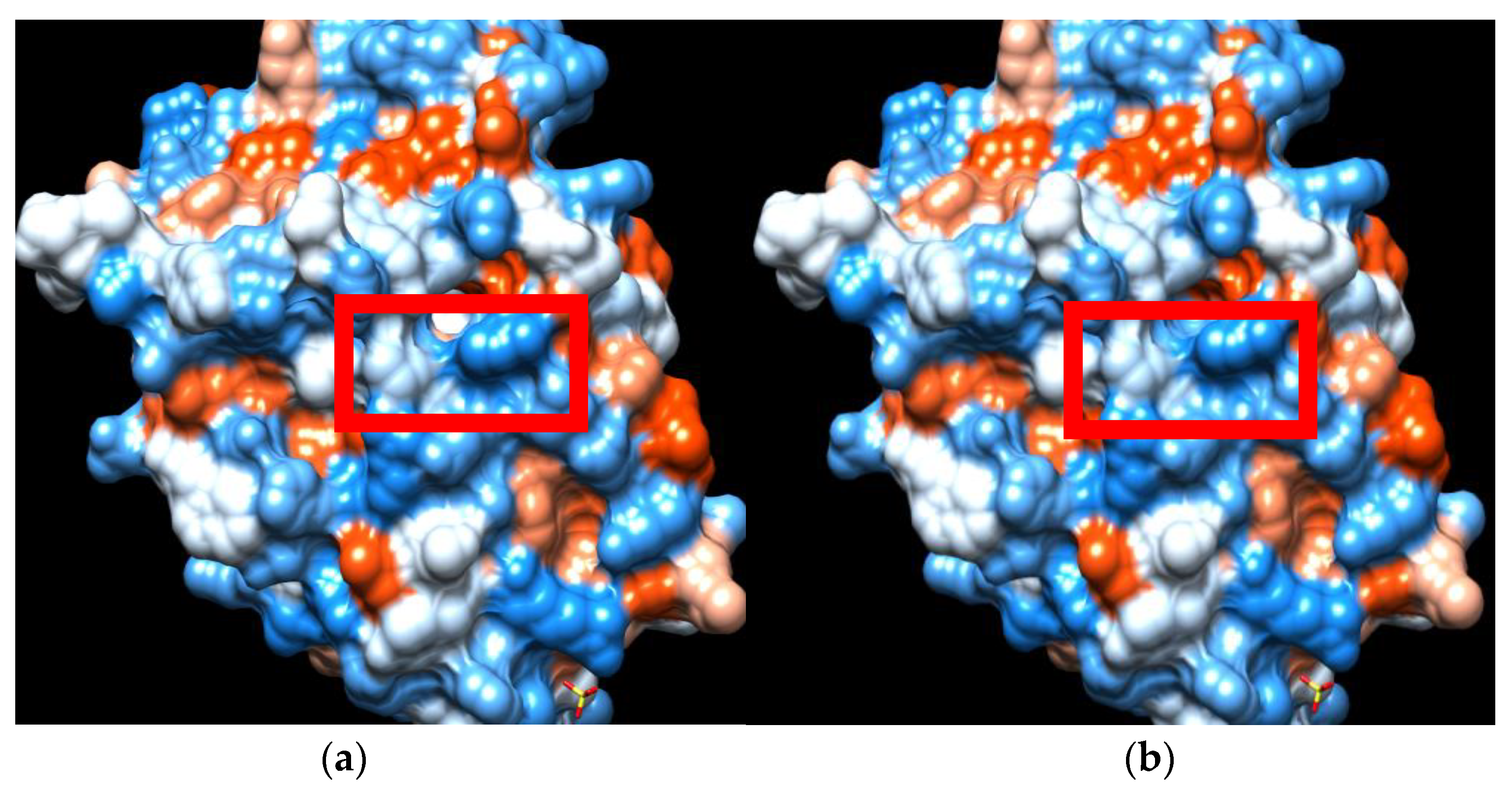{kind=link}
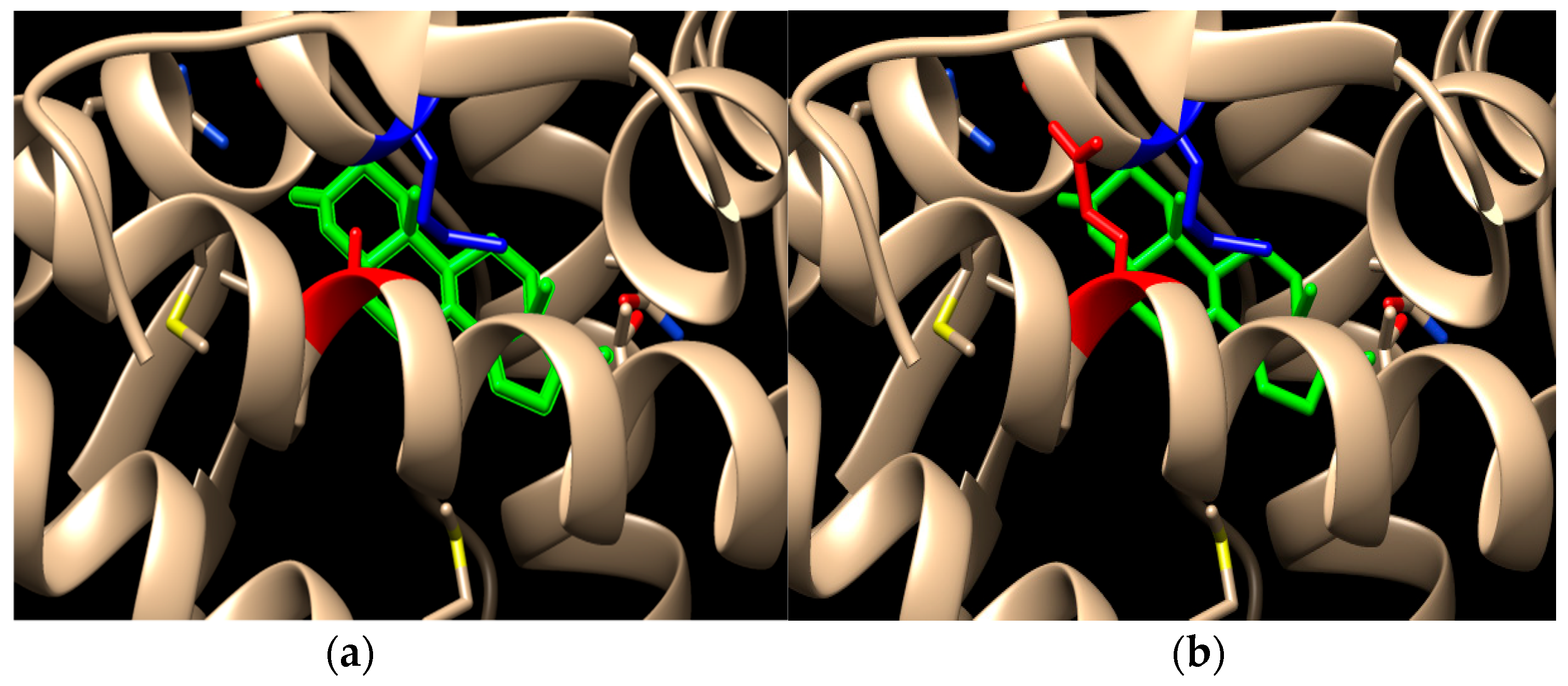{kind=link}
| At Diagnosis | 1 Month after Surgery, on Oral HRT | On Transdermal HRT | Reference Values | |
|---|---|---|---|---|
| FSH (IU/L) | 5.6 | 111.9 | 2.6 | 0.1–7.2 |
| LH (IU/L) | 18.2 | 41 | 0.2 | <11.9 (age 11–14) <14.6 (follicular phase <18 yrs) |
| Estradiol (pg/mL) | 30.7 | 21 | 476.3 | <60 |
| T (ng/dL) | 680 | 14 | N/A | 17–75 |
| DHT (pg/mL) | 400 | N/A | N/A | <300 |
| AMH (ng/mL) | 10.6 | N/A | N/A | 0.6–7.8 (age 15–19) |
| SHBG (nmol/L) | 57.2 | N/A | N/A | 18–87 |
| Phenotype | Age | Mutation | Reference |
|---|---|---|---|
| PAIS (Quigley scale 3) | 3 | Ala871Ala | [50] |
| PAIS (hypospadias and cryptorchidism) | Ala871Val | [51] | |
| PAIS (hypospadias and micropenis) | Ala871Val | [52] | |
| PAIS (hypospadias) | 4 | Ala871Val | [53] |
| MAIS (bilateral gynecomastia) | 24 | Ala871Val | [54] |
| PAIS (isolated micropenis) | 3 | Ala871Val | [55] |
| PAIS (hypospadias and gynecomastia) | 1 | Ala871Val | [10] |
| PAIS (hypospadias and Wilms tumor) | 3 | Ala871Val | [56] |
| PAIS (female with virilized external genitalia) | Ala871Gly | [52] | |
| CAIS (female with Sertoli cell tumor) | 60 and 57 | Ala871Glu | [57] |
| CAIS | 16 | Ala871Glu | This report |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapama, A.; Papadimitriou, D.T.; Mastorakos, G.; Vlahos, N.F.; Papagianni, M. Identification of the Rare Ala871Glu Mutation in the Androgen Receptor Gene Leading to Complete Androgen Insensitivity Syndrome in an Adolescent Girl with Primary Amenorrhea. Children 2022, 9, 1900. https://doi.org/10.3390/children9121900
Kapama A, Papadimitriou DT, Mastorakos G, Vlahos NF, Papagianni M. Identification of the Rare Ala871Glu Mutation in the Androgen Receptor Gene Leading to Complete Androgen Insensitivity Syndrome in an Adolescent Girl with Primary Amenorrhea. Children. 2022; 9(12):1900. https://doi.org/10.3390/children9121900
Chicago/Turabian StyleKapama, Aikaterini, Dimitrios T. Papadimitriou, George Mastorakos, Nikolaos F. Vlahos, and Maria Papagianni. 2022. "Identification of the Rare Ala871Glu Mutation in the Androgen Receptor Gene Leading to Complete Androgen Insensitivity Syndrome in an Adolescent Girl with Primary Amenorrhea" Children 9, no. 12: 1900. https://doi.org/10.3390/children9121900