
Clinical and Functional Characteristics of a New Phenotype of SMA Type I among a National Sample of Spanish Children: A Cross-Sectional Study
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
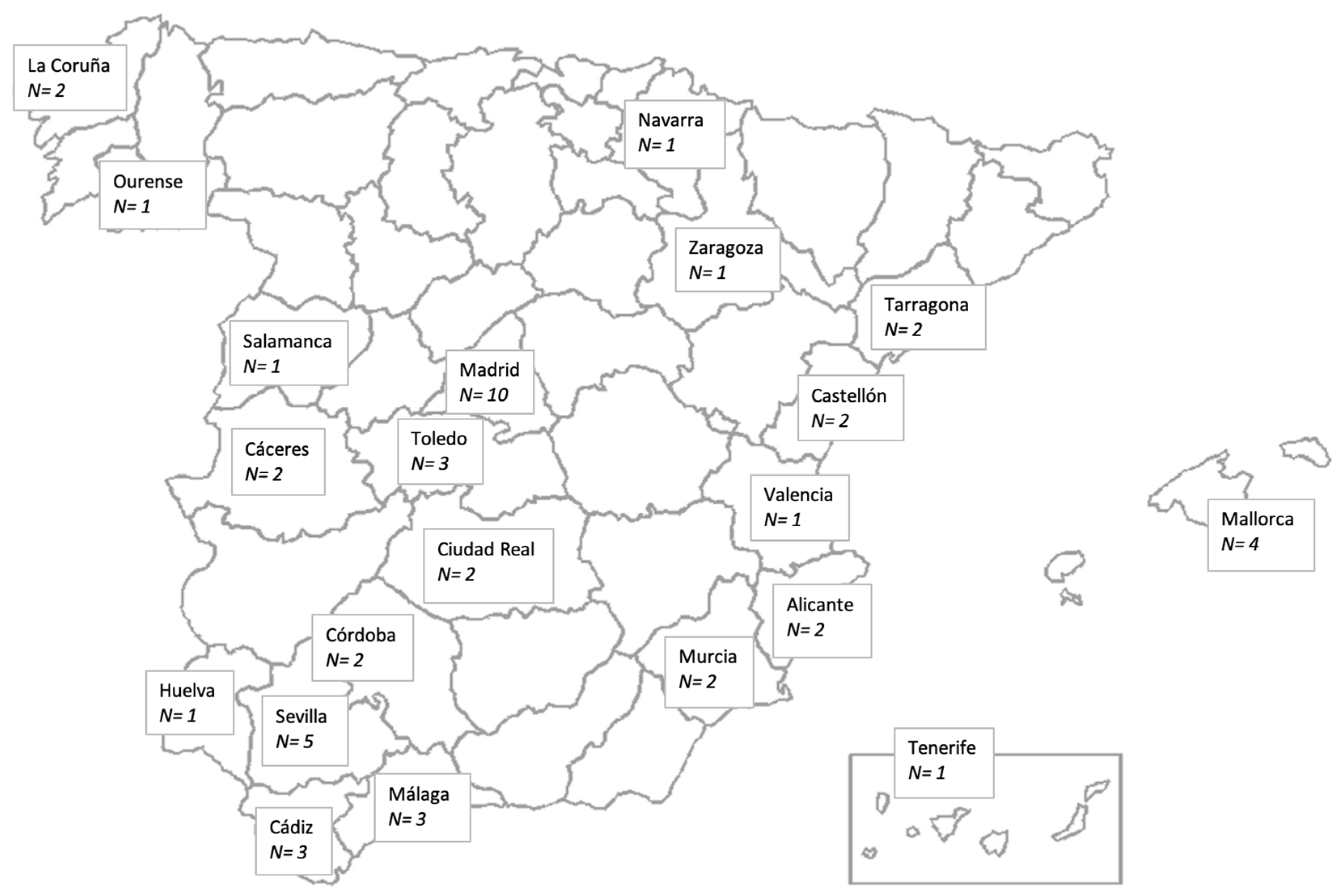
2.2. Participants and Recruitment
2.3. Data Collection
2.3.1. Sociodemographic and Physical Conditions
2.3.2. Functional Assessments
2.4. Analysis of Results
2.5. Ethical Considerations
3. Results
3.1. Patient Characteristics and Demographics
3.2. Clinical and Functional Outcomes
3.3. Standardized Assessments
3.4. Therapies and Technical Aids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lefebvre, S.; Bürglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol. Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.D.; Kassar, D.; Kissel, J.T. Spinal Muscular Atrophy: Diagnosis and Management in a New Therapeutic Era. Muscle Nerve 2015, 51, 157–167. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet. J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef]
- Arnold, E.S.; Fischbeck, K.H. Spinal muscular atrophy. Handb. Clin. Neurol. 2018, 148, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Darras, B.T. Spinal Muscular Atrophies. Pediatr. Clin. N. Am. 2015, 62, 743–766. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.H.; Dubowitz, V. The Natural History of Type I (Severe) Spinal Muscular Atrophy. Neuromuscul. Disord. 1994, 4, 497–502. [Google Scholar] [CrossRef]
- Zerres, K.; Rudnik-Schöneborn, S. Natural History in Proximal Spinal Muscular Atrophy. Clinical Analysis of 445 Patients and Suggestions for a Modification of Existing Classifications. Arch. Neurol. 1995, 52, 518–523. [Google Scholar] [CrossRef]
- Bach, J.R. Medical Considerations of Long-Term Survival of Werdnig-Hoffmann Disease. Am. J. Phys. Med. Rehabil. 2007, 86, 349–355. [Google Scholar] [CrossRef]
- Finkel, R.S.; McDermott, M.P.; Kaufmann, P.; Darras, B.T.; Chung, W.K.; Sproule, D.M.; Kang, P.B.; Reghan Foley, A.; Yang, M.L.; Martens, W.B.; et al. Observational Study of Spinal Muscular Atrophy Type I and Implications for Clinical Trials. Neurology 2014, 83, 810–817. [Google Scholar] [CrossRef]
- Kolb, S.J.; Coffey, C.S.; Yankey, J.W.; Krosschell, K.; Arnold, W.D.; Rutkove, S.B.; Swoboda, K.J.; Reyna, S.P.; Sakonju, A.; Darras, B.T.; et al. Natural History of Infantile-Onset Spinal Muscular Atrophy. Ann. Neurol. 2017, 82, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H. New and Developing Therapies in Spinal Muscular Atrophy: From Genotype to Phenotype to Treatment and Where Do We Stand? Int. J. Mol. Sci. 2020, 21, 3297. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, R.; Pane, M.; Coratti, G.; Palermo, C.; Leone, D.; Pera, M.C.; Abiusi, E.; Fiori, S.; Forcina, N.; Fanelli, L.; et al. Clinical Phenotypes and Trajectories of Disease Progression in Type 1 Spinal Muscular Atrophy. Neuromuscul. Disord. 2018, 28, 24–28. [Google Scholar] [CrossRef]
- Oskoui, M.; Levy, G.; Garland, C.J.; Gray, J.M.; O’Hagen, J.; De Vivo, D.C.; Kaufmann, P. The Changing Natural History of Spinal Muscular Atrophy Type 1. Neurology 2007, 69, 1931–1936. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.H.Y.; Wong, V.C.N.; Ip, P. Spinal Muscular Atrophy: Survival Pattern and Functional Status. Pediatrics 2004, 114, e548–e553. [Google Scholar] [CrossRef] [PubMed]
- Tizzano, E.F. La Atrofia Muscular Espinal En El Nuevo Escenario Terapéutico. Rev. Médica Clínica Las Condes 2018, 29, 512–520. [Google Scholar] [CrossRef]
- Mercuri, E.; Finkel, R.S.; Muntoni, F.; Wirth, B.; Montes, J.; Main, M.; Mazzone, E.; Vitale, M.; Snyder, B.; Quijano-Roy, S.; et al. Diagnosis and Management of Spinal Muscular Atrophy: Part 1: Recommendations for Diagnosis, Rehabilitation, Orthopedic and Nutritional Care. Neuromuscul. Disord. 2018, 28, 103–115. [Google Scholar] [CrossRef]
- Tizzano, E.F.; Finkel, R.S. Spinal Muscular Atrophy: A Changing Phenotype beyond the Clinical Trials. Neuromuscul. Disord. 2017, 27, 883–889. [Google Scholar] [CrossRef]
- Valencia, B.; Bach, J.R. Eighteen Years with Spinal Muscular Atrophy (SMA) Type 1. Tanaffos 2013, 12, 70–73. [Google Scholar] [PubMed]
- Haaker, G.; Fujak, A. Proximal Spinal Muscular Atrophy: Current Orthopedic Perspective. Appl. Clin. Genet 2013, 6, 113–120. [Google Scholar] [CrossRef]
- Mercuri, E.; Lucibello, S.; Pera, M.C.; Carnicella, S.; Coratti, G.; De Sanctis, R.; Messina, S.; Mazzone, E.; Forcina, N.; Fanelli, L.; et al. Long-Term Progression in Type II Spinal Muscular Atrophy: A Retrospective Observational Study. Neurology 2019, 93, E1241–E1247. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.; Drennan, J.; Russman, B. Functional Classification and Orthopaedic Management in Spinal Muscular Atrophy. J. Bone Jt. Surg. Br. 1981, 63, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Fujak, A.; Raab, W.; Schuh, A.; Richter, S.; Forst, R.; Forst, J. Natural Course of Scoliosis in Proximal Spinal Muscular Atrophy Type II and IIIa: Descriptive Clinical Study with Retrospective Data Collection of 126 Patients. BMC Musculoskelet. Disord. 2013, 14, 283. [Google Scholar] [CrossRef] [PubMed]
- Pechmann, A.; König, K.; Bernert, G.; Schachtrup, K.; Schara, U.; Schorling, D.; Schwersenz, I.; Stein, S.; Tassoni, A.; Vogt, S.; et al. SMArtCARE—A Platform to Collect Real-Life Outcome Data of Patients with Spinal Muscular Atrophy. Orphanet. J Rare Dis. 2019, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, J.P.; Von Elm, E.; Altman, D.G.; Gøtzsche, P.C.; Mulrow, C.D.; Pocock, S.J.; Poole, C.; Schlesselman, J.J.; Egger, M. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): Explanation and Elaboration. PLoS Med. 2007, 4, 1628–1654. [Google Scholar] [CrossRef]
- Glanzman, A.M.; Mazzone, E.; Main, M.; Pelliccioni, M.; Wood, J.; Swoboda, K.J.; Scott, C.; Pane, M.; Messina, S.; Bertini, E.; et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test Development and Reliability The CHOP INTEND Is a Reliable Measure of Motor Skills in Patients with SMA-I and Neuromuscular Disorders Presenting in Infancy. Neuromuscul. Disord. 2010, 20, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Glanzman, A.M.; McDermott, M.P.; Montes, J.; Martens, W.B.; Flickinger, J.; Riley, S.; Quigley, J.; Dunaway, S.; O’Hagen, J.; Deng, L.; et al. Validation of the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatr. Phys. Ther. 2011, 23, 322–326. [Google Scholar] [CrossRef]
- Haataja, L.; Mercuri, E.; Regev, R.; Cowan, F.; Rutherford, M.; Dubowitz, V.; Dubowitz, L. Optimality Score for the Neurologic Examination of the Infant at 12 and 18 Months of Age. J. Pediatr. 1999, 135, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, L.; Dubowitz, V.; Mercuri, E. The Neurological Assessment of the Preterm and Full-Term Newborn Infant; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- De Sanctis, R.; Coratti, G.; Pasternak, A.; Montes, J.; Pane, M.; Mazzone, E.S.; Young, S.D.; Salazar, R.; Quigley, J.; Pera, M.C.; et al. Developmental Milestones in Type I Spinal Muscular Atrophy. Neuromuscul. Disord. 2016, 26, 754–759. [Google Scholar] [CrossRef]
- O’Hagen, J.M.; Glanzman, A.M.; McDermott, M.P.; Ryan, P.A.; Flickinger, J.; Quigley, J.; Riley, S.; Sanborn, E.; Irvine, C.; Martens, W.B.; et al. An Expanded Version of the Hammersmith Functional Motor Scale for SMA II and III Patients. Neuromuscul. Disord. 2007, 17, 693–697. [Google Scholar] [CrossRef]
- Glanzman, A.M.; O’Hagen, J.M.; McDermott, M.P.; Martens, W.B.; Flickinger, J.; Riley, S.; Quigley, J.; Montes, J.; Dunaway, S.; Deng, L.; et al. Validation of the Expanded Hammersmith Functional Motor Scale in Spinal Muscular Atrophy Type II and III. J. Child Neurol. 2011, 26, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Montes, J.; Gordon, A.M.; Pandya, S.; De Vivo, D.C.; Kaufmann, P. Clinical Outcome Measures in Spinal Muscular Atrophy. J. Child Neurol. 2009, 24, 968–978. [Google Scholar] [CrossRef]
- Mazzone, E.S.; Mayhew, A.; Montes, J.; Ramsey, D.; Fanelli, L.; Young, S.D.; Salazar, R.; De Sanctis, R.; Pasternak, A.; Glanzman, A.; et al. Revised Upper Limb Module for Spinal Muscular Atrophy: Development of a New Module. Muscle Nerve 2017, 55, 869–874. [Google Scholar] [CrossRef]
- Ou, S.F.; Ho, C.S.; Lee, W.T.; Lin, K.L.; Jones, C.C.; Jong, Y.J. Natural History in Spinal Muscular Atrophy Type I in Taiwanese Population: A Longitudinal Study. Brain Dev. 2021, 43, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Cances, C.; Vlodavets, D.; Comi, G.P.; Masson, R.; Mazurkiewicz-Bełdzińska, M.; Saito, K.; Zanoteli, E.; Dodman, A.; El-Khairi, M.; Gorni, K.; et al. ANCHOVY Working Group. Natural History of Type 1 Spinal Muscular Atrophy: A Retrospective, Global, Multicenter Study. Orphanet. J. Rare Dis. 2022, 17, 300. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Pane, M.; De Rose, P.; Vasta, I.; Sorleti, D.; Aloysius, A.; Sciarra, F.; Mangiola, F.; Kinali, M.; Bertini, E.; et al. Feeding Problems and Malnutrition in Spinal Muscular Atrophy Type II. Neuromuscul. Disord. 2008, 18, 389–393. [Google Scholar] [CrossRef]
- Fujak, A.; Kopschina, C.; Forst, R.; Mueller, L.A.; Forst, J. Use of Orthoses and Orthopaedic Technical Devices in Proximal Spinal Muscular Atrophy. Results of Survey in 194 SMA Patients. Disabil. Rehabil. Assist. Technol. 2011, 6, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Zenios, M.; Sampath, J.; Cole, C.; Khan, T.; Galasko, C.S.B. Operative Treatment for Hip Subluxation in Spinal Muscular Atrophy. J. Bone Jt. Surg. Ser. B 2005, 87, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Granata, C.; Bonfiglioli, S.; Marini, M.L.; Cervellati, S.; Savini, R. Scoliosis in Spinal Muscular Atrophy: Natural History and Management. Dev. Med. Child Neurol. 1989, 31, 501–508. [Google Scholar] [CrossRef]
- Granata, C.; Merlini, L.; Magni, E.; Marini, M.L.; Stagni, S.B. Spinal Muscular Atrophy: Natural History and Orthopaedic Treatment of Scoliosis. Spine 1989, 14, 760–762. [Google Scholar] [CrossRef]
- Al Amrani, F.; Amin, R.; Chiang, J.; Xiao, L.; Boyd, J.; Law, E.; Nigro, E.; Weinstock, L.; Stosic, A.; Gonorazky, H.D. Scoliosis in Spinal Muscular Atrophy Type 1 in the Nusinersen Era. Neurol. Clin. Pract. 2022, 12, 279–287. [Google Scholar] [CrossRef]
- Sporer, S.M.; Smith, B.G. Hip Dislocation in Patients with Spinal Muscular Atrophy. J. Pediatr. Orthop. 2003, 23, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Wijngaarde, C.A.; Brink, R.C.; De Kort, F.A.S.; Stam, M.; Otto, L.A.M.; Asselman, F.L.; Bartels, B.; Van Eijk, R.P.A.; Sombroek, J.; Cuppen, I.; et al. Natural Course of Scoliosis and Lifetime Risk of Scoliosis Surgery in Spinal Muscular Atrophy. Neurology 2019, 93, E149–E158. [Google Scholar] [CrossRef]
- Canavese, F.; Sussman, M.D. Strategies of Hip Management in Neuromuscular Disorders: Duchenne Muscular Dystrophy, Spinal Muscular Atrophy, Charcot-Marie-Tooth Disease and Arthrogryposis Multiplex Congenita. Hip Int. 2009, 19 (Suppl. S6), S46–S52. [Google Scholar] [CrossRef] [PubMed]
- Granata, C.; Magni, E.; Merlini, L.; Cervellati, S. Hip Dislocation in Spinal Muscular Atrophy. Chir. Organi Mov. 1990, 75, 177–184. [Google Scholar]
- Townsend, E.L.; Simeone, S.D.; Krosschell, K.J.; Zhang, R.Z.; Swoboda, K.J. Stander Use in Spinal Muscular Atrophy: Results from a Large Natural History Database. Pediatr. Phys. Ther. 2020, 32, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Pane, M.; Coratti, G.; Sansone, V.A.; Messina, S.; Catteruccia, M.; Bruno, C.; Sframeli, M.; Albamonte, E.; Pedemonte, M.; D’Amico, A.; et al. Type I SMA “New Natural History”: Long-Term Data in Nusinersen-Treated Patients. Ann. Clin. Transl. Neurol. 2021, 8, 548–557. [Google Scholar] [CrossRef]
- De Vivo, D.C.; Bertini, E.; Swoboda, K.J.; Hwu, W.L.; Crawford, T.O.; Finkel, R.S.; Kirschner, J.; Kuntz, N.L.; Parsons, J.A.; Ryan, M.M.; et al. Nusinersen Initiated in Infants during the Presymptomatic Stage of Spinal Muscular Atrophy: Interim Efficacy and Safety Results from the Phase 2 NURTURE Study. Neuromuscul. Disord. 2019, 29, 842–856. [Google Scholar] [CrossRef]
- Bray, N.; Kolehmainen, N.; McAnuff, J.; Tanner, L.; Tuersley, L.; Beyer, F.; Grayston, A.; Wilson, D.; Edwards, R.T.; Noyes, J.; et al. Powered Mobility Interventions for Very Young Children with Mobility Limitations to Aid Participation and Positive Development: The Empower Evidence Synthesis. Health Technol. Assess. 2020, 24, 1–194. [Google Scholar] [CrossRef]
- Dunaway, S.; Montes, J.; O’Hagen, J.; Sproule, D.M.; Vivo, D.C.D.; Kaufmann, P. Independent Mobility after Early Introduction of a Power Wheelchair in Spinal Muscular Atrophy. J. Child Neurol. 2013, 28, 576–582. [Google Scholar] [CrossRef]


{kind=link}
| Ia | Ib | Ic | Total | |
|---|---|---|---|---|
| Number (%) | 16 (31.4) | 22 (43.1) | 13 (25.5) | 51 (100) |
| Sex, n (%) of children | ||||
| Male | 10 (62.5) | 12 (54.5) | 9 (69.2) | 31 (60.8) |
| Age of symptom onset, months, median (IR) † | 0.2 (0.71) | 2 (1.5) | 4 (1) | 2 (2.64) |
| Age at beginning of treatment, months, median (IR) † | 2 (3.35) | 4 (2.48) | 7 (8.25) | 4.5 (4) |
| Age at time of assessment, years, median (IR) | ||||
| 3.6 (2.69) | 3.2 (3.17) | 3.8 (2.99) | 3.6 (2.6) | |
| SMN2 Copies, n (%) | ||||
| 1 | 0 | 1 (4.5) | 0 | 1 (2) |
| 2 | 15 (93.8) | 21 (95.5) | 9 (69.2) | 45 (88.2) |
| 3 | 0 | 0 | 2 (15.4) | 2 (3.9) |
| Unknown | 1 (6.3) | 0 | 2 (15.4) | 3 (5.9) |
| Treatment, n (%) | ||||
| Nusinersen | 9 (56.3) | 20 (90.9) | 10 (76.9) | 39 (76.5) |
| Risdiplam | 1 (6.3) | 0 | 1 (7.7) | 2 (3.9) |
| Onasemnogene abeparvovec | 1 (6.3) | 0 | 1 (7.7) | 2 (3.9) |
| Onasemnogene abeparvovec + Nusinersen | 5 (31.3) | 2 (9.1) | 1 (7.7) | 8 (15.7) |
| Ia | Ib | Ic | Total | |
|---|---|---|---|---|
| Tracheostomy, n (%) | 5 (31.3) | 4 (18.2) | 2 (15.4) | 11 (21.6) |
| Ventilation ≥ 16 h/día, n (%) | 4 (25%) | 0 | 1 (7.69) | 5 (9.8) |
| Feeding, n (%) | ||||
| Mouth | 8 (50) | 13 (59.1) | 8 (61.5) | 29 (56.9) |
| Feeding tube/Button | 6 (37.5) | 8 (36.4) | 3 (23.1) | 17 (33.3) |
| Mixt | 2 (12.5) | 1 (4.5) | 2 (15.4) | 5 (9.8) |
| Orthopedic disorders | ||||
| Scoliosis n (%) | 14 (87.5) | 12 (54.5) | 8 (61.5) | 34 (66.7) |
| Degrees of Scoliosis, median (IR) | 40 (38) | 30 (12) | 33.5 (69) | 30 (23) |
| (n) | (n = 12) | (n = 11) | (n = 6) | (n = 29) |
| Hip subluxation/dislocation n (%) | 11 (68.8) | 14 (63.6) | 10 (76.9) | 35 (68.6) |
| Migration left hip, median (IR) | 72 (70) | 52.5 (80) | 34 (75) | 50 (72) |
| (n) | (n = 10) | (n = 8) | (n = 6) | (n = 24) |
| Migration right hip, median (IR) | 68 (71) | 52.5 (79) | 54.5 (63) | 52.5 (72) |
| (n) | (n = 10) | (n = 8) | (n = 6) | (n = 24) |
| Ia | Ib | Ic | Total | |
|---|---|---|---|---|
| Head control, n (%) | 10 (62.5) | 18 (81.8) | 11 (84.6) | 39 (76.5) |
| Independent sitting, n (%) | 10 (62.5) | 14 (63.6) | 10 (76.9) | 34 (66.7) |
| Standing with support, n (%) | 5 (31.3) | 11 (50) | 2 (15.4) | 18 (35.3) |
| Standing without support, n (%) | 1 (6.3) | 3 (13.6) | 0 | 4 (7.8) |
| Walking with support, n (%) | 3 (18.8) | 8 (36.4) | 1 (7.7) | 12 (23.5) |
| Walking unsupported, n (%) | 1 (6.3) | 0 | 0 | 1 (2) |
| Ia | Ib | Ic | Total | |
|---|---|---|---|---|
| CHOP-INTEND, median (IR) | 47 (16) | 51 (17) | 48 (12) | 48 (16) |
| HINE-2, median (IR) | 16 (14) | 17.5 (11) | 15 (9) | 16 (13) |
| Hammersmith, median (IR) (n) | 19 (22) (n = 13) | 22 (20) (n = 16) | 11.5 (18) (n = 10) | 19 (17) (n = 39) |
| RULM, median (IR) (n) | 19 (19) (n = 8) | 16 (8) (n = 13) | 15.5 (19) (n = 6) | 16 (8) (n = 27) |
| Physiotherapy | Speech Therapy | Occupational Therapy | Psychology | |
|---|---|---|---|---|
| n (%) | 51 (100) | 45 (88.2) | 13 (25.5) | 8 (15.7) |
| Frequency (h/s), median (IR) | 2.5 (1.5) | 1.5 (1.23) | 0.75 (0.5) | 0.75 (0.19) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de-Andrés-Beltrán, B.; Güeita-Rodríguez, J.; Palacios-Ceña, D.; Rodríguez-Fernández, Á.L. Clinical and Functional Characteristics of a New Phenotype of SMA Type I among a National Sample of Spanish Children: A Cross-Sectional Study. Children 2023, 10, 892. https://doi.org/10.3390/children10050892
de-Andrés-Beltrán B, Güeita-Rodríguez J, Palacios-Ceña D, Rodríguez-Fernández ÁL. Clinical and Functional Characteristics of a New Phenotype of SMA Type I among a National Sample of Spanish Children: A Cross-Sectional Study. Children. 2023; 10(5):892. https://doi.org/10.3390/children10050892
Chicago/Turabian Stylede-Andrés-Beltrán, Beatriz, Javier Güeita-Rodríguez, Domingo Palacios-Ceña, and Ángel Luis Rodríguez-Fernández. 2023. "Clinical and Functional Characteristics of a New Phenotype of SMA Type I among a National Sample of Spanish Children: A Cross-Sectional Study" Children 10, no. 5: 892. https://doi.org/10.3390/children10050892