
Evaluating Dysfunction in Fever-Induced Paroxysmal Weakness and Encephalopathy
 , ,
, ,
Abstract
:1. Introduction
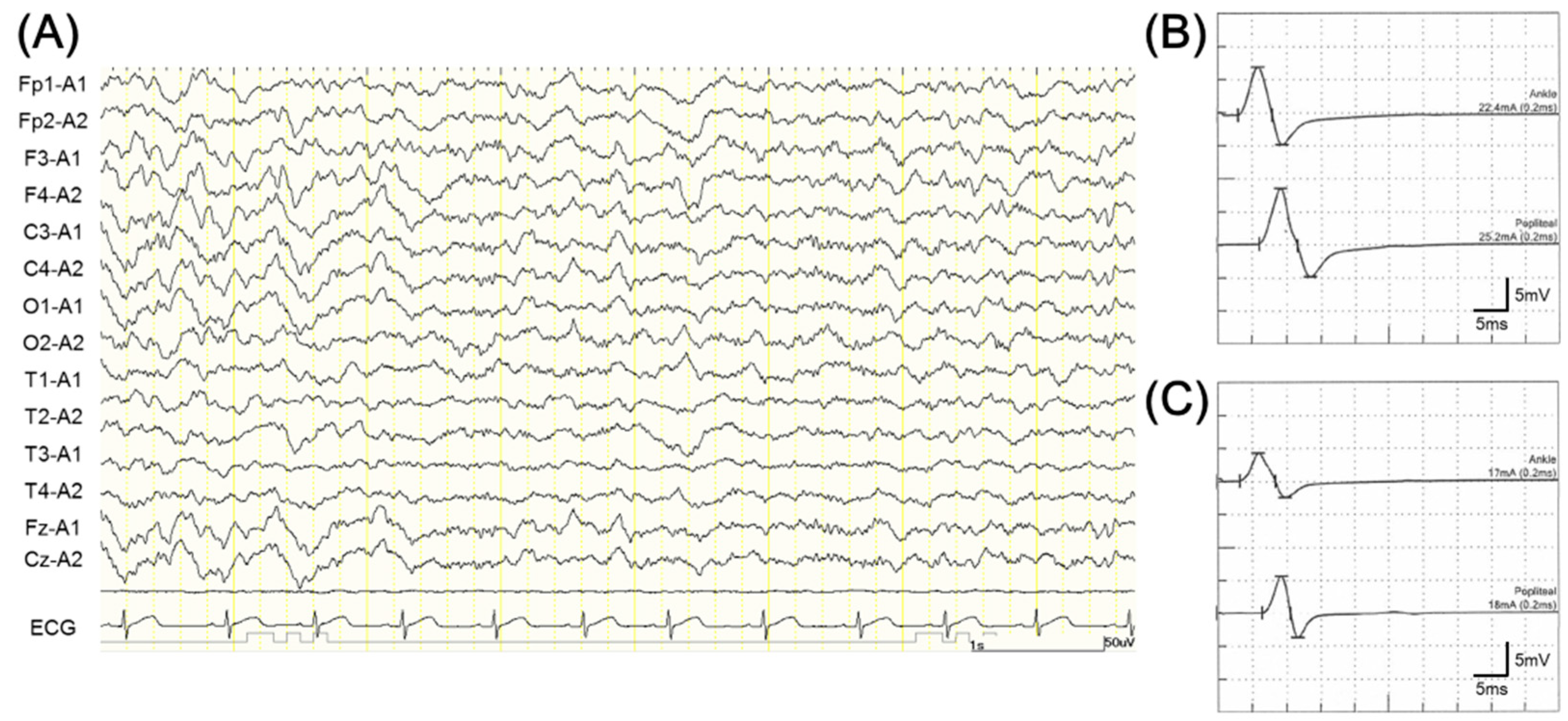
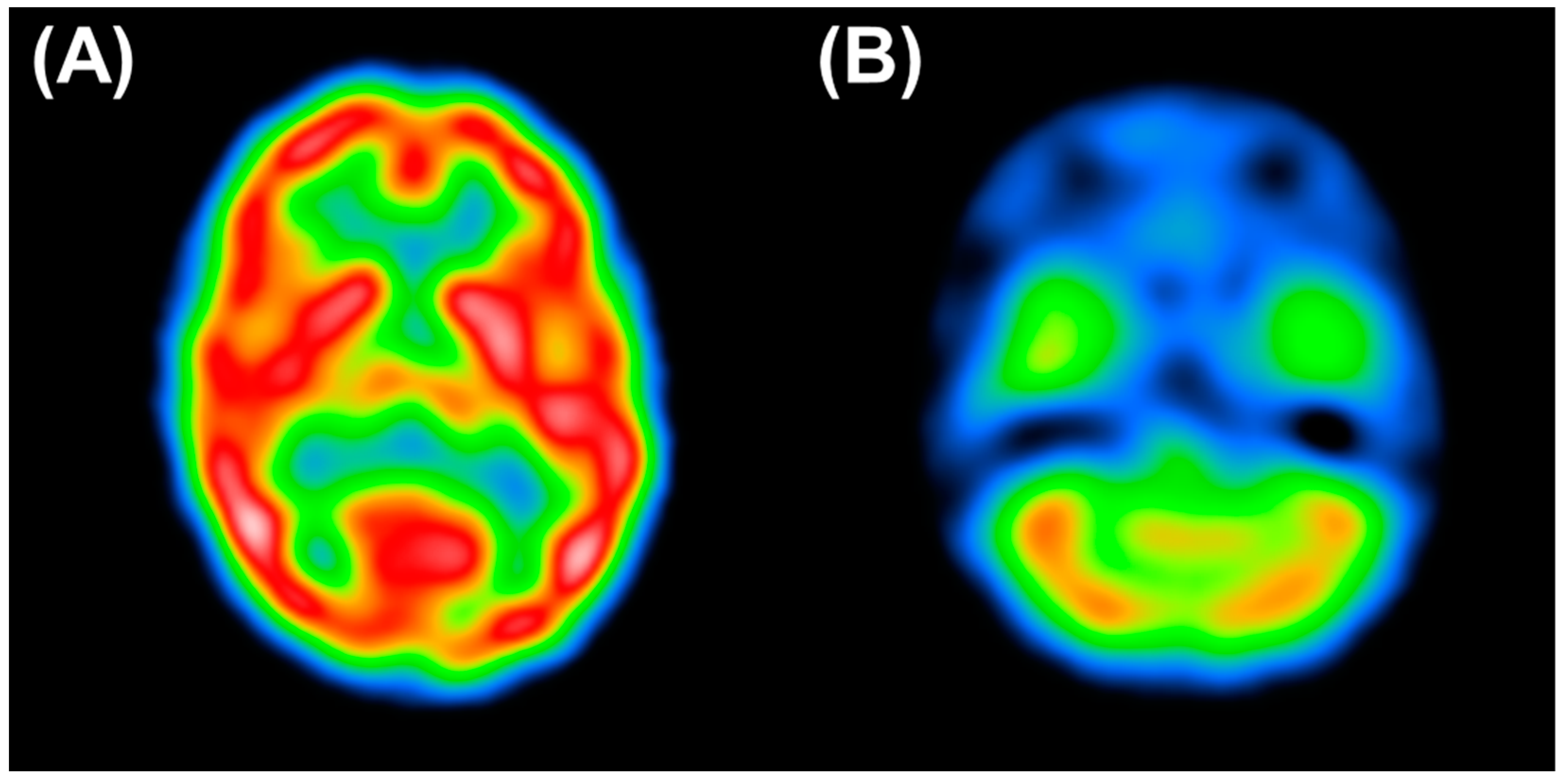
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carecchio, M.; Zorzi, G.; Ragona, F.; Zibordi, F.; Nardocci, N. ATP1A3-Related Disorders: An Update. Eur. J. Paediatr. Neurol. 2018, 22, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.T.; Silver, K.; Young, R.; DeBrosse, S.D.; Ebel, R.S.; Swoboda, K.J.; Acsadi, G. Fever-Induced Paroxysmal Weakness and Encephalopathy, a New Phenotype of ATP1A3 Mutation. Pediatr. Neurol. 2017, 73, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Dard, R.; Mignot, C.; Durr, A.; Lesca, G.; Sanlaville, D.; Roze, E.; Mochel, F. Relapsing Encephalopathy with Cerebellar Ataxia Related to an ATP1A3 Mutation. Dev. Med. Child Neurol. 2015, 57, 1183–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brashear, A.; Mink, J.W.; Hill, D.F.; Boggs, N.; McCall, W.V.; Stacy, M.A.; Snively, B.; Light, L.S.; Sweadner, K.J.; Ozelius, L.J.; et al. ATP1A3 Mutations in Infants: A New Rapid-Onset Dystonia-Parkinsonism Phenotype Characterized by Motor Delay and Ataxia. Dev. Med. Child Neurol. 2012, 54, 1065–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornarino, S.; Stagnaro, M.; Rinelli, M.; Tiziano, D.; Mancardi, M.M.; Traverso, M.; Veneselli, E.; De Grandis, E. Paroxysmal Features Responding to Flunarizine in a Child with Rapid-Onset Dystonia-Parkinsonism. Neurology 2014, 82, 2037–2038. [Google Scholar] [CrossRef] [PubMed]
- Kanemasa, H.; Fukai, R.; Sakai, Y.; Torio, M.; Miyake, N.; Lee, S.; Ono, H.; Akamine, S.; Nishiyama, K.; Sanefuji, M.; et al. De Novo p.Arg756Cys Mutation of ATP1A3 Causes an Atypical Form of Alternating Hemiplegia of Childhood with Prolonged Paralysis and Choreoathetosis. BMC Neurol. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicita, F.; Travaglini, L.; Sabatini, S.; Garavaglia, B.; Panteghini, C.; Valeriani, M.; Bertini, E.; Nardocci, N.; Vigevano, F.; Capuano, A. Childhood-Onset ATP1A3-Related Conditions: Report of Two New Cases of Phenotypic Spectrum. Park. Relat. Disord. 2016, 30, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Hattori, A.; Nakashima, M.; Ieda, D.; Hori, I.; Negishi, Y.; Ando, N.; Matsumoto, N.; Saitoh, S. A De Novo p.Arg756Cys Mutation in ATP1A3 Causes a Distinct Phenotype with Prolonged Weakness and Encephalopathy Triggered by Fever. Brain Dev. 2018, 40, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Sabouraud, P.; Riquet, A.; Spitz, M.A.; Deiva, K.; Nevsimalova, S.; Mignot, C.; Lesca, G.; Bednarek, N.; Doummar, D.; Pietrement, C.; et al. Relapsing Encephalopathy with Cerebellar Ataxia Are Caused by Variants Involving p.Arg756 in ATP1A3. Eur. J. Paediatr. Neurol. 2019, 23, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Dobretsov, M.; Hayar, A.; Kockara, N.T.; Kozhemyakin, M.; Light, K.E.; Patyal, P.; Pierce, D.R.; Wight, P.A. A Transgenic Mouse Model to Selectively Identify α3 Na,K-ATPase Expressing Cells in the Nervous System. Neuroscience 2019, 398, 274–294. [Google Scholar] [CrossRef] [PubMed]
- Biela, M.; Rydzanicz, M.; Szymanska, K.; Pieniawska-Smiech, K.; Lewandowicz-Uszynska, A.; Chruszcz, J.; Benben, L.; Kuzior-Plawiak, M.; Szyld, P.; Jakubiak, A.; et al. Variants of ATP1A3 in Residue 756 Cause a Separate Phenotype of Relapsing Encephalopathy with Cerebellar Ataxia (RECA)-Report of Two Cases and Literature Review. Mol. Genet. Genom. Med. 2021, 9, e1772. [Google Scholar] [CrossRef] [PubMed]
- Hully, M.; Ropars, J.; Hubert, L.; Boddaert, N.; Rio, N.; Bernadelli, M.; Desguerre, I.; Cornmier, D.; Aire, V.; Munnich, A.; et al. Mosaicism in ATP1A3-related disorders: Not just a theoretical risk. Neurogenetics 2017, 18, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.H.; Ozelius, L.J.; Brashear, A.; Lang, A.E.; Ahmad-Annuar, A.; Tan, C.T.; Lim, S.Y. Rapid-Onset Dystonia-Parkinsonism in a Chinese Girl with a De Novo ATP1A3 c.2267G>A (p.R756H) Genetic Mutation. Mov. Disord. Clin. Pract. 2015, 2, 74–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, A.L.; Alonso, I.; Magalhães, M. A Portuguese Rapid-Onset Dystonia-Parkinsonism Case with Atypical Features. Neurol. Sci. 2017, 38, 1713–1714. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, F.; Fawcett, K.; Sims, D.; Heger, A.; Houlden, H.; Hanna, M.G.; Kingston, H.; Sisodiya, S.M. Familial Childhood-Onset Progressive Cerebellar Syndrome Associated with the ATP1A3 Mutation. Neurol. Genet. 2017, 3, e145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirinzi, T.; Graziola, F.; Nicita, F.; Travaglini, L.; Stregapede, F.; Valeriani, M.; Curatolo, P.; Bertini, E.; Vigevano, F.; Capuano, A. Childhood Rapid-Onset Ataxia: Expanding the Phenotypic Spectrum of ATP1A3 Mutations. Cerebellum 2018, 17, 489–493. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J.; van de Laar, I.M.B.H.; de Rijk-van Andel, J.F.; Kamsteeg, E.J.; Kotsopoulos, I.A.W.; de Man, S.A. Expanding Phenotype of ATP1A3-Related Disorders: A Case Series. Child Neurol. Open 2021, 8, 2329048X211048068. [Google Scholar] [CrossRef] [PubMed]
- Zanotti-Fregonara, P.; Vidailhet, M.; Kas, A.; Ozelius, L.J.; Clot, F.; Hindié, E.; Ravasi, L.; Devaux, J.Y.; Roze, E. [123I]-FP-CIT and [99mTc]-HMPAO Single Photon Emission Computed Tomography in a New Sporadic Case of Rapid-Onset Dystonia-Parkinsonism. J. Neurol. Sci. 2008, 273, 148–151. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Reference | Present Case | [4] | [5] | [3] | [13] | [6] | [7] | [2] | [12] |
|---|---|---|---|---|---|---|---|---|---|
| Publication year | 2012 | 2014 | 2015 | 2015 | 2016 | 2016 | 2017 | 2017 | |
| Number of patients | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 5 | 2 |
| Mutation | R756C | R756H | R756H | R756C | R756H | R756C | R756C | R756H 3/5 R756L 2/5 | R756C |
| Age at onset | 1y7m | 9m | 9m | 1y1m | 10y | 1y5m | 11m | 13m–3y | 9m–1y10m |
| Symptoms | |||||||||
| Unresponsive | + | Anarthria | Anarthria | + | - | + | + | 5/5 | 1/2 |
| Hypotonia | + | + | + | + | - | + | + | 5/5 | 2/2 |
| Mutism | + | + | - | - | - | - | - | 0/5 | 0/2 |
| Dystonia | + | + | + | + | + | + | + | 3/5 | 2/2 |
| Ataxia | - | + | - | + | + | - | + | 5/5 | 2/2 |
| Electrophysiological examination | |||||||||
| EEG | Normal | NA | Normal | Normal | Normal | Normal | Normal | Normal 1/4 Slow wave 3/4 | NA |
| NCS | Normal | Normal | Normal | NA | NA | NA | NA | Normal 3/4 Absent F wave 1/4 | NA |
| ABR | Normal | NA | Normal | Normal | NA | NA | NA | NA | NA |
| VEP | NA | NA | NA | Normal | NA | NA | NA | NA | NA |
| SEP | NA | NA | Normal | Normal | NA | NA | NA | NA | NA |
| Reference | [14] | [15] | [16] | [8] | [9] | [11] | [17] | ||
| Publication year | 2017 | 2017 | 2018 | 2018 | 2019 | 2021 | 2021 | ||
| Number of patients | 1 | 3 | 2 | 1 | 3 | 2 | 2 | ||
| Mutation | R756H | R756H | R756C | R756C | R756H 2/3 R756C 1/3 | R756H | R756C | ||
| Age at onset | 1y2m | 8m–5y | 1y6m–1y7m | 9m | 1y5m–5y6m | 1y3m–1y7m | |||
| Symptoms | |||||||||
| Unresponsive | + | 0/3 | 1/2 | + | 1/3 | 2/2 | 0/2 | ||
| Hypotonia | + | 3/3 | 2/2 | + | 2/3 | 2/2 | 2/2 | ||
| Mutism | - | 0/3 | 0/2 | - | 3/3 | 1/2 | 0/2 | ||
| Dystonia | + | 2/3 | 1/2 | + | 1/3 | 0/2 | 1/2 | ||
| Ataxia | + | 3/3 | 2/2 | + | 2/3 | 2/2 | 2/2 | ||
| Electrophysiological examination | |||||||||
| EEG | generalized epileptic discharges | NA | NA | Normal | Normal 3/3 | NA | Normal 1/2 | ||
| NCS | NA | NA | Normal 1/2 | NA | NA | NA | NA | ||
| ABR | NA | NA | NA | NA | NA | NA | NA | ||
| VEP | NA | NA | NA | NA | NA | NA | NA | ||
| SEP | NA | NA | Normal 1/2 | NA | NA | NA | NA | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sano, F.; Fukao, T.; Yagasaki, H.; Kanemura, H.; Inukai, T.; Kaga, Y.; Nakane, T. Evaluating Dysfunction in Fever-Induced Paroxysmal Weakness and Encephalopathy. Children 2023, 10, 703. https://doi.org/10.3390/children10040703
Sano F, Fukao T, Yagasaki H, Kanemura H, Inukai T, Kaga Y, Nakane T. Evaluating Dysfunction in Fever-Induced Paroxysmal Weakness and Encephalopathy. Children. 2023; 10(4):703. https://doi.org/10.3390/children10040703
Chicago/Turabian StyleSano, Fumikazu, Toshimichi Fukao, Hideaki Yagasaki, Hideaki Kanemura, Takeshi Inukai, Yoshimi Kaga, and Takaya Nakane. 2023. "Evaluating Dysfunction in Fever-Induced Paroxysmal Weakness and Encephalopathy" Children 10, no. 4: 703. https://doi.org/10.3390/children10040703