
Mucopolysaccharidosis Type I in Mexico: Case-Based Review
 ,
,
Abstract
:1. Introduction
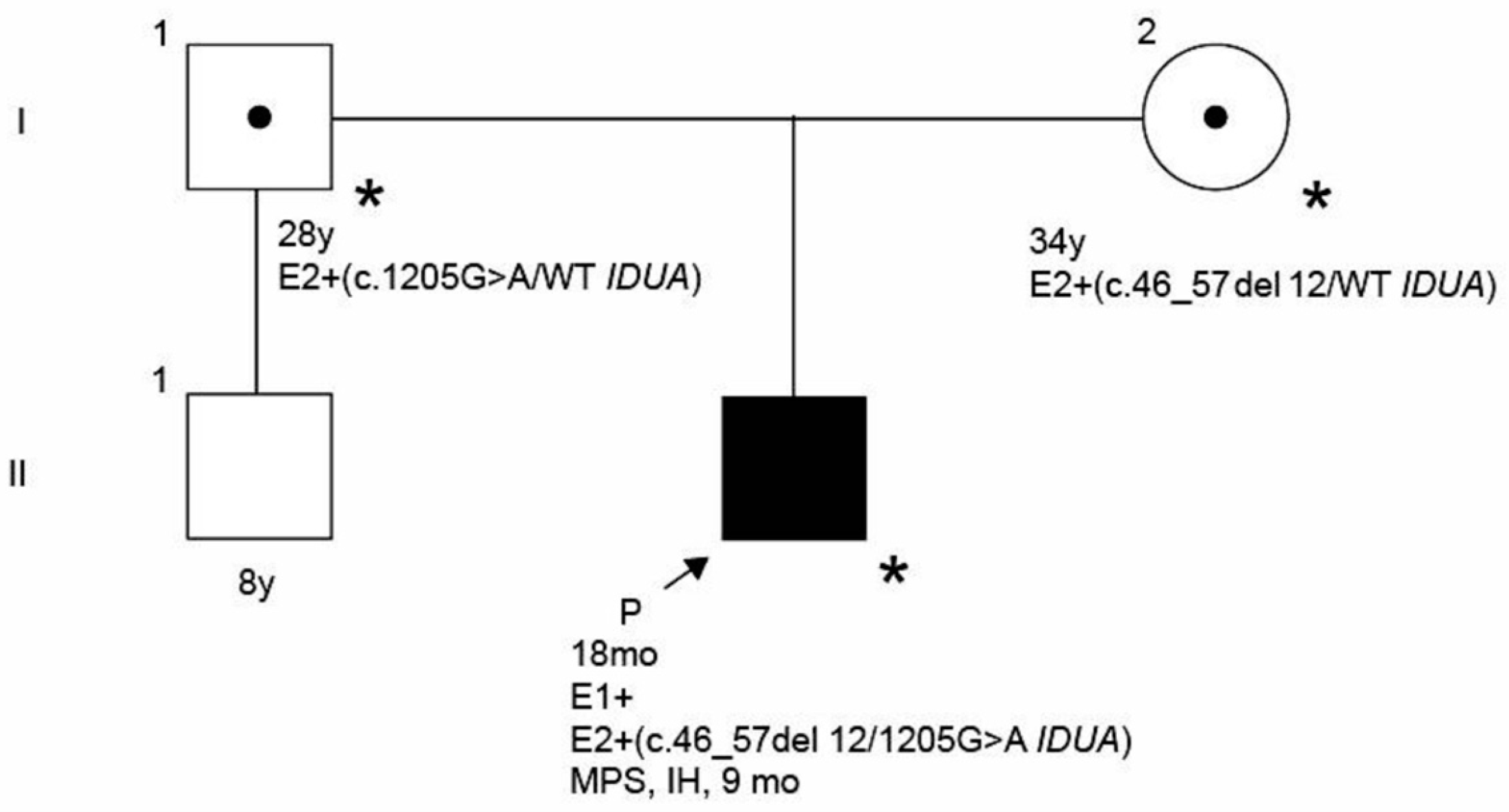
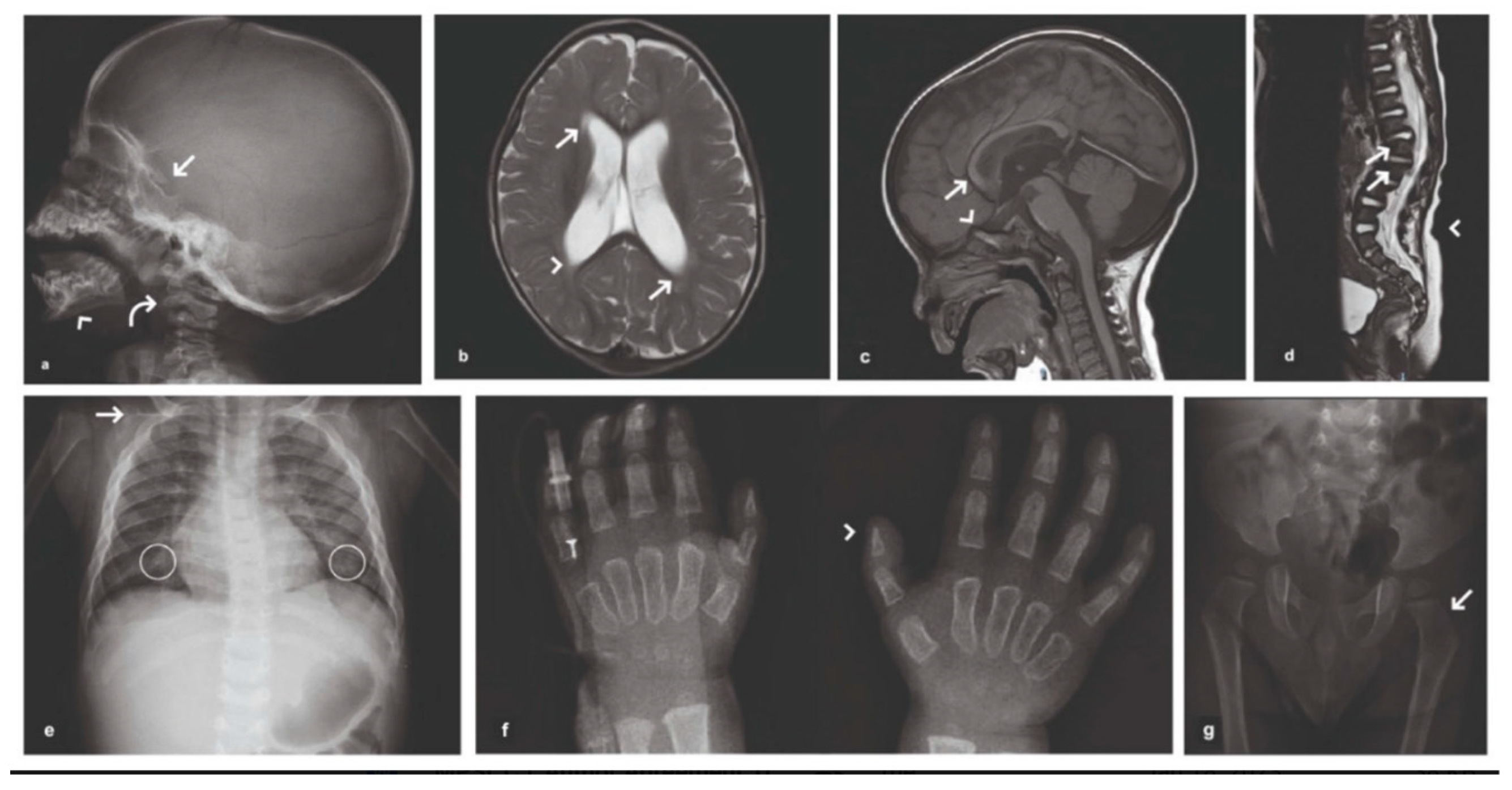
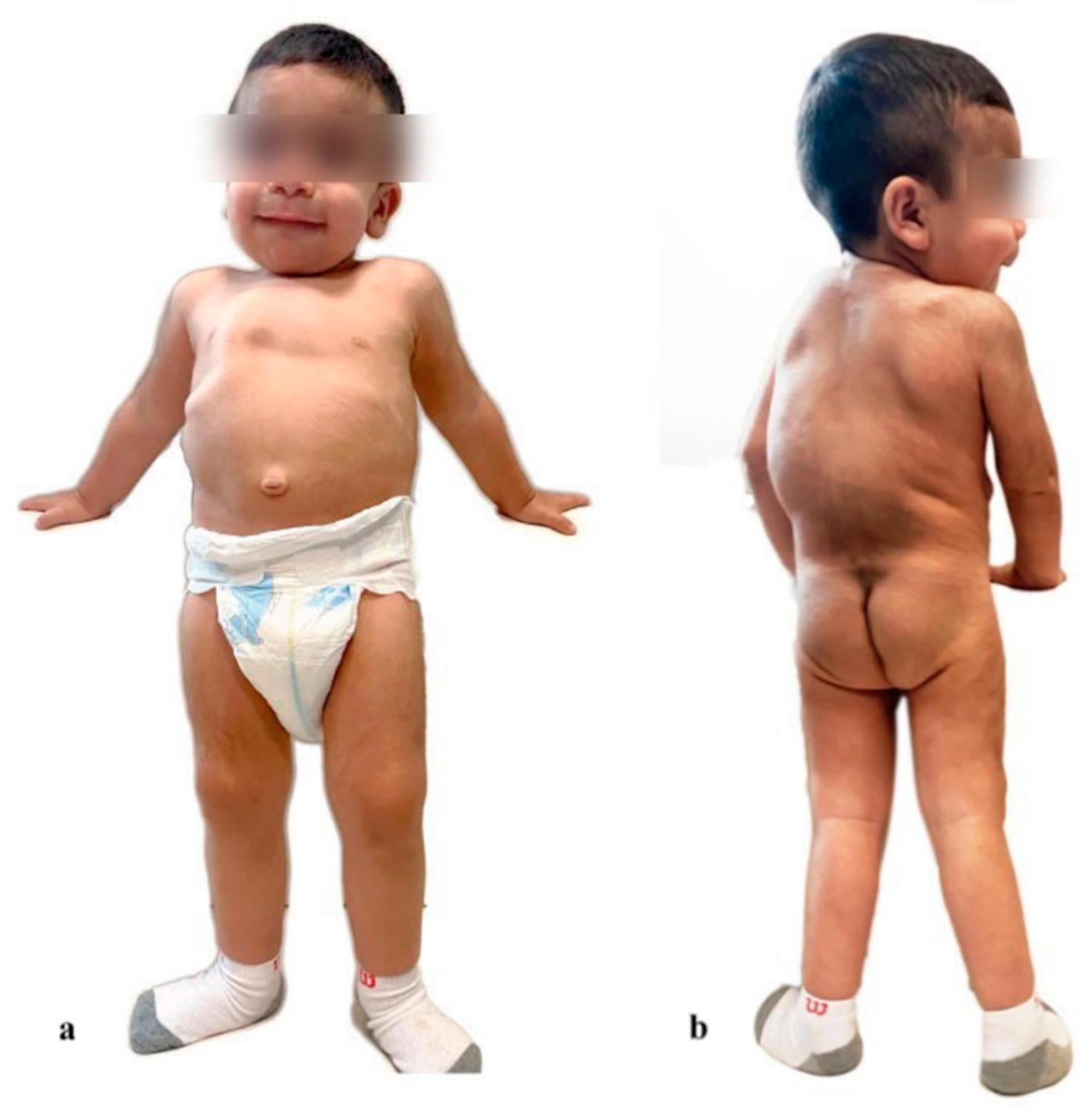
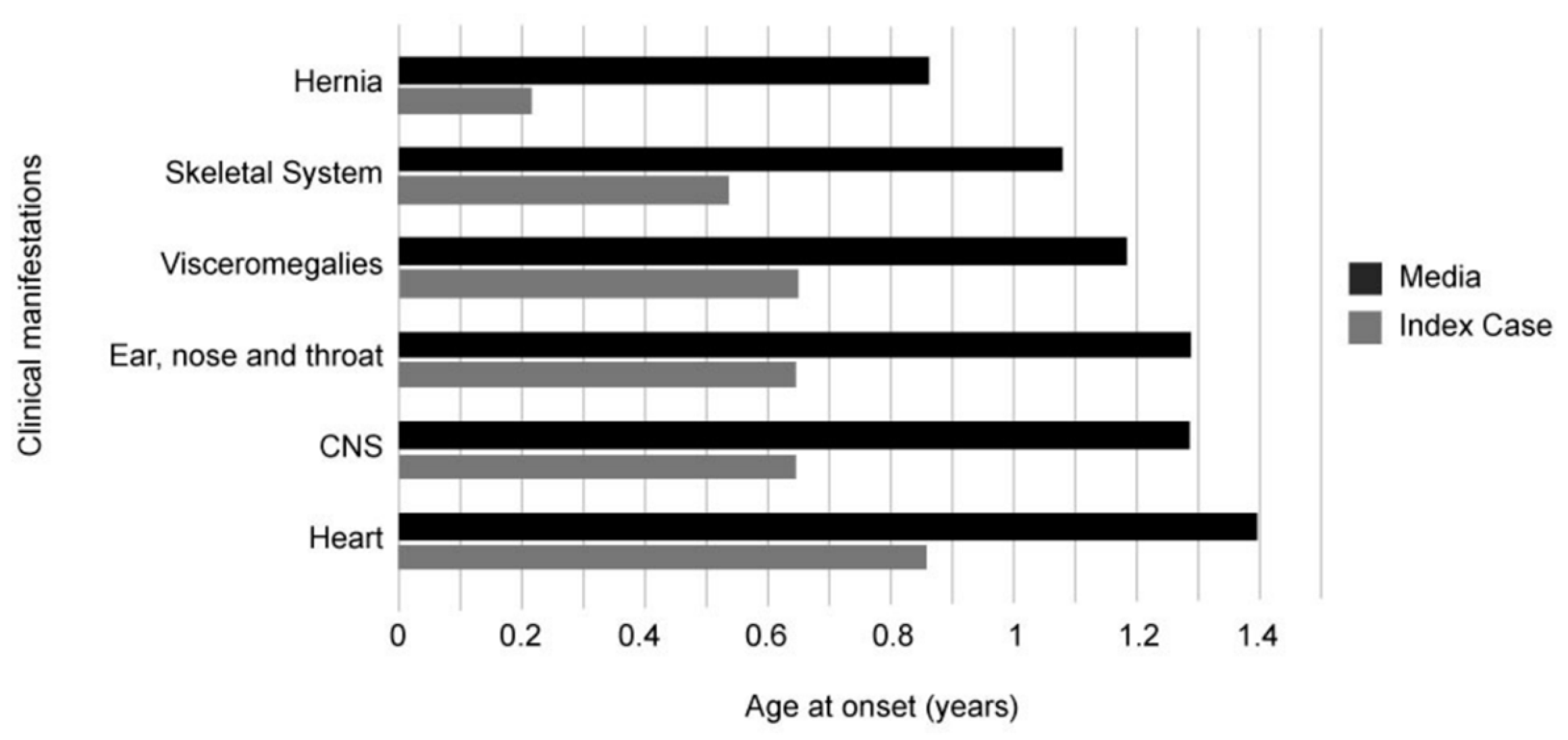
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, L.A.; Atherton, A.M.; Burton, B.K.; Day-Salvatore, D.L.; Kaplan, P.; Leslie, N.D.; Scott, C.R.; Stockton, D.W.; Thomas, J.A.; Muenzer, J. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J. Pediatr. 2017, 182, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, A.; D’Avanzo, F.; Rigon, L.; Rampazzo, A.; Concolino, D.; Barone, R.; Volpi, N.; Santoro, L.; Lualdi, S.; Bertola, F.; et al. Molecular diagnosis of patients affected by mucopolysaccharidosis: A multicenter study. Eur. J. Pediatr. 2019, 178, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Ruvalcaba, S.D.C.; Brambila-Tapia, A.J.L.; Juárez-Osuna, J.A.; Da Silva-José, T.D.; García-Ortiz, J.E. Biochemical diagnosis of mucopolysaccharidosis in a Mexican reference center. Genet. Mol. Biol. 2020, 43, e20180347. [Google Scholar] [CrossRef]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Anesth. Analg. 2014, 16, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide distribution of common IDUA pathogenic variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef]
- Pollard, L.M.; Jones, J.R.; Wood, T.C. Molecular characterization of 355 mucopolysaccharidosis patients reveals 104 novel mutations. J. Inherit. Metab. Dis. 2013, 36, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis. 2017, 12, 112. [Google Scholar] [CrossRef]
- Dornelles, A.D.; Artigalás, O.; da Silva, A.A.; Ardila, D.L.V.; Alegra, T.; Pereira, T.V.; Vairo, F.P.E.; Schwartz, I.V.D. Efficacy and safety of intravenous laronidase for mucopolysaccharidosis type I: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0184065. [Google Scholar] [CrossRef] [Green Version]
- Terlato, N.J.; Cox, G.F. Can mucopolysaccharidosis type I disease severity be predicted based on a patient’s genotype? A comprehensive review of the literature. Anesth. Analg. 2003, 5, 286–294. [Google Scholar] [CrossRef]
- Clarke, L.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Keenan, H.A.; Munoz-Rojas, M.V.; Okuyama, T.; Viskochil, D.; Whitley, C.B.; Wijburg, F.A.; et al. Genotype-phenotype relationships in mucopolysaccharidosis type I (MPS I): Insights from the International MPS I Registry. Clin. Genet. 2019, 96, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Meaney, C.A.; Greenland, G.; Adams, V.; Vellodi, A.; Young, E.P.; Winchester, B.G.; Beesley, C.E. Mutational analysis of 85 mucopolysaccharidosis type I families: Frequency of known mutations, identification of 17 novel mutations and in vitro expression of missense mutations. Hum. Genet. 2001, 109, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Reichert, R.; Campos, L.G.; Vairo, F.; de Souza, C.F.M.; Pérez, J.A.; Duarte, J.; Leiria, F.A.; Anés, M.; Vedolin, L.M. Neuroimaging Findings in Patients with Mucopolysaccharidosis: What You Really Need to Know. Radiographics 2016, 36, 1448–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmucci, S.; Attinà, G.; Lanza, M.L.; Belfiore, G.; Cappello, G.; Foti, P.V.; Milone, P.; Di Bella, D.; Barone, R.; Fiumara, A.; et al. Imaging findings of mucopolysaccharidoses: A pictorial review. Insights Imaging 2013, 4, 443–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Rojas, M.V.; Bay, L.; Sanchez, L.; Van Kuijck, M.; Ospina, S.; Cabello, J.F.; Martins, A.M. Clinical manifestations and treatment of mucopolysaccharidosis type I patients in Latin America as compared with the rest of the world. J. Inherit. Metab. Dis. 2011, 34, 1029–1037. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.M.; Dualibi, A.P.; Norato, D.; Takata, E.T.; Santos, E.S.; Valadares, E.R.; Porta, G.; de Luca, G.; Moreira, G.A.; Pimentel, H.; et al. Guidelines for the Management of Mucopolysaccharidosis Type I. J. Pediatr. 2009, 155, S32–S46. [Google Scholar] [CrossRef]
- Visigalli, I.; Delai, S.; Politi, L.S.; Di Domenico, C.; Cerri, F.; Mrak, E.; D'Isa, R.; Ungaro, D.; Stok, M.; Sanvito, F.; et al. Gene therapy augments the efficacy of hematopoietic cell transplantation and fully corrects mucopolysaccharidosis type I phenotype in the mouse model. Blood 2010, 116, 5130–5139. [Google Scholar] [CrossRef] [Green Version]
- Baxter, M.A.; Wynn, R.; Schyma, L.; Holmes, D.K.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. Marrow stromal cells from patients affected by MPS I differentially support haematopoietic progenitor cell development. J. Inherit. Metab. Dis. 2005, 28, 1045–1053. [Google Scholar] [CrossRef]
- De Ru, M.H.; Boelens, J.J.; Das, A.M.; Jones, S.A.; van der Lee, J.H.; Mahlaoui, N.; Mengel, E.; Offringa, M.; O’Meara, A.; Parini, R.; et al. Enzyme replacement therapy and/or hematopoietic stem cell transplantation at diagnosis in patients with mucopolysaccharidosis type I: Results of a European consensus procedure. Orphanet J. Rare Dis. 2011, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Grewal, S.; Krivit, W.; Defor, T.; Shapiro, E.; Orchard, P.; Abel, S.; Lockman, L.; Ziegler, R.; Dusenbery, K.; Peters, C. Outcome of second hematopoietic cell transplantation in Hurler syndrome. Bone Marrow Transpl. 2002, 29, 491–496. [Google Scholar] [CrossRef] [Green Version]
- Tolar, J.; Grewal, S.S.; Bjoraker, K.J.; Whitley, C.B.; Shapiro, E.G.; Charnas, L.; Orchard, P.J. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transpl. 2008, 41, 531–535. [Google Scholar] [CrossRef]
- Grewal, S.S.; Wynn, R.; Abdenur, J.E.; Burton, B.K.; Gharib, M.; Haase, C.; Hayashi, R.J.; Shenoy, S.; Sillence, D.; Tiller, G.E.; et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Anesth. Analg. 2005, 7, 143–146. [Google Scholar] [CrossRef] [Green Version]
- Sánchez Suarez, A.L.; Sánchez Sánchez, L.M. Mutaciones genéticas y su relación con el fenotipo clínico en pacientes con mucopo-lisacaridosis de tipo I en el noreste de México. Gac. Médica México 2014, 150, 289–296. [Google Scholar]
- Bertola, F.; Filocamo, M.; Casati, G.; Mort, M.; Rosano, C.; Tylki-Szymanska, A.; Tüysüz, B.; Gabrielli, O.; Grossi, S.; Scarpa, M.; et al. IDUA mutational profiling of a cohort of 102 European patients with mucopolysaccharidosis type I: Identification and characterization of 35 novel α-L-iduronidase (IDUA) alleles. Hum. Mutat. 2011, 32, E2189–E2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton-Navarro, P.; Pérez-Campos, E.; Pina-Canseco, M.D.S.; Fenton-Navarro, B. Gaucher’s Disease and Hurler’s Syndrome in Two First Cousins. Int. J. Hum. Genet. 2017, 17, 109–117. [Google Scholar] [CrossRef]
- Navarrete-Martínez, J.I.; Limón-Rojas, A.E.; Gaytán-García, M.D.J.; Reyna-Figueroa, J.; Wakida-Kusunoki, G.; Delgado-Calvillo, M.D.R.; Cantú-Reyna, C.; Cruz-Camino, H.; Cervantes-Barragán, D.E. Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: Three-year findings from a screening program in a closed Mexican health system. Mol. Genet. Metab. 2017, 121, 16–21. [Google Scholar] [CrossRef]
- Peña-Gomar, I.; Jiménez-Mariscal, J.L.; Cerón, M.; Rosas-Trigueros, J.; Reyes-López, C.A. c.1898C>G/p.Ser633Trp Mutation in Alpha-l-Iduronidase: Clinical and Structural Implications. Protein J. 2021, 40, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A.; the International Consensus Panel on the Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: Management and Treatment Guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Paatient | Genotype | Protein Variant | Expected Phenotype | Observed Clinical Phenotype | Genotype– Phenotype Correlation |
|---|---|---|---|---|---|
| 1–3 [22] | c.539G>C/c.1205G>A | p.Trp180Ser/p.Trp402Ter | Various phenotypes [6,22] | Hurler–Scheie | + |
| 4 [22] | c.46_57del12/c.385+1G>C | p.Ser16_Ala19del/intronic variant | Hurler b | Hurler–Scheie | − |
| 5 [22] | c.1598C>G/c.1598C>G | p.Pro533Arg/p.Pro533Arg | Various phenotypes [1,23] | Hurler–Scheie | + |
| 6 [22] | c.385+1G>C/c.1598C>G | Intronic variant/p.Pro533Arg | Hurler b | Hurler | + |
| 7 [22] | c.1205G>A/c.1587_1588insGC | p.Trp402Ter/p.Leu530ArgfsX31 | Hurler b | Hurler | + |
| 8 [24] | c.46_57del12/c.46_57del12 | p.Ser16_Ala19del/p.Ser16_Ala19del | Hurler–Scheie [23] | Hurler | − |
| 9 [25] | c.965T>A/c.1861C>T | p.Val322Glu/p.Arg621Ter | Hurler b | Detected by NBS | NA |
| 10 [25] | c.701G>C/c.965 T>A | p.Ser234Thr/p.Val322Glu | Hurler–Scheie b | Detected by NBS | NA |
| 11 [26] | c.1898C>G/c.1898 C>G | p.Ser633Trp/p.Ser633Trp | Hurler–Scheie | Hurler | − |
| Index patient | c.46_57del12/c.1205G>A | p.Ser16_Ala19del/p.Trp402Ter | Hurler [1] | Hurler | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cantú-Reyna, C.; Vazquez-Cantu, D.L.; Cruz-Camino, H.; Narváez-Díaz, Y.A.; Flores-Caloca, Ó.; González-Llano, Ó.; Araiza-Lozano, C.; Gómez-Gutiérrez, R. Mucopolysaccharidosis Type I in Mexico: Case-Based Review. Children 2023, 10, 642. https://doi.org/10.3390/children10040642
Cantú-Reyna C, Vazquez-Cantu DL, Cruz-Camino H, Narváez-Díaz YA, Flores-Caloca Ó, González-Llano Ó, Araiza-Lozano C, Gómez-Gutiérrez R. Mucopolysaccharidosis Type I in Mexico: Case-Based Review. Children. 2023; 10(4):642. https://doi.org/10.3390/children10040642
Chicago/Turabian StyleCantú-Reyna, Consuelo, Diana Laura Vazquez-Cantu, Héctor Cruz-Camino, Yuriria Arlette Narváez-Díaz, Óscar Flores-Caloca, Óscar González-Llano, Carolina Araiza-Lozano, and René Gómez-Gutiérrez. 2023. "Mucopolysaccharidosis Type I in Mexico: Case-Based Review" Children 10, no. 4: 642. https://doi.org/10.3390/children10040642